

Abstract
Two monofunctional Δ3, Δ2-enoyl-CoA isomerases, one in mitochondria (mECI) and the other in both mitochondria and peroxisomes (pECI), belong to the low-similarity isomerase/hydratase superfamily. Both enzymes catalyze the movement of a double bond from C3 to C2 of an unsaturated acyl-CoA substrate for re-entry into the β-oxidation pathway. Mutagenesis has shown that Glu165 of rat mECI is involved in catalysis; however, the putative catalytic residue in yeast pECI, Glu158, is not conserved in mECI. To elucidate whether Glu165 of mECI is correctly positioned for catalysis, the crystal structure of rat mECI has been solved. Crystal packing suggests the enzyme is trimeric, in contrast to other members of the superfamily, which appear crystallographically to be dimers of trimers. The polypeptide fold of mECI, like pECI, belongs to a subset of this superfamily in which the C-terminal domain of a given monomer interacts with its own N-terminal domain. This differs from that of crotonase and 1,4-dihydroxy-2-naphtoyl-CoA synthase, whose C-terminal domains are involved in domain swapping with an adjacent monomer. The structure confirms Glu165 as the putative catalytic acid/base, positioned to abstract the pro-R proton from C2 and reprotonate at C4 of the acyl chain. The large tunnel-shaped active site cavity observed in the mECI structure explains the relative substrate promiscuity in acyl-chain length and stereochemistry. Comparison with the crystal structure of pECI suggests the catalytic residues from both enzymes are spatially conserved but not in their primary structures, providing a powerful reminder of how catalytic residues cannot be determined solely by sequence alignments.
Keywords: domain swapping, low-similarity isomerase/hydratase superfamily, enoyl-CoA isomerase, crystal structure, fatty acid metabolism
To metabolize unsaturated fatty acids, eukaryotes require two auxiliary enzymes, 2,4-dienoyl-CoA reductase (DCR) and Δ3, Δ2-enoyl-CoA isomerase (ECI; EC 5.3.3.8). ECI catalyzes the movement of the double bond between carbons 3 and 4 of the acyl chain to carbons 2 and 3, allowing the acyl-CoA to re-enter the β-oxidation pathway at the second step. ECI differs from DCR in that it is essential for the metabolism of all fatty acids with double bonds at odd as well as even carbon positions (Schulz and Kunau 1987). However, like DCR, ECI is relatively promiscuous, catalyzing the isomerization of both cis and trans unsaturated fatty acyl chains (Stoffel et al. 1964; Palosaari et al. 1990; Zhang et al. 2002). Various isoforms of ECI are present in the mitochondria and peroxisomes of mammalian cells (Palosaari et al. 1990; Zhang et al. 2002), as well as in the peroxisomes of yeast (Geisbrecht et al. 1998; Gurvitz et al. 1998). Sequence analysis shows they belong to the low-similarity isomerase/hydratase superfamily (LSI/H) (Muller-Newen et al. 1995; Holden et al. 2001), with the mammalian forms grouped into four classes based on their substrate specificity, organelle location, and physical composition: mitochondrial enoyl-CoA isomerase (mECI), mitochondrial enoyl-CoA hydratase 1 (crotonase), peroxisomal enoyl-CoA isomerase (pECI), and multifunctional enzyme type-1 (MFE-1). Recent studies suggest that a previously characterized mitochondrial long-chain enoyl-CoA isomerase is the same as pECI, indicating that this isomerase is present in both mitochondria and peroxisomes (Zhang et al. 2002). Biochemical characterization of rat liver mECI shows that each subunit has a molecular weight of ~30 kDa, with a native molecular weight of 81 kDa (as determined by gel filtration) and 73 kDa (as determined by analytical ultracentrifugation), which led the investigators to suggest that the enzyme is dimeric (Palosaari et al. 1990).
Until very recently, the crystal structures of only two ECI family members had been solved: rat liver crotonase (Engel et al. 1996) and yeast pECI (Mursula et al. 2001). The structures show that both of these enzymes are composed of a dimer of trimers, with each subunit folded into two domains. The N-terminal α/β-domain, composed of approximately the first 200 amino acids, forms the core of the enzyme. The C terminus forms an α-helical domain made up of three α-helices and comprises about 60 amino acids. However, in the pECI structure the three α-helices wrap around theN-terminal domain, whereas in crotonase these α-helices protrude away from the core of the enzyme, with both domains forming extensive intra-trimer and inter-trimer interfaces. The structure of crotonase with a potent inhibitor, acetoacetyl-CoA, shows the substrate binds at the three intra-trimer interfaces, with Glu144 and Glu164 positioned to serve as catalytic residues and two backbone amide nitrogens from Ala98 and Gly141 forming an oxyanion hole that stabilizes the enolate intermediate formed during catalysis (Engel et al. 1996). The pECI structure lacks a bound substrate analog; however, site-directed mutagenesis and structural comparison with another member of the LSI/H superfamily, 4-chlorobenzoyl- CoA dehalogenase, suggest that Glu158 serves as the catalytic base (Mursula et al. 2001).
The difference between the two ECI isozymes is supported by studies that compared mECI’s and pECI’s catalytic efficiency with various substrates (Zhang et al. 2002). Results showed that both enzymes have a relatively broad substrate chain-length specificity, though pECI exhibits a slight preference for long-chain acyl-CoAs. However, pECI shows a much greater catalytic efficiency with 3-trans substrates, whereas mECI exhibits a preference for 3-cis substrates.
To gain further insight into the structure–function relationships of ECI, the crystal structure of rat mitochondrial ECI has been solved. The structure shows that some of the more general features of this superfamily of enzymes are conserved, such as the N-terminal domain fold, oxyanion hole, and CoA binding site. We also find that mECI shares, at both the sequence and structural level, the catalytic base of crotonase rather than that of the more functionally related peroxisomal isomerase, though pECI appears to share a spatially conserved catalytic residue. A similar conclusion was reached with human mECI, whose structure was reported while this manuscript was in preparation (Partanen et al. 2004). Our present studies complement and extend those observations by underscoring the significance of the three forms of domain-swapped subunit assembly found in the LSI/H superfamily and providing additional insights into the catalytic mechanism of the isomerization reaction of mECI.
Results
Overall protein structure
The final structure of mECI shows the enzyme to be composed of two domains. The N-terminal α/β domain is composed of four spiral turns that contain two β-strands and one α-helix, as seen in all structures of the LSI/H superfamily solved to date. Extension of the C terminus during model building and refinement showed rat mECI to have an α-helical domain composed of three α-helices that wrap around the body of the N-terminal domain (Fig. 1A,B ▶, right), giving rise to the term “self-association” fold. This is the same fold seen in human mECI (Partanen et al. 2004), 6-oxo camphor hydrolase (Whittingham et al. 2003), and pECI (Mursula et al. 2001) and is similar to that seen in methylmalonyl-CoA decarboxylase (Benning et al. 2000). In contrast, the V-shaped intra-trimer association fold, in which the C-terminal domain interacts with a neighboring subunit within the trimer, is seen in crotonase (Engel et al. 1996), the related AUH protein (AU binding homolog of enoyl-CoA hydratase) (Kurimoto et al. 2001), and 4-chlorobenzoyl-CoA dehalogenase (Benning et al. 1996), as well as in dienyol-CoA isomerase (Fig. 1B ▶, middle; Modis et al. 1998). An inter-trimer variation of the C-terminal crotonase-like fold, which spans the trimer–trimer interface, has also been recently reported for 1,4-dihydroxy- 2-naphtoyl-CoA synthase (MenB) (Fig. 1B ▶, right; Truglio et al. 2003).
Figure 1.
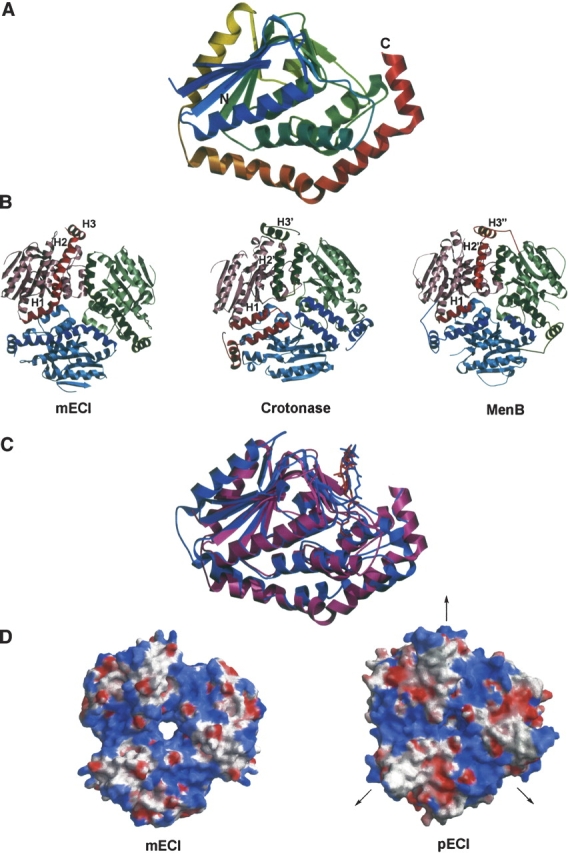
The overall fold of mECI and comparison with pECI. (A) Ribbon diagram of a single monomer, as found in the asymmetric unit. The color is ramped from blue, at the N terminus, to red, at the C terminus. Note the three α-helices of the C-terminal self-association fold at the bottom and right of the figure. (B) Comparison of the trimeric assemblies of mECI (left), crotonase (middle), and MenB (right). The trimeric forms of the enzymes are viewed down the threefold crystallographic axis, with each subunit colored differently. All three models are in the same orientation, with the trimer–trimer interfaces of crotonase and MenB pointing up. The C-terminal domain is colored a darker shade than the N-terminal domain. Comparison of the three figures clearly shows variations in the C-terminal topology: the self-association fold in mECI, intra-trimer domain swapping fold in crotonase, and inter-trimer domain swapping fold in MenB. However, the arrangement of α-helices within each monomer is similar. (C) Superimposition of the crystal structures of mECI, in purple, and pECI, colored light blue. Crotonyl-CoA molecules, as based on the octanoyl-CoA-crotonase complex, are included as darker colored sticks. (D) Comparison of the electrostatic surfaces of mECI (left) and pECI (right); contoured from + 10 kT/e, colored red, to +10 kT/e, colored blue. The models are in the same orientation, looking down the threefold axis, with the trimer–trimer interface of pECI facing up. The twofold axes that form the hexamer in pECI are perpendicular to the threefold axis. The distribution of negative charge between the two enzymes is very different.
Oligomeric state
Analysis of the crystal packing and the location of the threefold axis with respect to the asymmetric unit strongly suggests that, rather than being a dimer, as reported on the basis of analytical ultracentrifugation studies (Palosaari et al. 1990), mECI appears to be a trimer (Fig. 1B ▶, left), in agreement with a postulation made by Engel et al. (1996) and the recently reported structure of human mECI (Partanen et al. 2004). In contrast to pECI, the crystallographic twofold axes in the mECI structure fail to produce a dimer of trimers, as seen in the hexameric LSI/H enzymes. Therefore, even though mECI and pECI have similar folds (Fig. 1C ▶) (r.m.s. deviation of 1.3Å for 155 Cα atoms, ~ 60% of themodel), they exhibit relatively little sequence similarity (13% identity between rat mECI and yeast pECI) and have different oligomeric states. Comparison of the electrostatic potentials at the interface between the twotrimers of yeast pECI with the corresponding region in the rat mECI trimer shows significant differences, reflecting the relatively low sequence identity (Fig. 1D ▶). This region in pECI shows alternating regions of positively and negatively charged potential, with an ~4 Å deep and 10 Å wide cleft between each subunit of the trimer having significant positive character (>10 kT/e, or 6 kcal mol−1). This region corresponds to the first α-helix in the C-terminal α-helical domain (H1 in Fig. 1B ▶), although the basic amino acids in this region point in toward the N-terminal domain rather than out toward the trimer–trimer interface. The adjacent acidic patch is made up of residues that form parts of a β-strand and two α-helices on the surface of the N-terminal domain and the second α-helix of the C-terminal domain (H2 in Fig. 1B ▶). The positively charged regions of rat mECI correspond well with those on the pECI molecular surface, but lack similar sized acidic patches.
Although the arrangements of the C-terminal domain in each molecule of the trimeric assembly of the three classes of folds (self-association and the intra-/inter-trimer associations) are different, the overall assembly of these α-helices within a single subunit is similar (Fig. 1B ▶).Helix 1 (H1) of all three folds occupies the same position; helices 2 and 3 (H2 and H3) of the self-association fold from a single subunit occupy similar spatial positions to helices 2 and 3 (H2′ and H3′) of the neighboring subunit in the intra-trimer association fold and to helices H2″ and H3″ of a twofold symmetry related subunit on the other trimer in the intertrimer association fold. These are elegant variations of “domain swapping”, where α-helices between two neighboring subunits are swapped, as exemplified by the domain swapped dimeric form of bovine seminal RNase (Bennett et al. 1994).Figure 2B ▶ shows structure-based sequence alignment of the C-terminal domains, grouped according to which of the three classes of domain swapping folds they belong. There appears to be very little sequence homology within each group, though the intra-trimer domain swapping fold appears to have four well-conserved residues distributed across the entire regions (shown in red and yellow boxes). Interestingly, the conserved lysine and aspartate from the first and third helices of the C-terminal domain are also in the inter-trimer domain swapped group.
Figure 2.
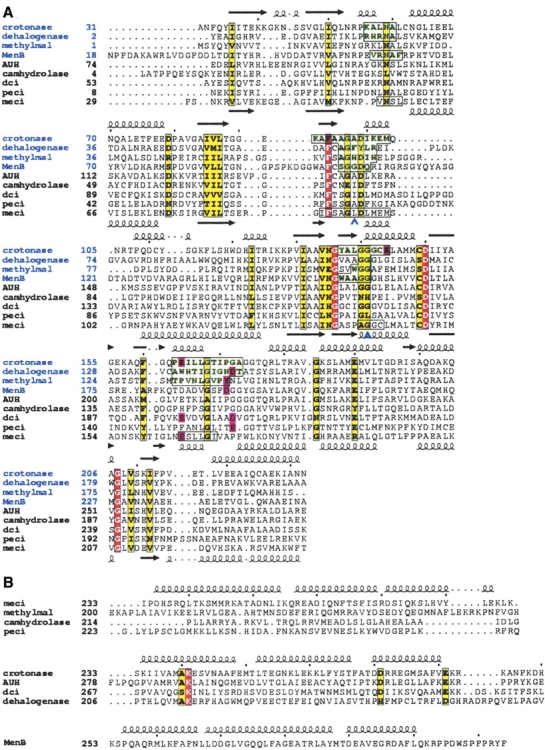
(A) Structure-based sequence alignment of the N-terminal domain of members of the LSI/H superfamily: crotonase, 4-chlorobenzoyl-CoA dehalogenase, methylmalonyl CoA-decarboxylase, 1,4-dihydroxy-2-naphtoyl-CoA synthase (MenB), AUH protein, 6-oxo camphor hydrolase, dienyol-CoA isomerase (dci), peroxisomal enoyl-CoA isomerase (pECI), and mitochondrial enoyl-CoA isomerase (mECI). Secondary structure elements from the crotonase and mECI structures are included on the top and bottom of each row of sequences, respectively. Red and yellow boxes show identical and homologous residues, respectively. The first four sequences (labeled in blue) are from crystal structures with CoA substrate analog bound. Regions of these peptides that are within 3.9 Å of these molecules are highlighted in green and boxed. The corresponding regions in the mECI sequence that are relatively conserved are shown in boxes. Blue chevrons denote residues that form the oxyanion hole. Pink boxes show primary catalytic residues for each enzyme. Also, no catalytic residue has been highlighted for 6-oxo camphor hydrolase sequence, as this residue is believed to reside in the C-terminal domain, which is not included in this alignment. Since the primary function of AUH protein is in binding RNA, no catalytic residues have been highlighted. (B) Sequence alignment of the C-terminal domains grouped according to the C-terminal fold; enzymes having the self-association fold (top); enzymes belonging to the intra-trimer domain swapping fold (middle); and MenB (bottom), the sole member having the inter-trimer domain swapping fold.
Substrate binding and active sites
Figure 2A ▶ shows a structure-based sequence alignment of the N-terminal domain of members of the LSI/H superfamily. Among the superfamily members, the first four structures (identified in blue type) have been solved with CoA substrate analogs. Superposition of these four structures show that the CoA moiety of the substrate binds to a conserved position on the enzyme, corresponding to four discrete regions of the primary structure (highlighted in green and boxed in Fig. 2A ▶). These regions are also moderately conserved in the mECI structure (boxed in Fig. 2A ▶). The second significant feature of this superfamily of enzymes is a conserved oxyanion hole that binds the carbonyl oxygen of the substrate acyl chain, stabilizing the enolate form of the thioester linkage during catalysis (Holden et al. 2001). The oxyanion hole is made up of two residues that hydrogen bond to the oxygen atom via their backbone amide groups. Structure-based sequence alignment shows that both of these residues are conserved in mECI (Leu95 and Gly140), with neither residue being a proline. Mutation of Gly141 to proline in the oxyanion hole of crotonase, causing the removal of one of the backbone amide nitrogens, results in a million-fold reduction in kcat (Bell et al. 2001), illustrating the importance of the interaction of the amide group with the carbonyl oxygen of substrate for catalysis. Finally, if one highlights the known primary catalytic residues of all LSI/H structures in the structure-based sequence alignment, there is an apparent clustering at three distinct loci (highlighted with pink boxes in Fig. 2A ▶). The first houses Glu144 of crotonase, which corresponds to Leu143 in the mECI structure. Since there are no other potential catalytic residues in this region, the fact that the functional homolog, pECI, has a similar arrangement of such innocuous amino acids (Ala129) and the knowledge that Glu144 is not necessary for isomerase activity in crotonase (Kiema et al. 1999) suggest that there is no corresponding catalytic residue for mECI at this locus. The second cluster, around Glu164 of crotonase, corresponds to Glu165 of mECI. Mutation of this residue in rat mECI has shown that this residue is essential for isomerase activity (Muller-Newen and Stoffel 1993). Surprisingly, this residue is not conserved in pECI, corresponding to Phe150. Instead, from substrate modeling and mutagenesis, Glu158, which corresponds to Pro173 in mECI, is proposed as the putative catalytic base in pECI (Mursula et al. 2001). Interestingly, this residue in pECI lies at the third cluster of catalytic residues. Other members whose catalytic residues lie at the third cluster include dienoyl-CoA isomerase, 4-chlorobenzoyl- CoA dehalogenase, MenB, and methylmalonyl-CoA decarboxylase. However, judging from the one amino acid offset position of Tyr140 in methylmalonyl-CoA decarboxylase and Asp192 in MenB, the spatial position of these residues does not appear to be absolutely conserved.
Further inspection of the structure-based sequence alignment shows four residues are conserved in all the LSI/H structures (red boxes, Fig. 2A ▶): Phe91, Gly135, Asp149, and Gly208 (all in mECI numbering). Mapping these positions onto the mECI, crotonase, and MenB structures shows these residues lie at positions remote from the active site, with the latter three residues lying at the intra-trimer interfaces, suggesting that they function in substrate binding and/or maintaining structural and oligomerization integrity rather than catalysis.
When crotonyl-CoA is modeled in the active site of both mECI and pECI, the structures show that the putative catalytic carboxylate moieties appear to approach the acyl chain from different angles (Fig. 3A ▶). Inspection of the active site of mECI shows Glu165 to be positioned to abstract the pro-R hydrogen from carbon 2 of the acyl chain, in agreement with a postulate made by Fillgrove and Anderson (2000), which was based on the stereospecificity of crotonase and consistent with the recent structural analysis of human mECI complexed with octanoyl- CoA (Partanen et al. 2004). Modeling studies with pECI also suggest that the carboxylate group of Glu158 is positioned for pro-R abstraction (Fig. 3A ▶).
Figure 3.
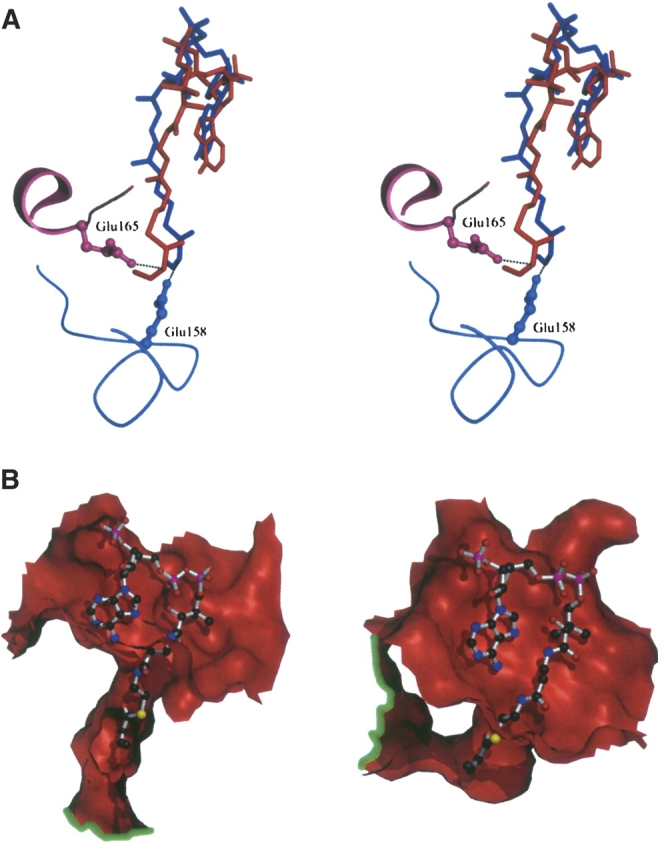
(A) Stereoview of the active site of mECI, shown in red, and pECI, in blue. The respective putative catalytic residues are included. Modeled product, crotonyl-CoA, is also included as balls-and-sticks. Dotted lines show the potential interaction between the carboxylate of the catalytic residue and the C2 atom of the acyl chain. Note the differing positions of the catalytic residue in mECI and pECI. The mECI model clearly suggests proton abstraction is pro-R specific, while the pECI model shows abstraction might also be pro-R specific. (B) Cutaway of the topology of the putative substrate binding site for mECI (left) and pECI (right). Crotonyl-CoA, which is modeled based on the crystal structure of the octanoyl-CoA–crotonase structure, is included as ball-and-sticks. The green lines highlight the lip of the tunnel on different regions of the monomer surface, suggesting both enzymes can accept a wide variety of acyl chain lengths. Dark gray balls represent carbon atoms, red are oxygen, blue are nitrogen, yellow are sulfur, and purple are phosphorus.
Substrate chain-length specificity
Crotonase, which can hydrate acyl-CoAs with acylchain lengths ranging from C4 to at least C16 (Fong and Schulz 1977), binds substrate using a flexible tunnel that runs from the CoA binding site to the trimer–trimer interface, passing via the active site (Engel et al. 1998). This tunnel is partly formed by a disordered loop between residues 113 and 120 in the N-terminal domain, corresponding to a well-ordered α-helix in the mECI structure (residues 110–119). This observation suggests that a binding mode for mECI similar to crotonase is unlikely. Examination of the putative substrate binding site for the mECI structure shows a large tunnel that starts at the CoA binding site and passes through to the other side of the enzyme via the catalytic site (Fig. 3B ▶, left). However, in contrast to crotonase, the “exit” of the tunnel on the face opposing the CoA binding site runs through a hole formed between a turn in the N-terminal domain and the second α-helix of the C-terminal domain, lined mostly by aromatic residues. Modeling of crotonyl-CoA into the active site suggests that acyl chains of up to ~ 14 carbon atoms in length can be easily accommodated in this tunnel. A similar scenario is seen in the pECI structure; however, the tunnel appears to be somewhat shorter than mECI’s, and, as with crotonase, terminates at the trimer–trimer interface rather than being exposed to solvent (Fig. 3B ▶, right).
Discussion
The low-sequence similarity isomerase/hydratase (LSI/H) superfamily is one of a growing number of enzyme superfamilies that demonstrate how polypeptide folds are used as scaffolds to catalyze a wide variety of reactions, with the LSI/H superfamily catalyzing reactions ranging from isomerization to dehalogenation and decarboxylation. The structures of LSI/H superfamily members determined to date show the large N-terminal core domain to fold in an almost isosteric manner, with the largest rmsd being 2.1 Å between 6-oxo camphor hydrolase and dienoyl-CoA isomerase for all Cα atoms in this domain. This domain is composed of ~ 200 amino acids, or ~ 75% of the mature peptide.
The 60-amino-acid α-helical C-terminal domain shows somewhat greater diversity, having one of three folds: the V-shaped intra-trimer domain swapping fold which is seen in crotonase, 4-chlorobenzoyl-CoA dehalogenase, dienoyl- CoA isomerase, and AUH protein; the inter-trimer domain swapping fold, which to date has only been observed in MenB; and the self-association fold, which wraps around the N-terminal domain, as observed in mECI, pECI, 6-oxo camphor hydrolase, and a longer form seen in methylmalonyl-CoA decarboxylase. It should be noted that while these folds are different, they all contain three α-helices of about equal length, but exhibit only limited sequence homology (Fig. 2B ▶). These different C-terminal folds provide elegant examples of variations of domain swapping, first identified in the domainswapping dimeric form of RNase (Bennett et al. 1994).
With the exception of mECI, whose molecular assembly is a trimer, all LSI/H structures determined to date appear crystallographically as hexameric proteins, composed of a dimer of trimers. The oligomeric state of 4-chlorobenzoyl- CoA dehalogenase has been reported to be a trimer; however, inspection of crystal packing shows that ~ 2100 Å2 (corresponding to 7.7% of the total trimer surface of 27,700 Å2) is buried between the two trimers. Therefore, for the purposes of this discussion, the dehalogenase will be treated as a dimerof trimers along with othermembers of the superfamily. Each type of C-terminal domain fold appears to form part of the interface between neighboring subunits within the trimer and also, with the exception of mECI, the trimer–trimer interface, by domain swapping, which places the α-helices in spatially conserved positions (Fig. 1B ▶). The trimer–trimer interface areas range from 5.4% of total hexameric molecular surface for AUH protein to 38.3% for MenB (Fig. 4 ▶). In all of these enzymes, the trimer exhibits threefold rotational symmetry, with the hexamer formed by twofold rotation perpendicular to the threefold axis.mECI, originally believed to form a dimer (Palosaari et al. 1990), appears to be a trimer in the crystal lattice with the twofold axes failing to form a hexamer. Whether a LSI/H superfamily member forms a trimer or a hexamer appears to be determined by factors far more complex than surface and electrostatic complementarity, as inspection of the electrostatic surface at the interface of the hexameric forms reveals few discernable patterns (Fig. 4 ▶). Furthermore, regions that form the interface appear to be dependent on the conditions in which the protein is present, as demonstrated by the “tight” and “loose” forms of pECI (Mursula et al. 2004). Further studies examining effects on ionic strength and oligomerization states should address these unusual properties found in pECI and the apparent oligomeric anomaly of mECI (being theonly trimer amongtheLSI/H superfamily), aswellas the true oligomeric state of the dehalogenase where the crystallographic trimer–trimer interface is rather high for its molecular ensemble to be a single trimer.
Figure 4.

Electrostatic potential maps of the trimer–trimer interfaces of different C-terminal folds found in LSI/H superfamily members. (A) Self-association fold; (B) intra-trimer domain swapping fold; (C) inter-trimer domain swapping fold. The threefold axis is projected out of the page as in Figure 1B ▶, and the three twofold axes that form each hexamer lie on the plane of the page. The trimers are orientated such that the trimer–trimer interface is facing the viewer. The percentage for the trimer–trimer interface out of the total hexamer surface area for each molecule is listed. Comparison of each picture shows there is little conservation in regions that form the interface or their electrostatic character.
Structures of all members of the LSI/H superfamily solved in the presence of a CoA substrate analog show the active site to be spatially conserved, being present at either the intra-trimer interface, as seen for enzymes that have the C-terminal domain swapping fold (crotonase, 4-chlorobenzoyl-CoA dehalogenase [Benning et al. 1996]) and the inter-trimer variation seen in MenB (Truglio et al. 2003), or within a single subunit seen in methylmalonyl- CoA decarboxylase (Benning et al. 2000) and recently reported for human mECI (Partanen et al. 2004), both of which have the C-terminal self-association fold. Among all members of the LSI/H superfamily, the most extensive structural studies have been done with crotonase. Structures with acetoacetyl-CoA (Engel et al. 1996; PDB accession code 1DUB), 4-(N,N-dimethylamino)cinnamoyl-CoA (Bahnson et al. 2002; PDB accession code 1EY3), hexanoyl- CoA (Bell et al. 2002; PDB accession code 1MJ3), and octanoyl-CoA (Engel et al. 1998; PDB accession code 2DUB) show that Glu144 and Glu164 are positioned for catalysis at carbon 3 of the acyl chain, with awater molecule positioned between the C2–C3 bond of the acyl chain of substrate and these two catalytic residues. The coordinates for the recently reported structure of human mECI in complex with octanoyl-CoA are presently unavailable for analysis; however, it was stated that the mode of the acyl chain binding to human mECI is different from that found in crotonase (Partanen et al. 2004).
Comparisons of the catalytic residues of mECI, pECI, and crotonase suggest that, despite the primary function of each enzyme, the N-terminal domain of mECI is more closely related to crotonase than pECI. For isomerase activity, both crotonase and mECI share the same spatially conserved catalytic residue, Glu164 in crotonase and Glu165 in mECI, whereas pECI’s catalytic residue, Glu158, suggests that this enzyme belongs to the subfamily of enzymes that place their primary catalytic residues at the third locus. These enzymes include 4-chlorobenzoyl-CoA dehalogenase and methylmalonyl- CoA decarboxylase, the latter of which has similar C-terminal self-association topology to the ECIs. One can therefore hypothesize that, although functionally dissimilar, pECI is more closely related to methylmalonyl-CoA decarboxylase than to mECI. The active site topology of the mECI crystal structure also supports the proposal that this enzyme is pro-R specific (Fillgrove and Anderson 2000). Structure-based sequence alignment shows that mECI, crotonase, and DCI share the same catalytic residue: Glu165 in mECI, Glu164 in crotonase, and E196 in DCI. Both crotonase and DCI have also been shown to be pro-R specific (Bucklers et al. 1970; Chen et al. 1994), lending further support to the proposed stereochemistry of this reaction. While there are no data supporting this hypothesis for the peroxisomal form, modeling studies suggest that this enzyme might also be pro-R specific.
Despite the lack of a conserved catalytic residue between the two ECIs, the catalytic mechanism of mECI appears to be much like that of pECI, with Glu 165 acting as a general base at C2 of the acyl chain, and the backbone amide nitrogens of Leu95 and Gly140 forming an oxyanion hole via hydrogen bonds to the thioester carbonyl oxygen (Fig. 5 ▶). Inspection of the crotonyl-CoA bound models of mECI and pECI suggests that Glu165 and Glu158, respectively, appear to function as the general acid as well, as there is no other residue that could perform this function. Minor torsion about the Cα–Cβ bond of the side chain of the catalytic glutamate would bring the carboxyl group into close association with the C4 atom of the acyl chain for reprotonation of the anionic reaction intermediate. Support for this mechanism comes from NMR studies that show [1H] 3-enoyl-CoA is converted to [1H] 2-enoyl-CoA in the presence of deuterated solvent (Fillgrove and Anderson 2000). However, the possibility that a water molecule may act as the general acid when substrate is bound cannot be excluded.
Figure 5.

Proposed reaction mechanism for mECI, with Glu165 acting as both the general base and conjugate acid. Although the current mECI structure has no substrate analog bound, crotonyl-CoA has been modeled in. The carboxylate of Glu165 is about 3 Å from the C2 atom of the acyl chain of modeled substrate. The Glu165 to C4 atom distance is also ~3 Å for the cis conformation and ~4 Å in the trans conformation, implying that Glu165 might undergo minor conformational changes before reprotonating the C4 atom during catalysis. Backbone amide groups from Leu95 and Gly140 form hydrogen bonds to the thioester carbonyl oxygen, stabilizing the enolate intermediate.
Finally, analysis of substrate binding, as shown by modeling of crotonyl-CoA, shows a long tunnel in both mECI and pECI structures that runs from one region of the molecular surface of the enzyme to another, via the catalytic site. This appears to sequester the hydrophobic acyl chain of the CoA substrate from solvent, while allowing a wide variety of chain lengths to bind for catalysis. This hypothesis is supported by kinetic studies that show micromolar Km values for substrates having chain lengths from C6 to C14 (Zhang et al. 2002). Peroxisomal ECI’s slight preference for longer chain acyl-CoAs compared with mECI’s might be due to the fact that the tunnel exit on the opposing face of the CoA binding site is sequestered from solvent by the trimer– trimer interface, whereas mECI’s is not. However, with the limits of structure interpretation imposed by the modeling studies presented here, the current structures for mECI and pECI fail to explain the stereochemical preference for each enzyme, mECI preferring 3-cis double bonds, and pECI preferring 3-trans. The recently determined human mECI structure also shows a large tunnel, suggesting that there are two alternative binding modes for the fatty acyl moiety of the substrate, depending on the length and stereochemistry of the acyl chain. Further studies including crystal structure determinations of both enzymes in the presence of both stereoisomeric analogs are needed.
The results presented here show that most members of the LSI/H superfamily enzymes contain one or more primary catalytic residues from three structurally conserved loci. These residues are further embellished by additional residues that line the active site and determine the chemistry of the reaction. Therefore, care must be taken when analyzing sequence alignments in the absence of structures, as one can easily come to wrong conclusions as to which residues participate in catalysis. This is exemplified by a fully conserved aspartate residue, which appears to be required for structural rather than functional reasons. Thus, the structures of the LSI/H superfamily members offer a powerful example of a catalytically essential residue that is not conserved in the primary structure of related enzymes, as also observed in some other superfamilies (Todd et al. 2002).
Materials and methods
Cloning, expression, and purification
Rat mitochondrial enoyl-CoA isomerase was cloned, expressed, and purified to homogeneity for crystallization. Briefly, the open reading frame corresponding to mature mECI (amino acids Ala26 through Gly289) was cloned from rat liver Marathon-Ready cDNA (CLONTECH) using the following primers: 5′-CAG GATCCCATATGGCGCGTCGCTTCTCTAACAAGC-3′ and 5′-CAGTAAGCTTATCAGCCCTTCTTTTGCTTGAGCTT-3′. The PCR product was subcloned into the BamHI-HindIII site of vector pND-1 (a gift from Dr. Didier Negre) to obtain an expression plasmid. This plasmid was used to transform Escherichia coli BL21(DE3)pLysS cells, which were cultured in LB media, and induced by addition of 0.6 mM IPTG. The enzyme was purified to homogeneity by chromatography on hydroxyapatite, CM-52, Matrix Gel-Red A, and Sephacryl S-200 HR columns. A typical preparation yielded about 10 mg of pure protein from a 2-L culture. The purified protein was concentrated to about 6 mg/mL in a buffer containing 20 mM KPi, pH 7.25, and 1 mM EDTA and stored at −80°C.
Protein crystallization
For screening, pure mECI was further concentrated to 10 mg/mL prior to the addition of crystallization agents. Conditions 19–28 from the Hampton Crystal Screen II were tested, and one of them, condition 27 (25% PEG-MME 550, 0.1 M MES [pH 6.5], 0.01 M zinc sulfate), yielded small cubic crystals overnight (0.1 × 0.1 × 0.1 mm). This condition was further optimized by addition of 8% glycerol to the original crystallization agent and dilution of the protein to 7 mg/mL. This resulted in much larger crystals (0.2 × 0.2 × 0.2 mm), which grew over a period of about 1 wk. The crystals belong to the space group I4132 with cell dimensions of a=b=c=170.6 Å.
Data collection and structure determination
The data used for molecular replacement, up to 2.8 Å resolution, were collected on an R-AXIS IIC image plate (Molecular Structure Corp.) equipped with a Rigaku RU 200 generator and Osmic confocal mirrors (Table 1). A high-resolution data set used for final refinement was collected to 2.2 Å resolution on a RU 300 generator equipped with an R-AXIS IV++ image plate, an MSC X-stream 2000 cooling system set at −180 °C, and Osmic confocal mirrors. HKL (Otwinowski and Minor 1997) was used for reduction of the preliminary low-resolution data, and d*trek (Pflugrath 1999) was used for high-resolution data reduction. For molecular replacement, a homology model was built using MODELLER (Såli and Blundell 1993), derived from the coordinates of three proteins of known structure that exhibited highest sequence homology with the mature rat mECI sequence. A FASTA search (Pearson and Lipman 1988) through the PDB database (Berman et al. 2000) showed AUH protein (Kurimoto et al. 2001; PDB accession code 1HZD), crotonase (Bahnson et al. 2002; PDB accession code 1EY3), and 4-chlorobenzoyl- CoA dehalogenase (Zhang et al. 2001; PDB accession code 1JXZ) to have 29%, 23%, and 24% sequence identities, respectively. Since the Matthews coefficient suggested one molecule per asymmetric unit (VM=3.44), the homology model, corresponding to one monomer, was used as a probe for crossrotational molecular replacement. Initial attempts at molecular replacement failed to find a solution. However, when the 60- amino-acid region corresponding to the C-terminal α-helical domain that is structurally conserved in the three template models but not in the pECI structure (Mursula et al. 2001, 2004; PDB accession codes 1HNO and 1PJH) was deleted, leaving only the N-terminal domain (residues 30–228) as the probe, a solution was obtained. This solution was confirmed to be correct by rigid body refinement and simulated annealing in CNS (Brunger et al. 1998), using the 2.8 Å data set, yielding reasonable R-factors (Rfree=50.6%, Rcryst=42.5%). This model was further refined and then used for solvent flattening in SOLOMON (Abrahams and Leslie 1996) in conjunction with phase combination of the model in SigmaA (Read 1986). The refined mECI model was then manually fitted to the modified map using the graphics program Xfit (McRee 1999), and the resulting model was subsequently annealed using the 2.2 Å data set. Several rounds of alternating positional refinement in CNS and manual model adjustment in O (Jones et al. 1991) were performed with the high-resolution data until the model was judged to satisfactorily fit the 2|Fo| − |Fc| map. Water molecules were initially included in the model using the DDQ water molecule search program (van den Akker and Hol 1999), and then added manually toward the end of refinement, with positional and B-factor refinement in the final stages until Rfree convergence was achieved. Final statistics for the 2.2Å model are listed in Table 2.
Table 1.
Data collection statistics of mECI
| Crystal | ECI (hi res.) | ECI (low res.) |
| Resolution (Å) | 30.0 (2.28)–2.20 | 30.0 (2.85)–2.80 |
| Space group | I4132 | I4132 |
| a=b=c (Å) | 169.9 | 170.6 |
| Mosaicity (°) | 0.48 | 1.22 |
| Completeness (%) | 99.9 (100) | 95.1 (87.1) |
| Total no. of ref. | 424,963 | 111,389 |
| Unique no. of ref. | 21,510 | 10,224 |
| Redundancy | 19.8 | 10.9 |
| <I/σI> | 11.2 (3.3) | 13.8 (2.4) |
| Rsym (%) | 8.9 (41.1) | 9.6 (30.5) |
| Detector | R-AXIS IV++ | R-AXIS II |
Values in parentheses represent the highest resolution shell.
Table 2.
Summary of refinement statistics for mECI
| Crystal | ECI (hi res.) |
| Resolution (Å)a | 30.032.20 |
| Rcryst/Rfree (%) | 23.9/26.5 |
| Average B-factors (Å2) | |
| Protein [no. of atoms] | 41.9 [2006] |
| Solvent [no. of atoms] | 45.3 [90] |
| Others [no. of atoms] | 48.9 [45] |
| r.m.s. deviation | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 1.4 |
| Ramachandran analysis | |
| Most favored (%) | 91.2 |
| Allowed (%) | 8.4 |
| Disallowed (%) | 0.4b |
aRfree, for 5% of randomly selected reflections omitted from refinement.
b Ala139 lies at the border between generously allowed and disallowed regions.
Structure-based sequence alignment was performed using INDONESIA (D. Madsen., P. Johansson, and G.J. Kleywegt, in prep.), employing the Levitt and Gerstein method (Levitt and Gerstein 1998), followed by some minor manual adjustments. Electrostatic and surface calculations were performed using GRASP (Nicholls et al. 1991) with an ionic strength of 0.1 M, and ligand–protein interactions were analyzed by LIGPLOT (Wallace et al. 1995). Interface surface area calculations were made using the “buried_surface_area” script in CNS (Brunger et al. 1998). Crotonyl-CoA modeling in the active site of both mECI and pECI was performed in the Insight2000 software package (Accelrys Inc.), using the atomic coordinates of octanoyl- CoA from 2DUB as a starting point (Engel et al. 1998), followed by conjugate gradient energy minimization in Discover3 (Accelrys Inc.).
Acknowledgments
The work was supported by grants from the NIH, GM29076 (J.-J.P.K.) and HL30847 and GM08168 (H.S.). The atomic coordinates and structure factors have been deposited at the RCSB Protein Data Bank, Rutgers University, New Brunswick, NJ (accession code 1XX4).
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041303705.
References
- Abrahams, J.P. and Leslie, A.G.W. 1996. Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr. D Biol. Crystallogr. 52 30–42. [DOI] [PubMed] [Google Scholar]
- Bahnson, B.J., Anderson, V.E., and Petsko, G.A. 2002. Structural mechanism of enoyl-CoA hydratase: Three atoms from a single water are added in either an E1cb stepwise or concerted fashion. Biochemistry 41 2621–2629. [DOI] [PubMed] [Google Scholar]
- Bell,A.F., Wu, J., Feng,Y., and Tonge, P.J. 2001. Involvement of glycine 141 in substrate activation by enoyl-CoA hydratase. Biochemistry 40 1725–1733. [DOI] [PubMed] [Google Scholar]
- Bell, A.F., Feng, Y., Hofstein, H.A., Parikh, S., Wu, J., Rudolph, M.J., Kisker, C., Whitty, A., and Tonge, P.J. 2002. Stereoselectivity of enoyl- CoA hydratase results from preferential activation of one of two bound substrate conformers. Chem. Biol. 9 1247–1255. [DOI] [PubMed] [Google Scholar]
- Bennett, M.J., Choe, S., and Eisenberg, D. 1994. Domain swapping: Entangling alliances between proteins. Proc. Natl. Acad. Sci. 91 3127–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benning, M.M., Taylor, K.L., Liu, R.-Q., Yang, G., Xiang, H., Wesenberg, G., Dunaway-Mariano, D., and Holden, H.M. 1996. Structure of 4-chlorobenzoyl coenzyme A dehalogenase determined to 1.8 Å resolution: An enzyme catalyst generated via adaptive mutation. Biochemistry 35 8103–8109. [DOI] [PubMed] [Google Scholar]
- Benning, M.M., Haller, T., Gerlt, J.A., and Holden, H.M. 2000. New reactions in the crotonase superfamily: Structure of methylmalonyl CoA decarboxylase from Escherichia coli. Biochemistry 39 4630–4639. [DOI] [PubMed] [Google Scholar]
- Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. 2000. The Protein Data Bank. Nucleic Acids Res. 28 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54 905–921. [DOI] [PubMed] [Google Scholar]
- Bucklers, L., Umani-Ronchi, A., Retey, J., and Arigoni, D. 1970. Stereochemistry of the enzymatic dehydration of butyryl-CoA [German]. Experientia 26 931–933. [DOI] [PubMed] [Google Scholar]
- Chen, L.S., Jin, S.J., and Tserng, K.Y. 1994. Purification and mechanism of Δ3, Δ 5-t-2,t-4-dienoyl-CoA isomerase from rat liver. Biochemistry 33 10527–10534. [DOI] [PubMed] [Google Scholar]
- Engel, C.K., Mathieu, M., Zeelen, J.P., Hiltunen, J.K., and Wierenga, R.K. 1996. Crystal structure of enoyl-coenzyme A (CoA) hydratase at 2.5 angstroms resolution: A spiral fold defines the CoA-binding pocket. EMBO J. 15 5135–5145. [PMC free article] [PubMed] [Google Scholar]
- Engel, C.K., Kiema, T.R., Hiltunen, J.K., and Wierenga, R.K. 1998. The crystal structure of enoyl-CoA hydratase complexed with octanoyl- CoA reveals the structural adaptations required for binding of a long chain fatty acid-CoA molecule. J. Mol. Biol. 275 847–859. [DOI] [PubMed] [Google Scholar]
- Fillgrove, K.L. and Anderson, V.E. 2000. Orientation of coenzyme A substrates, nicotinamide and active site functional groups in (Di)enoylcoenzyme A reductases. Biochemistry 39 7001–7011. [DOI] [PubMed] [Google Scholar]
- Fong, J.C. and Schulz, H. 1977. Purification and properties of pig heart crotonase and the presence of short chain and long chain enoyl coenzyme A hydratases in pig and guinea pig tissues. J. Biol. Chem. 252 542–547. [PubMed] [Google Scholar]
- Geisbrecht, B.V., Zhu, D., Schulz, K., Nau, K., Morrell, J.C., Geraghty, M., Schulz, H., Erdmann, R., and Gould, S.J. 1998. Molecular characterization of Saccharomyces cerevisiae Δ3, Δ2-enoyl-CoA isomerase. J. Biol. Chem. 273 33184–33191. [DOI] [PubMed] [Google Scholar]
- Gurvitz, A., Mursula, A.M., Firzinger, A., Hamilton, B., Kilpelainen, S.H., Hartig, A., Ruis, H., Hiltunen, J.K., and Rottensteiner, H. 1998. Peroxisomal Δ3-cis-Δ2-trans-enoyl-CoA isomerase encoded by ECI1 is required for growth of the yeast Saccharomyces cerevisiae on unsaturated fatty acids. J. Biol. Chem. 273 31366–31374. [DOI] [PubMed] [Google Scholar]
- Holden, H.M., Benning, M.M., Haller, T., and Gerlt, J.A. 2001. The crotonase superfamily: Divergently related enzymes that catalyze different reactions involving acyl coenzyme a thioesters. Acc.Chem.Res. 34 145–157. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kiema, T.R., Engel, C.K., Schmitz, W., Filppula, S.A., Wierenga, R.K., and Hiltunen, J.K. 1999. Mutagenic and enzymological studies of the hydratase and isomerase activities of 2-enoyl-CoA hydratase-1. Biochemistry 38 2991–2999. [DOI] [PubMed] [Google Scholar]
- Kurimoto, K., Fukai, S., Nureki, O., Muto, Y., and Yokoyama, S. 2001. Crystal structure of human AUH protein, a single-stranded RNA binding homolog of enoyl-CoA hydratase. Structure 9 1253–1263. [DOI] [PubMed] [Google Scholar]
- Levitt, M. and Gerstein, M. 1998. A unified statistical framework for sequence comparison and structure comparison. Proc. Natl. Acad. Sci. 95 5913–5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRee, D.E. 1999. Practical protein crystallography, 2nd ed. Academic Press, San Diego, CA.
- Modis, Y., Filppula, S.A., Novikov, D.K., Norledge, B., Hiltunen, J.K., and Wierenga, R.K. 1998. The crystal structure of dienoyl-CoA isomerase at 1.5 Å resolution reveals the importance of aspartate and glutamate sidechains for catalysis. Structure 6 957–970. [DOI] [PubMed] [Google Scholar]
- Muller-Newen, G. and Stoffel, W. 1993. Site-directed mutagenesis of putative active-site amino acid residues of 3,2-trans-enoyl-CoA isomerase, conserved within the low-homology isomerase/hydratase enzyme family. Biochemistry 32 11405–11412. [DOI] [PubMed] [Google Scholar]
- Muller-Newen, G., Janssen, U., and Stoffel, W. 1995. Enoyl-CoA hydratase and isomerase form a superfamily with a common active-site glutamate residue. Eur. J. Biochem. 228 68–73. [DOI] [PubMed] [Google Scholar]
- Mursula, A.M., van Aalten, D.M., Hiltunen, J.K., and Wierenga, R.K. 2001. The crystal structure of Δ (3)- Δ (2)-enoyl-CoA isomerase. J. Mol. Biol. 309 845–853. [DOI] [PubMed] [Google Scholar]
- Mursula, A.M., Hiltunen, J.K., and Wierenga, R.K. 2004. Structural studies on Δ (3)- Δ (2)-enoyl-CoA isomerase: The variable mode of assembly of the trimeric disks of the crotonase superfamily. FEBS Lett. 557 81–87. [DOI] [PubMed] [Google Scholar]
- Nicholls, A., Sharp, K.A., and Honig, B. 1991. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11 281–296. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Palosaari, P.M., Kilponen, J.M., Sormunen, R.T., Hassinen, I.E., and Hiltunen, J.K. 1990. Δ 3, Δ 2-enoyl-CoA isomerases. Characterization of the mitochondrial isoenzyme in the rat. J. Biol. Chem. 265 3347–3353. [PubMed] [Google Scholar]
- Partanen, S.T., Novikov, D.K., Popov, A.N., Mursula, A.M., Hiltunen, J.K., and Wierenga, R.K. 2004. The 1.3 Å crystal structure of human mitochondrial Δ3-Δ2-enoyl-CoA isomerase shows a novel mode of binding for the fatty acyl group. J. Mol. Biol. 342 1197–1208. [DOI] [PubMed] [Google Scholar]
- Pearson, W.R. and Lipman, D.J. 1988. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. 85 2444–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflugrath, J.W. 1999. The finer things in X-ray diffraction data collection. Acta Crystallogr. D Biol. Crystallogr. 55 (Pt. 10) 1718–1725. [DOI] [PubMed] [Google Scholar]
- Read, R.J. 1986. Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Crystallogr. A 42 140–149. [Google Scholar]
- Såli, A. and Blundell, T.L. 1993. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234 779–815. [DOI] [PubMed] [Google Scholar]
- Schulz, H. and Kunau, W.-H. 1987. β-oxidation of unsaturated fatty acids: A revised pathway. Trends Biochem. Sci. 12 403–406. [Google Scholar]
- Stoffel, W., Ditzer, R., and Caeser, H. 1964. Der stoffwechsel der ungesättigen fettsäuren. III. Zur β-oxidation der mono-und polyenfettsäuren. Der mechanismus der enzymatischen reaktionen an 3-cis-enoyl-CoA-verbindungen. Hoppe Seylers Z. Physiol. Chem. 339 167–181. [PubMed] [Google Scholar]
- Todd, A.E., Orengo, C.A., and Thornton, J.M. 2002. Plasticity of enzyme active sites. Trends Biochem. Sci. 27 419–426. [DOI] [PubMed] [Google Scholar]
- Truglio, J.J., Theis, K., Feng, Y., Gajda, R., Machutta, C., Tonge, P.J., and Kisker, C. 2003. Crystal structure of Mycobacterium tuberculosis MenB, a key enzyme in vitamin K2 biosynthesis. J. Biol. Chem. 278 42352–42360. [DOI] [PubMed] [Google Scholar]
- van den Akker, F. and Hol, W.G. 1999. Difference density quality (DDQ): A method to assess the global and local correctness of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 55 206–218. [DOI] [PubMed] [Google Scholar]
- Wallace, A.C., Laskowski, R.A., and Thornton, J.M. 1995. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 8 127–134. [DOI] [PubMed] [Google Scholar]
- Whittingham, J.L., Turkenburg, J.P., Verma, C.S., Walsh, M.A., and Grogan, G. 2003. The 2-Å crystal structure of 6-oxo camphor hydrolase. New structural diversity in the crotonase superfamily. J. Biol. Chem. 278 1744–1750. [DOI] [PubMed] [Google Scholar]
- Zhang, W., Wei, Y., Luo, L., Taylor, K.L., Yang, G., Dunaway-Mariano, D., Benning, M.M., and Holden, H.M. 2001. Histidine 90 function in 4-chlorobenzoyl-coenzyme a dehalogenase catalysis. Biochemistry 40 13474–13482. [DOI] [PubMed] [Google Scholar]
- Zhang, D., Yu, W., Geisbrecht, B.V., Gould, S.J., Sprecher, H., and Schulz, H. 2002. Functional characterization of Δ3, Δ2-enoyl-CoA isomerases from rat liver. J. Biol. Chem. 277 9127–9132. [DOI] [PubMed] [Google Scholar]