

Abstract
Calcitonin, a peptide hormone associated with medullary carcinoma of the thyroid, has the potential to form amyloid fibrils and may be a valuable model for investigating the role of peptide–membrane interactions in β-sheet and amyloid formation. Via a new model peptide system, bovine calcitonin, we found that the exposure of peptide to phospholipid membranes altered its structure relative to the structures formed in aqueous solutions. Of particular relevance to the amyloidoses, incubation of calcitonin with cholesterol-rich and ganglioside-containing membranes resulted in significant enrichment in the β-sheet and amyloid content of the peptide. The formation of amyloid was also accelerated in these systems. A correlation between the phospholipid-induced structural alterations and calcitonin binding affinities to phospholipid membranes was evident. Bovine calcitonin has considerably higher binding affinity for the phospholipid systems that enhanced its β-sheet and amyloid structure. Electrostatic forces were not the governing forces behind the observed behavior, as supported by the fact that the ionic strength did not affect the peptide structures or binding affinities. A Van’t Hoff analysis of the temperature-dependent peptide binding affinities indicated that binding led to an increase in enthalpy and possibly an increase in entropy of the peptide–membrane systems. Experiments with other amyloid-forming peptides such as β-amyloid of Alzheimer’s disease have also shown similar results and may indicate the need to manipulate peptide–membrane interactions in order to control amyloid formation and its associated disease.
Keywords: secondary structure, amyloid, calcitonin, phospholipid membranes
The amyloidoses are complex, multiform disorders characterized by the polymerization and aggregation of normally innocuous and soluble proteins into extracellular insoluble fibrillar species. At least 16 biochemically unique proteins have been found as the fibrillar components of disease-associated amyloid deposits (Kelly 1996; Wetzel 1996; Lansbury 1999). While these amyloidogenic proteins have little sequence or structural homology, they all generate similar, straight, unbranched amyloid fibrils with prominent cross β-sheet structures and the abilities to bind Congo red dye (Lansbury 1999; Murphy 2002; Nilsson 2004).
Several lines of evidence suggest a strong link between amyloid fibril formation and disease pathology. The amyloid fibrils or protofibrils derived from aggregated Aβ peptides, β2-macroglobulin, and amylin have been shown to be neurotoxic in vitro (Walsh et al. 1997; Ward et al. 2000; Olofsson et al. 2002; Ono et al. 2002). Researchers have indicated that the neurotoxic effects of these proteins or peptides were minimized by compounds that bind to amyloid fibrils or inhibit fibril formation (Ono et al. 2002; Rijkers et al. 2002; Perez et al. 2003; Bodles et al. 2004; De Felice et al. 2004; Gilead and Gazit 2004; Porat et al. 2004).
A peptide hormone, calcitonin, has been reported to have several important normal functions in the body. These functions include regulation of calcium-phosphorus metabolism and potentially serving as a neuromodulator and/or a neurotransmitter in the central nervous system (Fischer et al. 1981; Rizzo and Goltzman 1981; Inzerillo et al. 2004; Uyama et al. 2004). The peptide structure associated with its normal hormone functions appears to be an amphipathic helix (Epand et al. 1986a, b; Siligardi et al. 1994; Motta et al. 1998). In vivo, calcitonin was able to form amyloid fibrils associated with medullary carcinoma of the thyroid (Sletten et al. 1976), which is composed of ~7%–10% of thyroid carcinomas (Ljungberg 1966; Williams et al. 1966; Fletcher 1970; Hill et al. 1973; Hazard 1977; Ponder 1984). Both intracellular and extracellular amyloid fibril deposits are related to these tumors (DeLellis et al. 1979; Dammrich et al. 1984; Butler and Khan 1986; Berger et al. 1988; Silver et al. 1988; Byard et al. 1990). In vitro, human calcitonin forms amyloid fibrils in physiological buffer conditions, limiting the efficacy of human calcitonin in the treatment of osteoporosis (Arvinte et al. 1993; Bauer et al. 1994; Kanaori and Nosaka 1995). In addition, evidence showed that the fibrils of human calcitonin or bovine calcitonin were toxic in cell culture (Schubert et al. 1995; Liu and Schubert 1997; Rymer and Good 2001).
The mechanism by which amyloids form in vivo has yet to be elucidated. A number of in vitro studies have shown that the peptide structure and amyloid formation were affected by pH, solvent, chaperones, and/or peptide concentration (Atwood et al. 1998; Kirkitadze et al. 2001; Chi et al. 2003; Ohnishi and Takano 2004). In addition, a growing number of observations with the best-characterized amyloid, Aβ of Alzheimer’s disease, suggest the critical role of peptide–membrane interactions in aggregation and amyloid formation. Aβ has been observed to undergo a random-coil to β-sheet transition and accelerated amyloid formation upon binding to membranes of varying composition, with both hydrophobic and electrostatic interactions being implicated (McLaurin and Chakrabartty 1996; Choo-Smith et al. 1997; Terzi et al. 1997; Matsuzaki and Horikiri 1999). Results from our laboratory have suggested two membrane components, cholesterol and ganglioside, are of importance regarding the Aβ-induced neurotoxicity (Wang et al. 2001).
Up until now, the secondary and macromolecular structures of bovine calcitonin have not been explored. In this work, upon performing a careful biophysical characterization on bovine calcitonin, we first demonstrated that we were able to manipulate its secondary and macromolecular structures to produce amyloid and nonamyloid structures. Moreover, we utilized bovine calcitonin as a model peptide system with which to examine the role of phospholipid membranes on the structural transitions of amyloid-forming peptides and further elucidated the commonalities between this amyloid and the Aβ. With this new model system, we specifically addressed (1) the effect of phospholipid membranes of different compositions on the bovine calcitonin secondary structure and amyloid formation, (2) the affinity of bovine calcitonin for phospholipid membranes as a function of peptide structure and membrane composition, and (3) the nature of the forces involved in the peptide–membrane interaction. We found that membranes containing cholesterol and gangliosides promoted β-sheet and amyloid formation; that calcitonin had the highest binding affinities for these same membranes; that the peptide–membrane affinities were strongly correlated with the β-sheet contents of the calcitonin; and that entropic considerations, not electrostatic forces, probably governed the peptide–membrane interactions.
Results
The secondary solution structures of bovine calcitonin
As illustrated in Table 1, bovine calcitonin did not possess one dominant secondary structure in physiological buffers like PBS and phosphate buffer, but it contained approximately equal β-sheet and α-helix contents. Dissolution of calcitonin in acidic buffers such as acetate buffer, PBS containing TFA, or PBS containing acetonitrile did not significantly alter this secondary structure. However, dissolving bovine calcitonin in deionized water, 100% D2O, basic buffers such as borate buffer, and helix-promoting solvents such as TFE and HFIP produced predominantly α-helical structures. Also, suspending bovine calcitonin in unbuffered salt solutions of NaF, CaCl2, and MgCl2 yielded peptide solutions with enriched β-sheet content. In particular, a divalent salt solution composed of 0.005 M CaCl2 and 0.001 M MgCl2 in water induced calcitonin to adopt a structure with 55 ± 10% β-sheet and only 15 ± 10% α-helix content.
Table 1.
Estimates of the secondary structural components of bovine calcitonin under various solvation conditions as obtained from CD spectral analyses
| Solvent | % Random coil | % β-Turn | % β-Sheet | % β-Helix |
| Deionized water | 5 | 0 | 0 | 95 |
| Saline | 40 | 5 | 20 | 35 |
| Phosphate buffer (pH 7.4) | 40 | 10 | 30 | 20 |
| PBS (pH 7.4) | 30 | 20 | 30 | 20 |
| Phosphate buffer (pH 4.5) | 35 | 10 | 30 | 25 |
| Acetate buffer (pH 5.3) | 40 | 10 | 25 | 25 |
| Borate buffer (pH 8.5) | 5 | 0 | 0 | 95 |
| 0.001 M CaC12 in water | 45 | 0 | 25 | 30 |
| 0.005 M CaC12 and 0.001 M MgC12 in water | 25 | 5 | 55 | 15 |
| 2 M NaF in water | 35 | 5 | 25 | 40 |
| 100% D2O | 5 | 0 | 0 | 95 |
| 100% TFE | 5 | 0 | 0 | 95 |
| 100% HFIP | 5 | 0 | 0 | 95 |
| 0.1% TFA in PBS | 20 | 10 | 30 | 40 |
| 5% Acetonitrile in PBS | 35 | 10 | 25 | 30 |
The spectra were taken 2 h after sample preparation. The compositions of the solvents and buffers listed in the table are saline (0.15 M NaCl), phosphate buffer (0.01 M KH2PO4), PBS (0.1 M NaH2PO4, 0.14 M NaCl), acetate buffer (0.02 M C2H3O2Na, 0.15 M NaCl), borate buffer (0.05 M Na2B4O7•10 H2O), and divalent cations (0.005MCaCl2 and 0.001MMgCl2). The error associated with the structural estimates is taken to be ± 10%.
The influence of phospholipids on calcitonin secondary structure
As depicted in the CD spectra of Figure 1 ▶, all of the phospholipid environments influenced the bovine calcitonin secondary structure as compared with its structure in PBS. Figure 2 ▶ shows the change in β-sheet content of the peptide upon incubation with phospholipid vesicles of varying compositions. In the presence of DPPC vesicles only, calcitonin had essentially no β-sheet structure. When incubated with PBS or DOPS/DPPC vesicles, the β-sheet conformations of peptide solution were estimated to be ~30% and 5%, respectively. However, when exposed to cholesterol-rich DPPC vesicles, neuronal-like vesicles, or ganglioside-containing DPPC vesicles, calcitonin adopted structures with ~50%, 70%, and 80% β-sheet content, respectively. Varying the salt concentration (0.01–0.15 M NaCl) had no discernable effect on the calcitonin structure in any of the phospholipid environments.
Figure 1.
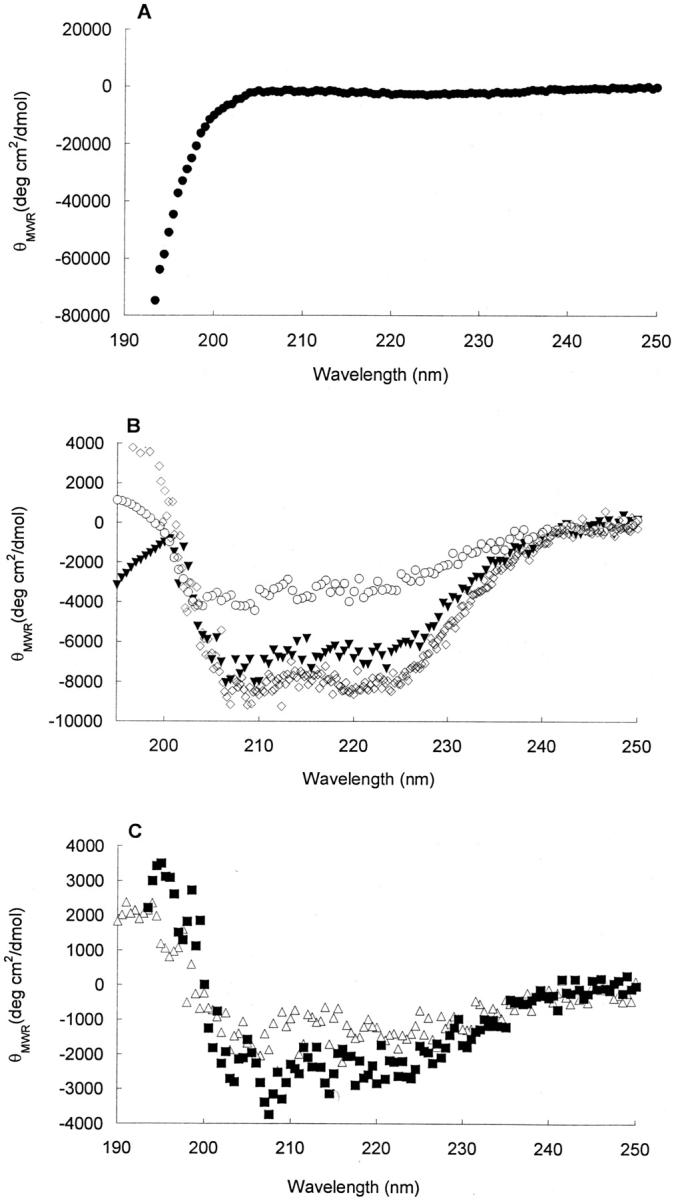
Representative CD spectra of bovine calcitonin incubated with different phospholipid membranes. For all of the spectra, the initial calcitonin solventwas PBS, and the incubation timewas 24 h. CD spectra of 80 μM bovine calcitonin were incubated with DPPC vesicles (circle) (A); DOPS/DPPC vesicles (triangle), PBS (open diamond), and cholesterol- rich DPPC vesicles (open circle) (B); and cholesterol-rich neuronal-like vesicles (square) and ganglioside-containing DPPC vesicles (open triangle) (C). The structural analysis of the peptide incubated with membranes is indicated by % α-helix, H; % β-sheet, S; % random-coil, R; and % β-turn, T: DPPC vesicles (0 H, 0 S, 50 R, 50 T), PBS (20 H, 30 S, 30 R, 20 T), DOPS/DPPC vesicles (45 H, 10 S, 20 R, 25 T), cholesterol-rich DPPC vesicles (10 H, 55 S, 20 R, 15 T), cholesterol-rich neuronal-like vesicles (5 H, 70 S, 20 R, 5 T), and ganglioside-containing DPPC vesicles (0 H, 80 S, 20 R, 0 T).
Figure 2.

The β-sheet contents of 80 μM bovine calcitonin in PBS incubated with different phospholipid membranes as estimated from CD spectra. The incubation time for all of the lipid samples was 24 h. The uncertainties associated with the structural determinations were estimated to be ± 10%.
The influence of phospholipids on the formation of calcitonin amyloid
Along with altering the calcitonin secondary structure, incubation with phospholipid membranes altered the extent of calcitonin amyloid formation. As illustrated by the Congo red difference spectra in Figure 3 ▶, the cholesterol-rich vesicles and the ganglioside-containing DPPC vesicles, which promoted β-sheet enrichment in the peptide, also induced significant amyloid formation. Exposure of the peptide to the DPPC, DOPS, and DOPS/DPPC vesicles that reduced the calcitonin β-sheet content relative to PBS resulted in solutions that did not bind Congo red and formed no detectable amyloid. The zero Congo red difference spectra are not shown.
Figure 3.

The amyloid content of the calcitonin-lipid suspensions after 24 h as indicated by Congo red binding. The absorbance difference spectra of bovine calcitonin incubated with the cholesterol-rich DPPC vesicles (open circle), the cholesterol-rich neuronal-like vesicles (square), and the ganglioside-containing DPPC vesicles (open triangle) relative to the spectrum of calcitonin dissolved in PBS alone (triangle).
The binding of bovine calcitonin to the phospholipid vesicles
As a complementary measure of the bovine calcitonin-phospholipid membrane interactions, equilibrium binding experiments with radioiodinated calcitonin were performed. The calcitonin secondary and tertiary structures were assumed to be represented by the CD and Congo red spectra taken under the sameconditions that the individual isotherms were measured. Spectral data from our laboratory indicated that the solution structures of calcitonin were not significantly altered by the iodination process (spectra not shown). As illustrated by the representative calcitonin binding isotherms in Figure 4 ▶, the isotherms were linear for the measured concentration ranges. Calcitonin had the highest binding affinity for the cholesterol-rich and ganglioside-containing membranes, the same membranes that strongly promoted β-sheet formation.
Figure 4.

Equilibrium binding of bovine calcitonin to phospholipid vesicles. The binding isotherms were obtained after incubating the vesicles with bovine calcitonin for 24 h. The initial calcitonin solvent was PBS. Representative equilibrium binding isotherms of 80 μM bovine calcitonin with DPPC vesicles (circle), DOPS/DPPC vesicles (triangle), cholesterol-rich DPPC vesicles (open circle), cholesterol-rich neuronal-like vesicles (square), and ganglioside-containing DPPC vesicles (open triangle).
The relationship between calcitonin β-sheet content and peptide–membrane binding affinity is further illustrated in Figure 5 ▶. More than a 10-fold difference in binding affinity was observed between the membrane systems that promoted calcitonin β-sheet formation and the membrane systems that inhibited β-sheet formation (p<0.01). The observed correlations between peptide structure and binding affinity were independent of salt concentration over the range of salt concentrations tested (0.01–0.15 M NaCl, p>0.3).
Figure 5.

The effect of calcitonin β-sheet content on the slopes of the equilibrium binding isotherms. The β-sheet contents of the lipid suspensions were determined by analyzing CD spectra. The initial calcitonin solvent in the lipid suspensions was PBS, and the binding curves were obtained after incubating the vesicles with calcitonin for 24 h. The binding isotherms contained a minimum of 10 data points, and their slopes were obtained by least-squares regressions. 1, DPPC vesicles; 2, DOPS/DPPC vesicles; 3, cholesterol-rich DPPC vesicles; 4, cholesterol-rich neuronal-like vesicles; and 5, ganglioside-containing DPPC vesicles.
The effect of initial calcitonin structure on binding
Using calcitonin dissolved in deionized water with an initial structure of almost 100% α-helix, we demonstrated that initial peptide structure did not determine the final peptide structure or binding affinity to cholesterol-rich neuronal-like vesicles. As seen in Figure 6 ▶, the calcitonin β-sheet content, Congo red binding, and binding affinity for the neuronal-like membranes increased with incubation time from negligible levels at 2 h to significant levels at 48 h (p<0.05).
Figure 6.

The influence of initial solvent on the calcitonin binding and conformational transitions. During these studies, the initial calcitonin solvent was deionized water. The calcitonin concentration was maintained at 80 μM. Peptide structure, α-helix (solid bars), and β-sheet (shaded bars) content was determined as a function of time incubated with cholesterol-rich neuronal-like membranes from analysis of CD spectra. Binding affinity (open bars) was determined from the slope of the binding isotherms taken as a function of incubation time: 2-h structure (95 H, 0 S, 5 R, 0 T), 24-h structure (20 H, 15 S, 40 R, 15 T), and 48-h structure (20 H, 55 S, 25 R, 0 T). At 48 h, the calcitonin suspension was also amyloidogenic (*, difference spectra not shown).
The effect of temperature on calcitonin binding
The binding affinities of bovine calcitonin to the DPPC vesicles, the cholesterol-rich DPPC vesicles, and the cholesterol-rich neuronal-like vesicles were examined as functions of temperature (Fig. 7 ▶). The calcitonin binding affinity increased with increasing temperature in all of the membrane systems (p<0.005). Van’t Hoff analyses of the temperature-dependent binding data indicated that the peptide–membrane bindings were endothermic interactions and that the enthalpy associated with binding was greater for the calcitonin-DPPC system than for either the calcitonin-cholesterol-rich DPPC system or the calcitonin-neuronal-like system (Table 2).
Figure 7.

The calcitonin binding affinities as a function of temperature. The initial calcitonin–lipid solvent was PBS. The binding isotherms were obtained after incubating the vesicles with calcitonin at the respective temperatures for 24 h. The calcitonin binding affinitie were obtained by least-squares regressions of the binding isotherms. The binding affinities for the DPPC vesicles (circle), the cholesterol-rich DPPC vesicles (open circle), and the cholesterol-rich neuronal-like vesicles (square).
Table 2.
A listing of all of the binding affinities of bovine calcitonin to the various phospholipid vesicles
| Lipid system | Temperature | Binding affinity (mmol bound calcitonin/mg lipid × M calcitonin) |
| DPPC (0.15 M NaCl) | 20°C | 0.018 ± 0.001 |
| DPPC (0.15 M NaCl) | 30°C | 0.242 ± 0.004 |
| DOPS (0.15 M NaCl) | 20°C | 0.124 ± 0.003 |
| DOPS/DPPC (0.15 M NaCl) | 20°C | 0.085 ± 0.002 |
| DPPC + cholesterol (0.15 M NaCl) | 4°C | 0.092 ± 0.002 |
| DPPC + cholesterol (0.15 M NaCl) | 20°C | 0.50 ± 0.01 |
| DPPC + cholesterol (0.15 M NaCl) | 30°C | 0.93 ± 0.03 |
| DPPC + ganglioside (0.15 M NaCl) | 20°C | 1.34 ± 0.04 |
| Neuronal-like (0.15 M NaCl) | 4°C | 0.148 ± 0.003 |
| Neuronal-like (0.15 M NaCl) | 20°C | 0.89 ± 0.02 |
| Neuronal-like (0.15 M NaCl) | 30°C | 1.59 ± 0.02 |
The initial calcitonin solvent in the lipid suspensions was PBS (0.15 M NaCl) or phosphate buffer (0.01 M NaCl), and the binding isotherms were obtained after incubating the vesicles with 80 μM calcitonin at the specified temperatures for 24 h. The binding isotherms contained a minimum of 10 data points, and their slopes (mmol bound calcitonin)/(mg lipid × M calcitonin in solution), which were taken as the binding affinities, were obtained by least-squares regressions.
Discussion
As of yet, no careful or thorough studies have been conducted to investigate the secondary and macromolecular structures of bovine calcitonin peptides and their link to the amyloid fibrils. In addition, we believe that bovine calcitonin, which is easily obtained and whose structure appears to be relatively easy to control, may prove to be a useful tool in elucidating the commonalities between this amyloid and Aβ. In order to demonstrate the usefulness of bovine calcitonin as a new model peptide for studying the process of amyloidoses, we first established the effects of solvent, salt concentration, and pH on the secondary structure of bovine calcitonin in solutions and determined the conditions essential to the production of amyloid and nonamyloid structures.
A series of evidence suggests that peptide–lipid interactions are important in the role of calcitonin as a hormone involved in the regulation of bone resorption and in the formation and biological activity of amyloids such as the calcitonin amyloids associated with medullary carcinoma of the thyroid. Therefore, we then examined how the peptide structure of bovine calcitonin was altered by incubation with phospholipid vesicles.
Several biophysical techniques including X-ray crystallography, nuclear magnetic resonance spectroscopy (NMR), Fourier transform infrared spectroscopy (FTIR), and CD spectroscopy have been suggested for examining the structural changes of proteins. Since amyloid fibrils are typically insoluble, heterogeneous, and noncrystalline, they are not amenable to conventional methods such as X-ray crystallography and solution NMR. Both FTIR and CD spectroscopies are nondestructive, requiring only a small amount of protein sample, and can accurately probe the relative changes resulting from the influence of the environment. However, FTIR has a few relatively stringent constraints on sample preparation: use of IR compatible ion-pairing agent, interference from residual water, and requirement of concentrated protein solution. As a result, CD spectroscopy is the most commonly used technique for deciphering the structural aspects of the amyloid formation mechanism (Thompson 2003; Nilsson 2004).
Our CD spectra indicate that bovine calcitonin did not possess one dominant secondary structure in physiological buffers like PBS, but contained approximately equal β-sheet and α-helix contents. However, exposing the peptide to helix-promoting solvents or incubating with divalent cation solutions shifted the peptide secondary structure to predominantly α-helix or β-sheet structures, respectively. The secondary solution structures of bovine calcitonin have not been previously reported by others; nevertheless, our structural data are similar to that reported for human calcitonin despite the appreciable sequence differences between the two species (Epand et al. 1983; Arvinte and Drake 1993; Siligardi et al. 1994). Other calcitonins such as salmon calcitonin, which are more commonly used in treating osteoporosis, have smaller tendencies to form β-sheet and fibril structures in physiological buffers (Motta et al. 1989, 1998; Arvinte and Drake 1993; Siligardi et al. 1994).
While aqueous buffers had a limited ability to alter calcitonin secondary structure, the phospholipid environments significantly altered both the peptide secondary and macromolecular structure. These conformational transitions did not appear to arise from surface electrostatic interactions since alterations in the ionic strength of the peptide–phospholipid suspensions produced no discernible effects on the obtained conformations. In addition, the charged phospholipid, DOPS, either alone or in combination with DPPC, had little affect on calcitonin structure, suggesting that charge or electrostatic forces were not the primary molecular-level interactions governing the structural transitions.
Of particular relevance to the amyloidoses are the phospholipid environments in which enhanced β-sheet and amyloid formation occurred. Incubation of bovine calcitonin with cholesterol-rich DPPC vesicles, cholesterol- rich neuronal-like vesicles, and ganglioside-containing DPPC vesicles always resulted in substantial β-sheet enrichments and augmented amyloid contents. The acceleration of amyloid fibril formation is particularly significant because a correlation appears to exist between the biological effects of amyloid-forming peptides and their aggregation state (Seilheimer et al. 1997; Ward et al. 2000; Rymer and Good 2001). Newly formed membrane-associated calcitonin microaggregates could act as templates or seeds that could recruit other calcitonin molecules from solution to the membrane environment, further promoting β-sheet and fibril formation (Choo-Smith et al. 1997; Lansbury 1999). Membrane-associated acceleration of amyloid formation has been seen in other amyloid-forming peptides (Matsuzaki and Horikiri 1999; Koppaka and Axelsen 2000; Kakio et al. 2003; Mandal and Pettegrew 2004).
The observed increases in β-sheet and amyloid structure upon incubation with phospholipid vesicles were highly correlated with our membrane binding results. We found that bovine calcitonin only bound significantly to the lipid systems that enhanced its β-sheet and amyloid structure such as the cholesterol-rich vesicles and the ganglioside-containing vesicles. The binding affinities of calcitonin for the DPPC, DOPS, and DPPC/DOPS vesicles, which did not promote β-sheet and amyloid formation, were minimal. We believe that this effect is due, in part, to the selective affinity of the peptide for cholesterol and gangliosides, and not due to electrostatic interactions. With other peptides, such as Aβ, structure-dependent peptide-cholesterol (Avdulov et al. 1997; Wang et al. 2001; Yanagisawa and Matsuzaki 2002; Tashima et al. 2004), and peptide-ganglioside, interactions have also been observed (Matsuzaki and Horikiri 1999; Wang et al. 2001; Yanagisawa and Matsuzaki 2002; Kurganov et al. 2004; Tashima et al. 2004).
To further our understanding of the nature of the calcitonin–phospholipid interactions, we examined the temperature dependence of the calcitonin binding to the DPPC vesicles, the cholesterol-rich DPPC vesicles, and the cholesterol-rich neuronal-like vesicles. In all phospholipid systems, the binding affinities increased with increasing temperature. A Van’t Hoff analysis of the data indicated that binding was endothermic. Thus, in order for appreciable equilibrium binding to occur, the entropy associated with the binding had to be positive or the disorder in the system had to increase upon binding. Given that the peptide appears to order more upon binding, having greater β-sheet and amyloid structure, we assume that the membrane must disorder in order to have a positive change in entropy of the system. In our experiments, the membranes containing cholesterol, for which calcitonin had greater binding affinities, would have been easier to disorder than membranes of pure phospholipids, for which lower binding affinities were observed. While our data suggest that membrane disordering occurs upon binding, others have demonstrated that interactions with amyloid-forming proteins caused changes in membrane structure (Matsuzaki and Horikiri 1999), the disordering of phospholipid acyl chains (Surewicz et al. 1987), and protein insertion into the membrane hydrophobic core (Mason et al. 1996).
Collectively, the preceding binding and structural results suggest the importance of membrane order/fluidity, cholesterol, and sialic acid residues to the calcitonin–lipid interactions that result in enhanced β-sheet and amyloid formation. While no previous structural and binding studies have been conducted with bovine calcitonin and lipids, our results are consistent with those of several studies conducted with salmon and human calcitonin (Epand et al. 1983; Viani et al. 1992; Bradshaw 1997). One group of investigators similarly reported that neither human nor salmon calcitonin significantly interacted with zwitterionic, phosphatidylcholine, or phosphatidylserine lipids (Epand et al. 1983). Salmon calcitonin has been observed to insert into DMPG bilayers as demonstrated by neutron diffraction (Bradshaw 1997) and to significantly broaden the DMPG phase transition (Epand et al. 1983). While we did not perform studies with DMPG, our structural and temperature-dependent binding data with bovine calcitonin and cholesterol-rich vesicles also show conformational alterations and suggest membrane perturbations that could result in phase transition broadening. Other researchers reported that salmon calcitonin modified the structural properties of bilayered membranes containing the glycosphingolipid sulfatide (Viani et al. 1992), which is consistent with our ganglioside containing membrane data.
Analogous to our bovine calcitonin results, the importance of peptide–lipid interactions to the conformation and amyloid formation of Aβ has been well documented. Some researchers have demonstrated that Aβ (25–35) and Aβ (1–40) interact with acidic phospho-lipids electrostatically and that they undergo a random-coil to β-sheet transition upon binding to these lipids (McLaurin and Chakrabartty 1996; Terzi et al. 1997). Alternative studies have illustrated that Aβ selectively binds and disorders the bilayers of ganglioside-containing vesicles and that these processes are often accompanied by accelerated β-sheet and amyloid formation (Choo-Smith and Surewicz 1997; Matsuzaki and Horikiri 1999; Koppaka and Axelsen 2000; Kakio et al. 2002, 2003; Mandal and Pettegrew 2004).
In conclusion, bovine calcitonin conformation is highly affected by membrane environments. Cholesterol-rich DPPC vesicles, cholesterol-rich neuronal-like vesicles, and ganglioside-containing DPPC vesicles induced substantial β-sheet and amyloid structure in the peptide that is characteristic of the amyloidoses. The observed calcitonin structural alterations were highly correlated to the calcitonin phospholipid binding, with significant binding only occurring with the membranes that promoted β-sheet structure. The calcitonin structural transitions and binding did not appear to arise from electrostatic interactions, but were probably entropically driven and associated with disordering of the membrane bilayers. Other amyloid-forming peptides, most notably Aβ of Alzheimer’s disease, show similar types of peptide–lipid interactions. Collectively, these results imply that the interactions of amyloid-forming peptides with membranes could enhance their β-sheet and amyloid character and could cause local membrane perturbations, initiating the cascade of cellular events leading to cell death. Our ability to manipulate protein–membrane interactions may lead to novel treatments for the amyloidoses.
Materials and methods
Materials
Bovine calcitonin was obtained from Sigma. Its sequence and molecular mass are CSNLSTCVLSAYWKDLNNYHR FSGMGFGPETP and 3596 Da, respectively. Na125I and iodobeads were purchased from Dupont NEN and Pierce, respectively. For vesicle preparation, the purified phospholipids and cholesterol were purchased from Avanti Polar Lipids. The mixed bovine brain gangliosides were obtained from Calbiochem. All other chemicals were purchased from Sigma.
Vesicle preparation
Phospholipid vesicles were prepared by mixing the specified phospholipids and/or cholesterol in chloroform with or without the gangliosides in methanol and then evaporating off the solvent under nitrogen at 50°C in a 421–4000 Micro Rotary Evaporator (Labconco). PBS (0.01 M NaH2PO4, 0.14 M NaCl [pH 7.4]) or phosphate buffer (0.01 M KH2PO4 [pH 7.4]) was then added to suspend the lipid films of phospholipid/gangliosides or phospholipids, producing final lipid concentrations of 20 mg/mL. The resulting suspensions were sonicated for 10 min and then frozen and thawed several times to obtain a more homogeneous distribution of small unilamellar vesicles. Vesicles consisted either of pure DPPC; a mixture of 18% (w/w) cholesterol and 82% (w/w) DPPC specified as cholesterol-rich vesicles; a mixture of 10% (w/w) DOPS and 90% (w/w) DPPC specified as DOPS/DPPC vesicles; a mixture of 36% (w/w) DPPC, 36% (w/w) DPPE, 10% (w/w) DOPS, and 18% (w/w) cholesterol specified as cholesterol-rich, neuronal-like vesicles (Bretscher 1973); or a mixture of 67% (w/w) DPPC and 33% (w/w) mixed gangliosides (predominantly GM1 and GD1a with the remainder consisting of GT1b and GD1b) specified as ganglioside-containing vesicles.
Circular dichroism
CD spectra of the calcitonin solutions were recorded on a Model 62DS spectrometer (Aviv Instruments) at 25°C using a bandwidth of 1.0 nm, a step interval of 0.5 nm, and a collection time of 20 sec/step. Either a 0.01-cm or a 0.001-cm quartz cell was used for far-UV (190–250 nm) measurements. The instrument was calibrated using D(+)-10-camphorsulfonic acid. Three scans each of duplicate samples were measured and averaged. Control buffer, solvent, or vesicle suspension scans were run in triplicate, averaged, and then subtracted from the sample spectra. Spectra were analyzed using the secondary structural parameters reported by Chang et al. (1978) to ascertain the sample percentages of α-helix, β-sheet, β-turn, and random-coil. Uncertainties of the percentage of a structural feature were estimated to be ± 10%.
The exact methods for peptide sample preparation for CD follow: For the nonlipid measurements, bovine calcitonin was dissolved in the respective buffers or solvents at the specified concentrations. The CD spectra were recorded between 2 and 24 h after sample preparation, and the samples were rotated on a model RD4524 rotator (Glascol) at 60 revolutions/min at 25°C prior to recording their spectra. The bovine calcitonin was observed to be fairly soluble under the experimental solvation conditions. Final calcitonin concentrations were verified with the bicinchoninic acid assay (Smith et al. 1985). For the phospholipid-containing samples, bovine calcitonin was first dissolved in PBS, phosphate buffer, or deionized water and then it was diluted in the respective vesicles to produce a calcitonin concentration of 80 μM and a lipid concentration of 2.5 mM. The peptide–lipid solutions were allowed to rotate at 60 revolutions/min at 25°Cfor 2, 24, or 48 h prior to recording their spectra to ensure the formation of a stable structure. The rotation times were 24 h, unless otherwise specified.
Congo red dye binding
To assess the presence of amyloid fibrils in the calcitonin solutions, Congo red binding studies were performed. Congo red dye was dissolved in PBS to a final concentration of 112 μM. The calcitonin solutions were prepared analogously as described for the CD analyses. Congo red absorbances of these calcitonin solutions and the free dye controls were determined by adding Congo red to a final concentration of 12 μM and acquiring spectral measurements from 300 to 900 nm at 25°C on a Model 420 UV-Vis spectrophotometer (Spectral Instruments) (Klunk et al. 1989a, b). Both the calcitonin solutions and the control solutions were allowed to interact with Congo red for 1 h prior to recording their spectra.
Radiolabeling of calcitonin
Bovine calcitonin was 125I-labeled at the tyrosine residues using iodobeads. Four hundred microcuries of Na125I was incubated with two iodobead catalyst beads in 100 μL of PBS. After a 10-min activation period, 0.482 μmol of bovine calcitonin dissolved in PBS, phosphate buffer, or deionized water was added to the catalyst beads. These calcitonin solutions had been rotated for 24 h prior to radiolabeling. The iodination reaction was allowed to proceed at room temperature for 20 min. Then, the catalyst beads were removed, and the unreacted Na125Iwas separated from the labeled peptide by dialysis against PBS, phosphate buffer, or deionized water, respectively.
Calcitonin membrane binding experiment
The DPPC, DOPS, DOPS/DPPC, ganglioside-containing DPPC, and neuronal-like vesicles were incubated with the 125I-labeled calcitonin at room temperature (20°C), 30°C, or 4°C for 2, 24, or 48 h. The resulting suspensions were centrifuged for 5 min at 10,000g to separate the free from the bound peptide. The activities of the supernatants and the pellets were measured in a Topcount Microplate Scintillation Counter (Packard) to determine the concentrations of the free and the bound peptide, respectively.
Estimates of the calcitonin binding affinities to the membranes were obtainedbyfitting straight lines through the binding isotherms using least-squares regressions. A minimum of 10 data points was used for each isotherm. The slopes of the lines fitted through the isotherm data were taken as the calcitonin binding affinities.
Van’t Hoff analyses of the binding affinities were performed by plotting the natural logarithm of the slopes of the binding isotherms for a particular membrane system as a function of the inverse absolute temperature. Then, linear least-squares regressions through the data in the Van’t Hoff plots were performed. The negative of the slopes of the fitted lines multiplied by the gas constant, R, were taken as estimates of the enthalpies of binding.
Statistical analysis
The significance of results was determined using a student’s t-test on n independent measurements, where n is specified in the figure legend. Unless otherwise indicated, significance was taken as p<0.05.
Acknowledgments
This research was supported by a grant from the NSF, USA (T.A.G.), a fellowship from the Welch Foundation (D.L.R.), and a grant from the National Science Council, Taiwan (S.S.W.).
Abbreviations
Aβ, β-amyloid
CD, circular dichroism
DPPC, 1,2-dipalmytoyl-sn-glycero-3-phosphocholine
DOPS, 1,2-dioleoylsn-glycero-3-[phospho-L-serine]
DPPE, 1,2-dipalmytoyl-sn-glycero-3-phosphoethanolamine
DMPG, dimyristoylphosphatidylglycerol
HFIP, 1,1,1,3,3,3-hexafluoro-2-propanol
PBS, phosphate buffered saline
TFA, trifluoroactic acid
TFE, 1,1,1-trifluoroethanol
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041240105.
References
- Arvinte, T. and Drake, A.F. 1993. Comparative study of human and salmon calcitonin secondary structure in solutions with low dielectric constants. J. Biol. Chem. 268 6408–6414. [PubMed] [Google Scholar]
- Arvinte, T., Cudd, A., and Drake, A.F. 1993. The structure and mechanism of formation of human calcitonin fibrils. J. Biol. Chem. 268 6415–6422. [PubMed] [Google Scholar]
- Atwood, C.S., Moir, R.D., Huang, X.D., Scarpa, R.C., Bacarra, N.M.E., Romano, D.M., Hartshorn, M.K., Tanzi, R.E., and Bush, A.I. 1998. Dramatic aggregation of Alzheimer Aβ by Cu(II) is induced by conditions representing physiological acidosis. J. Biol. Chem. 273 12817–12826. [DOI] [PubMed] [Google Scholar]
- Avdulov, N.A., Chochina, S.V., Igbavboa, U., Warden, C.S., Vassiliev, A.V., and Wood, W.G. 1997. Lipid binding to amyloid β-peptide aggregates: Preferential binding of cholesterol as compared with phosphatidylcholine and fatty acids. J. Neurochem. 69 1746–1752. [DOI] [PubMed] [Google Scholar]
- Bauer, H.H., Muller, M., Goette, J., Merkle, H.P., and Fringeli, U.P. 1994. Interfacial adsorption and aggregation associated changes in secondary structure of human calcitonin monitored by ATR-FTIR spectroscopy. Biochemistry 33 12276–12282. [DOI] [PubMed] [Google Scholar]
- Berger, G., Berger, N., Guillaud, M.H., Trouillas, J., and Vauzelle, J.L. 1988. Calcitonin-like immunoreactivity of amyloid fibrils in medullary thyroid carcinomas. An immunoelectron microscope study. Virchows Arch. A Pathol. Anat. Histopathol. 412 543–551. [DOI] [PubMed] [Google Scholar]
- Bodles, A.M., El-Agnaf, O.M., Greer, B., Guthrie, D.J., and Irvine, G.B. 2004. Inhibition of fibril formation and toxicity of a fragment of α-synuclein by an N-methylated peptide analogue. Neurosci. Lett. 359 89–93. [DOI] [PubMed] [Google Scholar]
- Bradshaw, J.P. 1997. Phosphatydylglycerol promotes bilayer insertion of salmon calcitonin. Biophys. J. 72 2180–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher, M.S. 1973. Membrane structure: Some general principles. Science 181 622–629. [DOI] [PubMed] [Google Scholar]
- Butler, M. and Khan, S. 1986. Immunoreactive calcitonin in amyloid fibrils of medullary carcinoma of the thyroid gland. An immunogold staining technique. Arch. Pathol. Lab. Med. 110 647–649. [PubMed] [Google Scholar]
- Byard, R.W., Thorner, P.S., Chan, H.S., Griffiths, A.M., and Cutz, E. 1990. Pathological features of multiple endocrine neoplasia type IIb in childhood. Pediatr. Pathol. 10 581–592. [DOI] [PubMed] [Google Scholar]
- Chang, C.T., Wu, C.S., and Yang, J.T. 1978. Circular dichroic analysis of protein conformation: Inclusion of the β-turns. Anal. Biochem. 91 13–31. [DOI] [PubMed] [Google Scholar]
- Chi, E.Y., Krishnan, S., Randolph, T.W., and Carpenter, J.F. 2003. Physical stability of proteins in aqueous solution: Mechanism and driving forces in nonnative protein aggregation. Pharm. Res. 20 1325–1336. [DOI] [PubMed] [Google Scholar]
- Choo-Smith, L.P. and Surewicz, W.K. 1997. The interaction between Alzheimer amyloid β (1–40) peptide and ganglioside GM1-containing membranes. FEBS Lett. 402 95–98. [DOI] [PubMed] [Google Scholar]
- Choo-Smith, L.P., Garzon-Rodriguez, W., Glabe, C.G., and Surewicz, W.K. 1997. Acceleration of amyloid fibril formation by specific binding of Aβ-(1–40) peptide to ganglioside-containing membrane vesicles. J. Biol. Chem. 272 22987–22990. [DOI] [PubMed] [Google Scholar]
- Dammrich, J., Ormanns, W., and Schaffer, R. 1984. Electron microscopic demonstration of calcitonin in human medullary carcinoma of thyroid by the immuno gold staining method. Histochemistry 81 369–372. [DOI] [PubMed] [Google Scholar]
- De Felice, F.G., Vieira, M.N., Saraiva, L.M., Figueroa-Villar, J.D., Garcia-Abreu, J., Liu, R., Chang, L., Klein, W.L., and Ferreira, S.T. 2004. Targeting the neurotoxic species in Alzheimer’s disease: Inhibitors of Aβ oligomerization. FASEB J. 18 1366–1372. [DOI] [PubMed] [Google Scholar]
- DeLellis, R.A., Nunnemacher, G., Bitman, W.R., Gagel, R.F., Tashjian Jr., A.H., Blount, M., and Wolfe, H.J. 1979. C-cell hyperplasia and medullary thyroid carcinoma in the rat. An immunohistochemical and ultrastructural analysis. Lab. Invest. 40 140–154. [PubMed] [Google Scholar]
- Epand, R.M., Epand, R.F., Orlowski, R.C., Schlueter, R.J., Boni, L.T., and Hui, S.W. 1983. Amphipathic helix and its relationship to the interaction of calcitonin with phospholipids. Biochemistry 22 5074–5084. [DOI] [PubMed] [Google Scholar]
- Epand, R.M., Epand, R.F., Orlowski, R.C., Seyler, J.K., and Colescott, R.L. 1986a. Conformational flexibility and biological activity of salmon calcitonin. Biochemistry 25 1964–1968. [DOI] [PubMed] [Google Scholar]
- Epand, R.M., Seyler, J.K., and Orlowski, R.C. 1986b. The hydrophobic moment of the amphipathic helix of salmon calcitonin and biological potency. Eur. J. Biochem. 159 125–127. [DOI] [PubMed] [Google Scholar]
- Fischer, J.A., Tobler, P.H., Kaufmann, M., Born, W., Henke, H., Cooper, P.E., Sagar, S.M., and Martin, J.B. 1981. Calcitonin: Regional distribution of the hormone and its binding sites in the human brain and pituitary. Proc. Natl. Acad. Sci. 78 7801–7805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher, J.R. 1970. Medullary (solid) carcinoma of the thyroid gland. A review of 249 cases. Arch. Surg. 100 257–262. [DOI] [PubMed] [Google Scholar]
- Gilead, S. and Gazit, E. 2004. Inhibition of amyloid fibril formation by peptide analogues modified with α-aminoisobutyric acid. Angew Chem. Int. Ed. Engl. 43 4041–4044. [DOI] [PubMed] [Google Scholar]
- Hazard, J.B. 1977. The C cells (parafollicular cells) of the thyroid gland and medullary thyroid carcinoma. A review. Am. J. Pathol. 88 213–250. [PMC free article] [PubMed] [Google Scholar]
- Hill Jr., C.S., Ibanez, M.L., Samaan, N.A., Ahearn, M.J., and Clark, R.L. 1973. Medullary (solid) carcinoma of the thyroid gland: An analysis of the M.D. Anderson Hospital experience with patients with the tumor, its special features, and its histogenesis. Medicine (Baltimore) 52 141–171. [PubMed] [Google Scholar]
- Inzerillo, A.M., Zaidi, M., and Huang, C.L.H. 2004. Calcitonin: Physiological actions and clinical applications. J. Pediatr. Endocrinol. Metab. 17 931–940. [DOI] [PubMed] [Google Scholar]
- Kakio, A., Nishimoto, S., Yanagisawa, K., Kozutsumi, Y., and Matsuzaki, K. 2002. Interactions of amyloid β-protein with various gangliosides in raft-like membranes: Importance of GM1 ganglioside-bound form as an endogenous seed for Alzheimer amyloid. Biochemistry 41 7385–7390. [DOI] [PubMed] [Google Scholar]
- Kakio, A., Nishimoto, S., Kozutsumi, Y., and Matsuzaki, K. 2003. Formation of a membrane-active form of amyloid β-protein in raft-like model membranes. Biochem. Biophys. Res. Commun. 303 514–518. [DOI] [PubMed] [Google Scholar]
- Kanaori, K. and Nosaka, A.Y. 1995. Study of human calcitonin fibrillation by proton nuclear magnetic resonance spectroscopy. Biochemistry 34 12138–12143. [DOI] [PubMed] [Google Scholar]
- Kelly, J.W. 1996. Alternative conformations of amyloidogenic proteins govern their behavior. Curr. Opin. Struct. Biol. 6 11–17. [DOI] [PubMed] [Google Scholar]
- Kirkitadze, M.D., Condron, M.M., and Teplow, D.B. 2001. Identification and characterization of key kinetic intermediates in amyloid β-protein fibrillogenesis. J. Mol. Biol. 312 1103–1119. [DOI] [PubMed] [Google Scholar]
- Klunk, W.E., Pettegrew, J.W., and Abraham, D.J. 1989a. Quantitative evaluation of congo red binding to amyloid-like proteins with a β-pleated sheet conformation. J. Histochem. Cytochem. 37 1273–1281. [DOI] [PubMed] [Google Scholar]
- Klunk, W.E., Pettegrew, J.W., and Abraham, D.J. 1989b. Two simple methods for quantifying low-affinity dye-substrate binding. J. Histochem. Cytochem. 37 1293–1297. [DOI] [PubMed] [Google Scholar]
- Koppaka, V. and Axelsen, P.H. 2000. Accelerated accumulation of amyloid β proteins on oxidatively damaged lipid membranes. Biochemistry 39 10011–10016. [DOI] [PubMed] [Google Scholar]
- Kurganov, B., Doh, M., and Arispe, N. 2004. Aggregation of liposomes induced by the toxic peptides Alzheimer’s Aβs, human amylin and prion (106–126): Facilitation by membrane-bound GM1 ganglioside. Peptides 25 217–232. [DOI] [PubMed] [Google Scholar]
- Lansbury Jr., P.T. 1999. Evolution of amyloid: What normal protein folding may tell us about fibrillogenesis and disease. Proc. Natl. Acad. Sci. 96 3342–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. and Schubert, D. 1997. Cytotoxic amyloid peptides inhibit cellular 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction by enhancing MTT formazan exocytosis. J. Neurochem. 69 2285–2293. [DOI] [PubMed] [Google Scholar]
- Ljungberg, O. 1966. Medullary carcinoma of the human thyroid gland. Autoradiographic localization of radioiodine. Acta Pathol. Microbiol. Scand. 68 476–480. [DOI] [PubMed] [Google Scholar]
- Mandal, P.K. and Pettegrew, J.W. 2004. Alzheimer’s disease:NMR studies of asialo (GM1) and trisialo (GT1b) ganglioside interactions with Aβ (1–40) peptide in a membrane mimic environment. Neurochem. Res. 29 447–453. [DOI] [PubMed] [Google Scholar]
- Mason, R.P., Estermyer, J.D., Kelly, J.F., and Mason, P.E. 1996. Alzheimer’s disease amyloid beta peptide 25–35 is localized in the membrane hydrocarbon core: X-ray diffraction analysis. Biochem. Biophys. Res. Commun. 222 78–82. [DOI] [PubMed] [Google Scholar]
- Matsuzaki, K. and Horikiri, C. 1999. Interactions of amyloid β-peptide (1– 40) with ganglioside-containing membranes. Biochemistry 38 4137–4142. [DOI] [PubMed] [Google Scholar]
- McLaurin, J. and Chakrabartty, A. 1996. Membrane disruption by Alzheimer β-amyloid peptides mediated through specific binding to either phospholipids or gangliosides. Implications for neurotoxicity. J. Biol. Chem. 271 26482–26489. [DOI] [PubMed] [Google Scholar]
- Motta, A., Morelli, M.A., Goud, N., and Temussi, P.A. 1989. Sequential 1H NMR assignment and secondary structure determination of salmon calcitonin in solution. Biochemistry 28 7996–8002. [DOI] [PubMed] [Google Scholar]
- Motta, A., Andreotti, G., Amodeo, P., Strazzullo, G., and Castiglione Morelli, M.A. 1998. Solution structure of human calcitonin in membrane-mimetic environment: The role of the amphipathic helix. Proteins 32 314–323. [PubMed] [Google Scholar]
- Murphy, R.M. 2002. Peptide aggregation in neurodegenerative disease. Annu. Rev. Biomed. Eng. 4 155–174. [DOI] [PubMed] [Google Scholar]
- Nilsson, M.R. 2004. Techniques to study amyloid fibril formation in vitro. Methods 34 151–160. [DOI] [PubMed] [Google Scholar]
- Ohnishi, S. and Takano, K. 2004. Amyloid fibrils from the viewpoint of protein folding. Cell. Mol. Life Sci. 61 511–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olofsson, A., Ostman, J., and Lundgren, E. 2002. Amyloid: Morphology and toxicity. Clin. Chem. Lab. Med. 40 1266–1270. [DOI] [PubMed] [Google Scholar]
- Ono, K., Hasegawa, K., Yamada, M., and Naiki, H. 2002. Nicotine breaks down preformed Alzheimer’s β-amyloid fibrils in vitro. Biol. Psychiatry 52 880–886. [DOI] [PubMed] [Google Scholar]
- Perez, M., Sadqi, M., Munoz, V., and Avila, J. 2003. Inhibition by Aplidine of the aggregation of the prion peptide PrP 106–126 into β-sheet fibrils. Biochim. Biophys. Acta 1639 133–139. [DOI] [PubMed] [Google Scholar]
- Ponder, B.A. 1984. Screening for familial medullary thyroid carcinoma: A review. J. R. Soc. Med. 77 585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porat, Y., Mazor, Y., Efrat, S., and Gazit, E. 2004. Inhibition of islet amyloid polypeptide fibril formation: A potential role for heteroaromatic interactions. Biochemistry 43 14454–14462. [DOI] [PubMed] [Google Scholar]
- Rijkers, D.T., Hoppener, J.W., Posthuma, G., Lips, C.J., and Liskamp, R.M. 2002. Inhibition of amyloid fibril formation of human amylin by N-alkylated amino acid and α-hydroxy acid residue containing peptides. Chemistry 8 4285–4291. [DOI] [PubMed] [Google Scholar]
- Rizzo, A.J. and Goltzman, D. 1981. Calcitonin receptors in the central nervous system of the rat. Endocrinology 108 1672–1677. [DOI] [PubMed] [Google Scholar]
- Rymer, D.L. and Good, T.A. 2001. The role of G protein activation in the toxicity of amyloidogenic Aβ-(1–40), Aβ-(25–35), and bovine calcitonin. J. Biol. Chem. 276 2523–2530. [DOI] [PubMed] [Google Scholar]
- Schubert, D., Behl, C., Lesley, R., Brack, A., Dargusch, R., Sagara, Y., and Kimura, H. 1995. Amyloid peptides are toxic via a common oxidative mechanism. Proc. Natl. Acad. Sci. 92 1989–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seilheimer, B., Bohrmann, B., Bondolfi, L., Muller, F., Stuber, D., and Dobeli, H. 1997. The toxicity of the Alzheimer’s β-amyloid peptide correlates with a distinct fiber morphology. J. Struct. Biol. 119 59–71. [DOI] [PubMed] [Google Scholar]
- Siligardi, G., Samori, B., Melandri, S., Visconti, M., and Drake, A.F. 1994. Correlations between biological activities and conformational properties for human, salmon, eel, porcine calcitonins and Elcatonin elucidated by CD spectroscopy. Eur. J. Biochem. 221 1117–1125. [DOI] [PubMed] [Google Scholar]
- Silver, M.M., Hearn, S.A., Lines, L.D., and Troster, M. 1988. Calcitonin and chromogranin A localization in medullary carcinoma of the thyroid by immunoelectron microscopy. J. Histochem. Cytochem. 36 1031–1036. [DOI] [PubMed] [Google Scholar]
- Sletten, K., Westermark, P., and Natvig, J.B. 1976. Characterization of amyloid fibril proteins from medullary carcinoma of the thyroid. J. Exp. Med. 143 993–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, P.K., Krohn, R.I., Hermanson, G.T., Mallia, A.K., Gartner, F.H., Provenzano, M.D., Fujimoto, E.K., Goeke, N.M., Olson, B.J., and Klenk, D.C. 1985. Measurement of protein using bicinchoninic acid. Anal. Biochem. 150 76–85. [DOI] [PubMed] [Google Scholar]
- Surewicz, W.K., Epand, R.M., Orlowski, R.C., and Mantsch, H.H. 1987. Structural properties of acidic phospholipids in complexes with calcitonin: A Fourier transform infrared spectroscopic investigation. Biochim. Biophys. Acta 899 307–310. [DOI] [PubMed] [Google Scholar]
- Tashima, Y., Oe, R., Lee, S., Sugihara, G., Chambers, E.J., Takahashi, M., and Yamada, T. 2004. The effect of cholesterol and monosialoganglioside (GM1) on the release and aggregation of amyloid β-peptide from liposomes prepared from brain membrane-like lipids. J. Biol. Chem. 279 17587–17595. [DOI] [PubMed] [Google Scholar]
- Terzi, E., Holzemann, G., and Seelig, J. 1997. Interaction of Alzheimerβ-amyloid peptide(1–40)with lipid membranes. Biochemistry 36 14845–14852. [DOI] [PubMed] [Google Scholar]
- Thompson, L.K. 2003. Unraveling the secrets of Alzheimer’s β-amyloid fibrils. Proc. Natl. Acad. Sci. 100 383–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyama, N., Geerts, A., and Reynaert, H. 2004. Neural connections between the hypothalamus and the liver. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 280A 808–820. [DOI] [PubMed] [Google Scholar]
- Viani, P., Cervato, G., Gatti, P., and Cestaro, B. 1992. Calcitonin-induced changes in the organization of sulfatide-containing membranes. Biochim. Biophys. Acta 1106 77–84. [DOI] [PubMed] [Google Scholar]
- Walsh, D.M., Lomakin, A., Benedek, G.B., Condron, M.M., and Teplow, D.B. 1997. Amyloid β-protein fibrillogenesis. Detection of a protofibrillar intermediate. J. Biol. Chem. 272 22364–22372. [DOI] [PubMed] [Google Scholar]
- Wang, S.S., Rymer, D.L., and Good, T.A. 2001. Reduction in cholesterol and sialic acid content protects cells from the toxic effects of beta-amyloid peptides. J. Biol. Chem. 276 42027–42034. [DOI] [PubMed] [Google Scholar]
- Ward, R.V., Jennings, K.H., Jepras, R., Neville, W., Owen, D.E., Hawkins, J., Christie, G., Davis, J.B., George, A., Karran, E.H., et al. 2000. Fractionation and characterization of oligomeric, protofibrillar and fibrillar forms of β-amyloid peptide. Biochem. J. 348 (Pt. 1) 137–144. [PMC free article] [PubMed] [Google Scholar]
- Wetzel, R. 1996. For protein misassembly, it’s the “I” decade. Cell 86 699–702. [DOI] [PubMed] [Google Scholar]
- Williams, E.D., Brown, C.L., and Doniach, I. 1966. Pathological and clinical findings in a series of 67 cases of medullary carcinoma of the thyroid. J. Clin. Pathol. 19 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa, K. and Matsuzaki, K. 2002. Cholesterol-dependent aggregation of amyloid β-protein. Ann. N. Y. Acad. Sci. 977 384–386. [DOI] [PubMed] [Google Scholar]