

Abstract
The presence of alanine (Ala) or acetyl serine (AcSer) instead of the normal Val residues at the N-terminals of either the α- or the β-subunits of human adult hemoglobin confers some novel and unexpected features on the protein. Mass spectrometric analysis confirmed that these substitutions were correct and that they were the only ones. Circular dichroism studies indicated no global protein conformational changes, and isoelectric focusing showed the absence of impurities. The presence of Ala at the N-terminals of the α-subunits of liganded hemoglobin results in a significantly increased basicity (increased pKa values) and a reduction in the strength of subunit interactions at the allosteric tetramer–dimer interface. Cooperativity in O2 binding is also decreased. Substitution of Ala at the N-terminals of the β-subunits gives neither of these effects. The substitution of Ser at the N terminus of either subunit leads to its complete acetylation (during expression) and a large decrease in the strength of the tetramer–dimer allosteric interface. When either Ala or AcSer is present at the N terminus of the α-subunit, the slope of the plot of the tetramer–dimer association/dissociation constant as a function of pH is decreased by 60%. It is suggested that since the network of interactions involving the N and C termini of the α-subunits is less extensive than that of the β-subunits in liganded human hemoglobin disruptions there are likely to have a profound effect on hemoglobin function such as the increased basicity, the effects on tetramer strength, and on cooperativity.
Keywords: hemoglobin, acetylation, subunit interfaces, tetramer stability, protein protonation, tetramer disassembly
The N-terminal Val residues of both the α- and β-subunits of human adult hemoglobin are involved in a number of well-known important functions as the tetramer undergoes the transition between the deoxy (T-state) and oxy (R-state) conformations associated with the cooperative release or uptake of O2, respectively (Perutz 1989). Among these functions are the alkaline Bohr effect involving the uptake or release of protons during this conformational switch, the binding of the allosteric effectors chloride (at Val-1[α]) and 2,3-DPG (at Val-1[β]) in the T-state to promote O2 release, and the binding of CO2. However, there are other processes involving N-terminal residues that are less well-understood but are also likely to be important. Among these are (1) the role of N-terminal acetylation in a fraction of the γ-subunit of fetal Hb, in the ζ subunits of some of the embryonic hemoglobins and in some fish hemoglobins; (2) the function of the greatly increased tetramer strength5 of fetal Hb (HbF) compared to that of adult Hb (HbA), which is due to a specific N-terminal protonation of the γ-subunit; and (3) the mechanism and possible physiological role of the pH dependence of tetramer–dimer association/ dissociation. We have sought in this communication to elucidate these functions further using site-directed mutagenesis of N-terminal residues of α- or β-subunits separately.
Acetylation of protein amino groups (N-terminal α-amino groups or lysine ɛ-amino groups) is one of the most common types of post-translational protein modifications (Tsunasawa and Sakiyama 1984; Bradshaw et al. 1998; Polevoda and Sherman 2003). N-terminal α-amino acetylation has been estimated to occur in about 90% of nascent eukaryotic proteins during biosynthesis (Yan et al. 1989) followed by further N-terminal processing to expose the new N-terminal residues present in mature proteins (Sheff and Rubenstein 1992). The function of α-amino group acetylation is incompletely defined, although its role in the protein kinase A system (Caesar and Blomberg 2004) and the myosin/tropomyosin/actin systems (Urbancikova and Hitchcock-De Gregori 1994) shows that it modulates protein–protein interactions. We reported a similar function for the N-terminal AcGly residue of the minor component of fetal Hb, HbF1 (Manning and Manning 2001). The acetylation of ɛ-amino groups of certain lysine residues occurs in some proteins, especially in histones where it is strongly correlated with gene expression (Allfrey et al. 1964), although the mechanism is still unclear (Kouzarides 2000).
We have focused on N-terminal Ala and Ser substitutions in this communication since these are subject to various extents of acetylation in vivo, whereas the natural N-terminal Val residue of human adult Hb is not. For example, the N-terminal Ser that occurs in some subunits of the embryonic hemoglobins is completely acetylated (Huehns et al. 1961; Randhawa et al. 1984), although its function is unknown. The ability to study the effects of a specific substitution at one or the other type of subunit greatly enhances the ability of site-directed mutants to provide new information on Hb function (Yagami et al. 2002). However, such recombinant mutants must be rigorously characterized by various methods, especially mass spectrometry, in order for the conclusions derived from their study to be valid. We have previously reported that the recombinant hemoglobins expressed in yeast (Wagenbach et al. 1991) faithfully retain the properties of native, natural hemoglobin (Martin de Llano et al. 1993, 1994; Martin de Llano and Manning 1994; Yagami et al. 2002).
Protonation of certain side chains in hemoglobin is involved in many of its functions (Perutz 1989). We demonstrated, by examining the pH dependence of tetramer–dimer association/dissociation over the pH 6.5–9.0 range, that the more favorable protonation of the N terminus of the γ-subunit of HbF (due to the higher pKa value of 8.1 of Gly-1[γ] compared to the pKa of 7.1 for Val-1[β] of HbA) was a major factor in conferring HbF with its enhanced tetramer strength (Manning and Manning 2001). We expand that experimental approach with other hemoglobins as described here and correlate it with specific N-terminal acetylation.
Results
Mutagenesis and expression
The same procedures used for previous site-directed mutagenesis studies (Martin de Llano et al. 1993, 1994; Martin de Llano and Manning 1994; Dumoulin et al. 1997, 1998; Yagami et al. 2002) were employed with the following exceptions: Oligonucleotides were prepared by Gene Link. After mutagenesis, the entire cDNA sequence for each mutant was confirmed as correct at the Molecular Biology Core Facility of the Forsyth Center. Expression of recombinant hemoglobins in yeast was performed in a New Brunswick Bio Flo IV fermenter at the Chemical Engineering Facility of Northeastern University. The hemoglobin yields were improved to 75–150 mg per 15 L fermentation through an initial screening of colonies that showed the best growth (Yagami et al. 2002).
Purification and characterization of hemoglobins
These procedures have been described previously (Martin de Llano et al. 1993, 1994; Dumoulin et al. 1997, 1998; Yagami et al. 2002) and provide pure hemoglobins after CM-52 chromatography and Mono S chromatography by FPLC. The pure hemoglobins were carefully characterized by one of the authors (J.C.P.) using mass spectrometry (Beavis and Chait 1996; Li et al. 1999). Circular dichroism measurements and isoelectric focusing were performed as described previously (Martin de Llano and Manning 1994). The results from these three analyses are described below.
Extents of acetylation
The presence of Ala at the N terminus of either the α- or the β-subunits of Hb (in separate studies) led to its acetylation in a fraction of the sample; the β-subunit N terminus was acetylated more than the α-subunit N terminus (65% vs. 33% of the total, respectively) with the remainder being unacetylated in each case. These acetylated and unacetylated forms were successfully separated by a combination of chromatography on CM-52 followed by FPLC on Mono S as described in Materials and Methods. We elected to study the unacetylated Ala subunits and to use the Ser replacements (described below) to study the effects of acetylation, since Ser was completely acetylated in the yeast expression system, thus permitting a simpler separation with higher yields. The unacetylated N-terminal Ala on the α- or β-subunits contained the normal Val on the other subunit, i.e., the terminology α2Aβ2 or α2β2A is used, where the N-terminal Ala is denoted by a superscript A on that subunit; the absence of a superscript indicates the presence of the natural Val residue at that site, e.g., α2β2, the usual terminology for HbA, is understood to have Val at both subunit N-terminals.
When Ser is present at the N terminus of the α- or β-subunits, the extent of its acetylation by the yeast expression system was essentially complete resulting in a less complex profile on CM-52 compared to that for the N-terminal Ala substitutions. The increased acetylation of Ser compared to Ala is consistent with the catalytic properties of the N-terminal acetyltransferase system in eukaryotes, i.e., N-terminal residues are acetylated in order of decreasing catalytic efficiency Ser > Ala > Gly (Polevoda and Sherman 2003). The terminology α2AcSβ2 and α2β2AcS is used to designate which subunit has N-terminal AcSer and which one has the normal N-terminal Val, as described above for the Ala substitutions. All samples were shown to be the desired materials, to be homogenous, and to be correctly folded as described next.
Mass spectrometry analysis
A three-step strategy was used in the characterization of the globin chains as previously published (Li et al. 1999) with modifications described herein. Initially, the average molecular masses of the intact globin α and β subunits were determined by ESI/MS and the results for the five hemoglobin samples are shown in Table 1. A typical mass spectrum is presented in Figure 1A ▶ for the α2AcSβ2 sample with its corresponding deconvoluted spectrum shown in the inset. The mass errors obtained from the measurements are below 100 ppm for all five recombinant hemoglobins, and the average molecular masses are consistent with the expected amino acids substitutions in each subunit as well as with the nature of the amino acid modifications where appropriate (Table 1). In each of the five samples only a single component for each subunit was present. Minor peaks, observed to the right of each major peak in the deconvoluted spectrum (inset to Fig. 1A ▶), correspond to sodium, potassium, and iron adducts and are present in relatively small amounts.
Table 1.
Mass spectrometry analysis of intact subunits
| Average molecular mass/μ | ||||||
| α-Chain | β-Chain | |||||
| Sample | Expected | Measured | ΔM | Expected | Measured | ΔM |
| α2Aβ2 | 15098.3 | 15098.5 ± 0.2 | 0.2 | 15867.2 | 15867.1 ± 0.5 | −0.1 |
| α2β2A | 15126.3 | 15126.6 ± 0.1 | 0.3 | 15839.1 | 15839.0 ± 0.3 | −0.1 |
| α2Aβ2A | 15098.3 | 15098.5 ± 0.2 | 0.2 | 15839.1 | 15838.9 ± 0.4 | −0.2 |
| α2AcSβ2 | 15156.3 | 15156.3 ± 0.4 | 0.0 | 15867.2 | 15867.3 ± 0.6 | 0.1 |
| α2β2AcS | 15126.3 | 15126.2 ± 0.3 | −0.1 | 15897.2 | 15897.5 ± 0.5 | 0.3 |
Average molecular masses for the intact globin chains are measured by ESI/MS in an ion trap mass spectrometer. Molecular masses were measured from the ion envelope in the charge state interval of 11+ through 20+ (see Fig. 1 ▶ for an example). Uncertainties represent the standard error of the mean (σ, n=10). δM values were calculated as expected mass subtracted from the measured value, and the expected values were calculated from the protein sequences.
Figure 1.
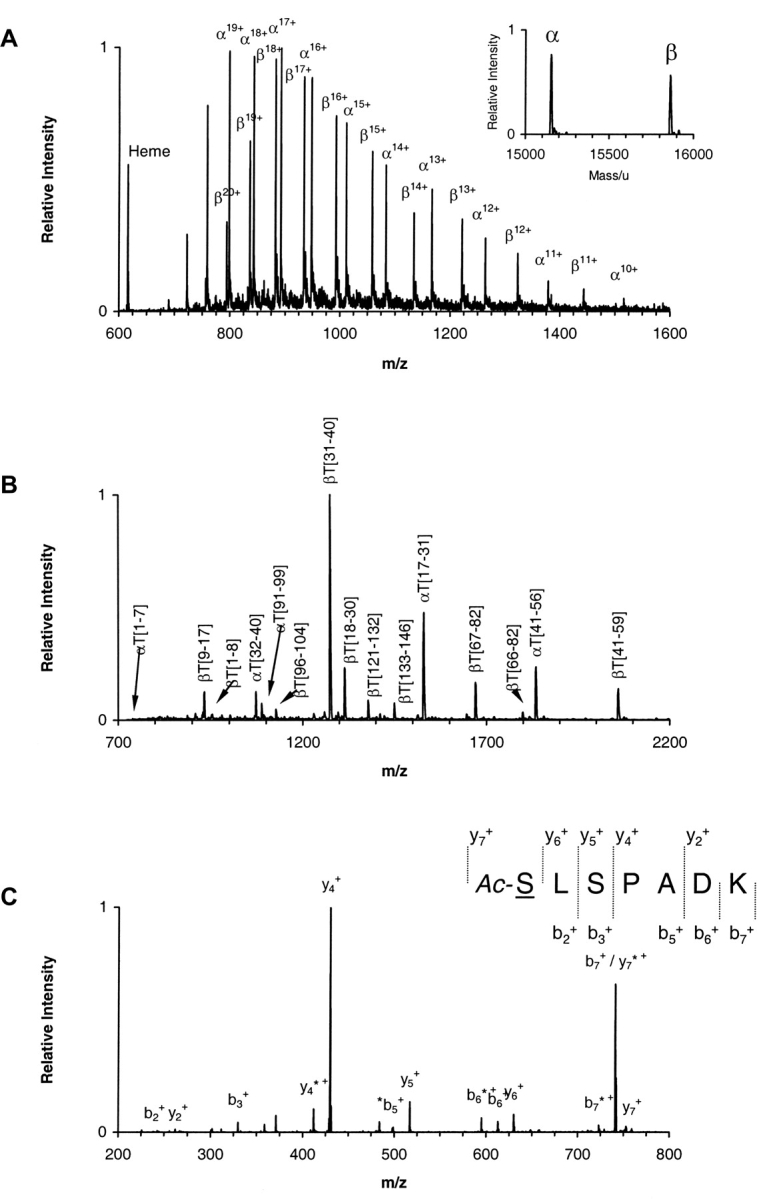

Mass spectrometry analysis. (A) ESI/MS analysis of hemoglobin sample α2AcSβ2. The ion envelopes for α and β chains are shown in the raw m/z spectrum. (Inset) Transformed mass spectrum after deconvolution of the ion envelopes encompassing the region of charge states 11+ through 20+ (m/z range 750–1500). (Refer to text for experimental details.) (B) MALDI-QqTOF/MS spectrum of a tryptic digest of hemoglobin sample α2AcSβ2. Peaks are labeled as α- or β-chain peptides followed by T for trypsin and the number of the first and last residues in the peptide in accordance with the numbering system for the primary structure of the entire polypeptide chain. The combined MALDI data yielded information covering 100% of both α and β globin chains. (C) ESI/MS/MS analysis of the singly charged peak of the α-chain N-terminal tryptic peptide αT(1–7) from hemoglobin sample α2AcSβ2. The underlined residue was mutated. Only b and y ions are labeled for clarity. Unidentified peaks are labeled with an asterisk. The mutated residue was assigned based on the occurrence of ions y6+ and y7+ and the PTC-derivative of the same peptide. (Refer to text for experimental details.) (D) ESI/MS/MS hypothesis-driven analysis of the N-terminal tryptic peptide αT(1–7) of human hemoglobin (row a) and recombinant hemoglobin samples α2Aβ2 (row b), α2β2A (row c) and α2Aβ2A (row d). Spectra show the isolated precursor ions selected based on either the presence or absence of the Val → Ala modification. The hypothetical precursor ions were subsequently fragmented.
The tryptic peptide mixtures were analyzed by both MALDI-TOF/MS andMALDI-QqTOF/MS. Figure 1B ▶ is a representative example of a peptide map obtained by MALDI-QqTOF/MS for α2AcSβ2. Although this technique produced mass errors around 10 ppm for internally calibrated spectra, the sequence coverage was not complete due to limitations in the scanning range, particularly above m/z 3400. MALDI-TOF/MS was thus used to extend the coverage to 100% for both chains from each hemoglobin sample (data not shown).
We have also applied MALDI-TOF/MS to ascertain the nature of the N-terminal residue in the peptides bearing the expected Val → AcSer substitution, which produces a mass difference that is indistinguishable from a Val → Glu substitution. However, the Val → Glu substitution would have a free N terminus and thus be amenable to further chemical modification, whereas the Val → AcSer, being already modified, would not. MALDI-TOF/MS analysis of the PITC-derivatized peptides from sample α2AcSβ2 showed that the N-terminal peptides αT(1–7) and βT(1–8) were modified by one and two phenylthiocarbamyl groups (PTC), respectively (data not shown). Since the PTC group reacts only with free primary amines, it is expected to react only with the ɛ-amino group on Lys7 in the α-chain peptide and with both the ɛ-amino group on Lys8 and the α-amino group on Val1 in the β-chain peptide. In the case of the recombinant hemoglobin α2β2AcS, two and one PTC groups were found respectively for the α- and β-chain peptides. These results are in agreement with the presence of a blocked N-terminal peptide only in the α-chain of sample α2AcSβ2 and a blocked N-terminal peptide only in the β-chain in sample α2β2AcS.
Finally, the positioning of the amino acid modifications within the primary structure of the peptides was confirmed by mass spectrometric fragmentation. As a representative example, Figure 1C ▶ shows the MS/MS spectrum of the precursor ion 759.38 for peptide αT(1–7) of expected sequence AcSLSPADK. Two major series of ions, b and y, are observed in this spectrum. Confirmation of the amino acid substitution and modification Val → AcSer is provided by the presence of ions y6+ and y7+. The combined set of data from this three-step approach leads to the unambiguous confirmation of the putative single amino acid substitution and modification.
A variation of the third step was employed in the analysis of samples α2Aβ2, α2β2A, and α2Aβ2A, which were expected to contain the amino acid substitution Val → Ala. Figure 1D ▶ shows MS/MS spectra obtained by hypothesis-driven mass spectrometry (Kalkum et al. 2003). In this case, precursor ions selected for fragmentation were not obtained from the MALDI-TOF peptide maps, but were calculated from the protein sequences using the hypothesis that the modification Val → Ala was either present or not. The same was done for HbA used as a control. The results show only the single species Val → Ala to be present in α2Aβ2, the single species Val β1 → Ala in α2β2A, and both species in sample α2Aβ2A as observed from the measurements of the average molecular mass of the intact chains and the peptide maps. The peptides were subsequently fragmented and a sequence was produced with the expected amino acid substitution in the correct position (data not shown).
Electrophoretic properties of substitutions at N-terminal residues
Purity was established by isoelectric focusing (IEF), as shown in Figure 2 ▶, although the mobilities introduced by some of these substitutions was unexpected. For example, valine and alanine as the free amino acids have nearly the same pKa for their α-amino groups (9.62 and 9.69, respectively) (Meister 1965). Generally, an α-NH2 group at the N terminus of a protein has a pKa value that is lower than that of the corresponding free amino acid, e.g., the Val α-NH2 groups at the N-terminals of the Hb subunits have pKa values of 6.95 for Val-1(α) and 7.05 for Val-1(β) (Garner et al. 1975). Surprisingly, as shown in Figure 2 ▶, the presence of Ala-1 on the α-subunits (α2Aβ2) leads to a significant isoelectric shift toward the cathode, indicative of an increased basicity, i.e., higher pKa, compared to the migration of α2β2, which has Val at the N-terminals of its α-subunits. From the relative IEF positions of the standard HbA and HbS, which have a difference of two charged residues per tetramer (Glu-6[β] → Val-6[β]), we estimate that the increased positive charge of Ala-1(α) amounts to about one positive charge per tetramer. The presence of Ala at the N terminus of the β-subunit (α2β2A) leads to only a very slight cathodal shift. The mutant with Ala on the N-terminals of both α- and β-subunits (α2Aβ2A) behaves electrophoretically in an additive manner for α2Aβ2 and α2β2A.
Figure 2.

Isoelectric focusing of purified recombinant Hb mutants. Approximately 5 μg of each Hb was applied to a pH 6.0–8.0 range gel (Hb Resolve, Perkin-Elmer Life Sciences). Electrophoresis was performed for 30 min at 600 V and then for 45 min at 900 V at 10°C. The gel was stained with the JBZ stain (Perkin-Elmer-Wallach). The anode is at the top and the cathode is at the bottom. The standard hemoglobins on the far left and the far right are hemoglobins A, F, S, and C. The identity of each recombinant hemoglobin is shown under each lane. A small superscript “A” indicates an N-terminal Ala on that subunit. A small superscript “AcS” indicates an N-terminal acetylserine on that subunit. Subunits without superscripts contain N-terminal Val, e.g., α2β2 is HbA.
Upon isoelectric focusing, the presence of AcSer at the N-terminal residues of the β-subunits (α2β2AcS) results in a slightly greater migration toward the anode than the same substitution on the α-subunit (α2AcSβ2) (Fig. 2 ▶). Since α2β2 migrates close to these positions, we conclude that there is very little positive charge on Val-1 in the subunits of adult HbA.
Circular dichroism
Circular dichroism (CD) studies on a variety of recombinant hemoglobins expressed in yeast had previously shown that their spectral properties in the far-ultraviolet, the nearultraviolet, and the visible regions were practically the same as those in the corresponding regions of natural HbA (Martin de Llano and Manning 1994). For the hemoglobins studied here, only the far-ultraviolet spectra reflecting global protein conformation are reported. Thus, for α2β2A and α2β2AcS (Fig. 3A ▶), α2AcSβ2 (Fig. 3B ▶), α2Aβ2 (Fig. 3C ▶), and the CD profiles in the far UV were practically super-imposable on that for HbA. These results are completely consistent with earlier conclusions, i.e., that overall global folding of these recombinant hemoglobins expressed in this yeast system is normal. CD spectra in the other spectral regions showed no anomalies other than minor fluctuations (data not shown).
Figure 3.
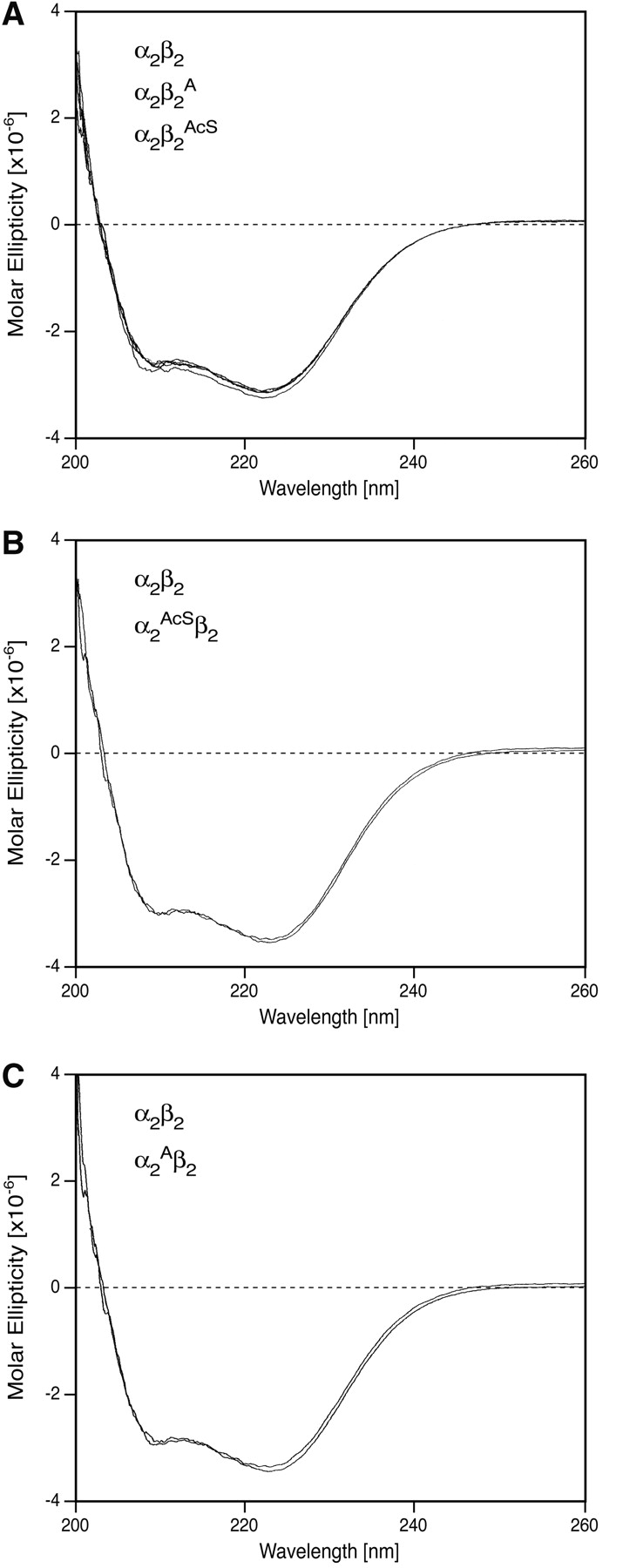
Circular dichroism spectra were recorded as described in the text. An Hb concentration of 10 μM as the tetramer was used in the far-ultraviolet region. (A) Samples α2β2, α2β2A, and α2β2AcS; (B) samples α2β2 and α2AcSβ2; (C) samples α2β2 and α2Aβ2.
Effects of N-terminal Ala and AcSer substitutions on the tetramer–dimer equilibrium constant of liganded hemoglobin (tetramer strength)
Previous reports from this laboratory have described a sensitive, reliable, and simple procedure to evaluate the dissociation properties of the tetramer–dimer allosteric interface of hemoglobins in the liganded state (Manning et al. 1996, 1999). This rapid gel filtration procedure employs the Pharmacia FPLC Director software for data analysis and provides equilibrium constants for liganded natural adult HbA that agree very well with published values obtained using other procedures (Manning et al. 1996, 1999).
The tetramer–dimer association/dissociation profiles for α2Aβ2, α2β2A, α2AcSβ2, and α2β2AcS at pH 7.5 are shown in Figure 4 ▶. The Kd can be read directly from the insets at the intersection of the experimental line with the horizontal line drawn at the value of 1 on the Y-axis (see Manning et al. [1996, 1999] for derivation of equation and explanation). Compared to liganded HbA (α2β2) with a Kd value of 0.68 μM, only α2β2A (Fig. 4 ▶, upper left panel) has similar tetramer strength with a Kd of 0.49 μM. Of particular interest, α2Aβ2 (Fig. 4 ▶, lower left panel) dissociates nearly an order of magnitude more (Kd=3.75 μM) than α2β2A or α2β2. The tetramer with Ala at the N-terminals of both subunits, α2Aβ2A, has a Kd value of 1.10 μM, which is less than the average Kd values found for α2Aβ2 and α2β2A and closer to that of the latter. This result could be due to communication between the subunit types with the β-subunit influence predominating. However, more study is needed on this point.
Figure 4.

Tetramer–dimer dissociation constants at pH 7.5. The gel filtration procedure used to determine these tetramer–dimer dissociation constants (Kd values) and the treatment of the data have been described in detail in Manning et al. (1996, 1999). The plots show the amounts of Hb present on the column during the analysis to attain a certain percent of tetramer, the functional O2-carrying structure of Hb. Further treatment of the data is shown in the inset where the Kd value can be read directly as the intersection point of the horizontal line at 1 on the Y axis with the experimental line.
Both N-terminal AcSer tetramers, α2AcSβ2 and α2β2AcS, dissociate extensively with Kd values of 7.67 μM and 2.92 μM, respectively (Fig. 4 ▶, two right panels). This result indicates that removal of the positive charge at the N-terminals of either subunit decreases the strength of the network of interactions in these regions. The detailed structural changes in α2Aβ2 and α2AcSβ2 are not known, so we cannot be certain why each disrupts the allosteric interface, although we speculate below on possible reasons.
Effects of protonation on tetramer strength
Understanding the effects of slightly acidic pH on Hb could be important in deducing the mechanism of the Root effect (Root 1931) exhibited by some fish hemoglobins; the Root effect is the ability of some hemoglobins to bind protons at low pH (down to pH 5.6) to promote the continuous release of O2 to provide such fish with a physiological advantage at low O2 tensions. In contrast, adult human hemoglobin exhibits an acid Bohr effect, i.e., oxygen affinity decreases from pH 9 to pH 6.5 but then increases at lower pH. Atha and Riggs (1976), Rollema et al. (1980), and Chu and Ackers (1981) have previously reported the effects of pH on the tetramer–dimer association/ dissociation properties at the α1β2 (α2β1) allosteric interface of liganded human HbA. They found that as the pH decreased, tetramer dissociation to dimers increased. We later expanded these studies to include HbF which showed a similar profile for the pH-dependence of dissociation as did HbA from pH 6.5 to pH 8.0, although the overall extent of dissociation was decreased (Dumoulin et al. 1997; Manning and Manning 2001). The slope of the dissociation/association profiles between pH 6.5 and 8.0 for both hemoglobins was about 1, meaning that for each 10-fold change in H+ concentration (1 pH unit) there was a 10-fold change in the Ka (or Kd). Above pH 8.0, however, HbF reversed its profile but HbA did not (Manning and Manning 2001). We attributed this behavior between pH 8.0–9.0 for HbF as due to the selective protonation of Gly-1 of the γ-subunit, which has a pKa of 8.1 and is protonated an order of magnitude more readily than Val-1(β) of the β-subunit of HbA (pKa=7.1), thus providing HbF with its increased tetramer strength.
In this communication we have studied the effects of pH on tetramer–dimer association of the N-terminal modified recombinant samples over the range of 6.5– 9.0 and report the results as Ka values, which are the reciprocals of Kd values, in order to compare the results (Fig. 5 ▶) with previously reported data. Earlier results (Manning and Manning 2001) of the pH profile versus Ka are also shown in Figure 5 ▶ for α2γ2 (HbF) as the dashed line; α2β2 (HbA) and α2γ2Ac (HbF1) as the middle solid line. Comparing the Ka versus pH profile for the β-subunit substitutions, α2β2A and α2β2AcS, the slopes of about 1 for the lines between pH 8.5 and 6.5 are superimposable on that for HbA. However, the slopes for α2Aβ2 and α2AcSβ2 with substitutions on the α-subunits are about 0.4. Since Ala-1(α) in α2Aβ2 is likely already predominantly protonated (see IEF gel in Fig. 2 ▶) and AcSer-1(α) in α2AcSβ2 cannot be protonated, neither residue contributes further to the protoninduced association/dissociation process. Thus, in naturally-occurring liganded HbA, Val-1(α) is inferred to be implicated as a site of protonation involved in tetramer strength and dissociation/association. The profile for α2Aβ2AcA also has a slope of 0.4, consistent with this interpretation (data not shown).
Figure 5.

Association constants for tetramer–dimer assembly as a function of pH. Dissociation constants (Kd) measured as in Figure 4 ▶, were determined at each pH indicated and then converted to association constants (Ka=1/Kd). The data for α2γ2 (HbF) (dashed line), α2β2 (HbA), and α2γ2AcG (HbF1) have been reported previously (Manning and Manning 2001). The symbols for each sample are α2γ2, ▴; α2β2, ▵; α2γ2AcG, x; α2β2A,•; α2β2AcS, ▪; α2AcSβ2, □, α2Aβ2, ○.
Effects of Ala and AcSer substitutions on O2-binding and cooperativity
The oxygen affinity (P50 values) and cooperativity (n values, Hill coefficients) for the recombinant hemoglobins with Ala or AcSer N-terminal substitutions are given in Table 2. Values are reported for eachHb alone, Hb with chloride, and Hb with DPG. Each Hb retained a normal ability to bind O2 and to respond to allosteric regulators, but did so with distinct differences in the degree of subunit cooperativity which depended on the location of the substitution. Decreases in cooperativity were consistently observed for the Ala and the AcSer substitutions on the α-subunits either in the presence or absence of allosteric effectors. This can be readily seen by comparing the slopes in each left and right vertical pairs of upper and lower panels in Figure 6 ▶. For those with either Ala or AcSer at the N terminus of the α-subunit, the Hill coefficients were lowered significantly and reproducibly to values< 2 either alone (Table 2) or in the presence of chloride (Table 2) or 2,3-DPG (upper panels in Fig. 6 ▶; Table 2). In contrast, the hemoglobins with the same substitutions on the β-subunit had normal cooperativity either alone or with chloride or 2,3-DPG (Table 2; lower panels in Fig. 6 ▶).
Table 2.
Summary of P50and n values
| No additions | +NaCl (200 mM) | +DPG (4 mM) | ||||
| Hemoglobin | P50 | n | P50 | n | P50 | n |
| α2β2 (HbA) | 7 | 2.5 | 16 | 2.7 | 24 | 2.7 |
| α2Aβ2 | 7 | 1.8 | 16 | 2.1 | 25 | 1.8 |
| α2β2A | 8 | 2.5 | 17 | 2.5 | 28 | 2.5 |
| α2Aβ2A | 7 | 2.1 | 15 | 2.0 | 24 | 1.7 |
| α2AcSβ2 | 9 | 1.8 | 17 | 1.9 | 31 | 1.8 |
| α2β2AcS | 9 | 2.3 | 20 | 2.3 | 23 | 2.3 |
Figure 6.

Hill Plots for N-terminal substituted mutant hemoglobins. The samples are identified on each panel: (upper left) α2Aβ2, (upper right) α2AcSβ2, (lower left) α2β2A, (lower right) α2β2AcS. These Hill plots were calculated from the O2-binding curve in the presence of 2,3-DPG. The points on each line were read from the HemOScan chart paper at each 5% O2-tension between 20% and 70% O2 saturation. The best fit line shown was then drawn through these points with weighting on the mid-range values. These data are from the O2-binding data in the presence of 2,3-DPG at pH 7.5. A similar lowering of the Hill coefficient for the N-terminal α-subunit substitutions was found for the O2-binding data for Hb alone and for Hb with chloride.
The two α-subunit substituted tetramers with lowered cooperativity are also those found to lower the slope of the association/dissociation profile as a function of pH as described above (Fig. 5 ▶). We speculate below regarding a possible relationship between these effects. It is interesting to note that the embryonic hemoglobins Gower-1 and Portland-2, where α-subunits are replaced by ζ-subunits containing N-terminal Ac-Ser, also have lowered cooperativity, whereas hemoglobin Gower-2, which has α-subunits with N-terminal Val, has normal cooperativity (He and Russell 2001). The mechanism of this effect and its significance are unknown.
Discussion
N-terminal protonation and basicity changes
The significant increase in basicity that accompanies the substitution of Ala for Val at the N terminus of the α-subunits (α2Aβ2) together with its decreased tetramer–dimer interface strength, suggest that the substituted Ala has probably been displaced from the α–α network of interactions normally present in liganded HbA (Fig. 7A ▶). This network, which involves the N-terminal region of one α-subunit and the C-terminal region of the other α-subunit, is not extensive in liganded Hb (Perutz 1989) and is very close to the tetramer–dimer allosteric interface whose dissociation/association constants are measured in Figures 4 ▶ and 5 ▶. Displacement of N-terminal Ala in α2Aβ2 may be more disruptive than the same substitution in α2β2A in the β–β network, and could explain some of the effects on Hb described in this communication. The β–β network of interactions is shown in Figure 7B ▶. It has more extensive interactions than does the α–α network. The replacement of Val-1(β) byAla in α2β2A leads to little change in basicity compared to that in α2Aβ2 (see Fig. 2 ▶) perhaps because the β–β network still retains other strong interactions preventing displacement of the substituted Ala-1. This suggestion is consistent with the tetramer strength of α2β2A being close to that for HbA (Figs. 4 ▶, 5 ▶). In contrast, the α2β2AcS substitution negates the N-terminal basicity and is disruptive to tetramer strength. The data for N-terminal α- and β-substitutions suggest that both α–α and β–β interactions play a major role in generating tetramer strength in Hb.
Figure 7.

N-terminal/C-terminal interactions for liganded human hemoglobin A. (A) α–α interactions; (B) β–β interactions. The coordinates used are from PDB=5 HHO by Shaanan (1983). The α-subunits in α–α are designated as chains A and C and the β-subunits in β–β are designated as chains B and D. In α–α the white lines with numbers (in angstroms) are H-bonds. The 2.24 Å distance is from one carboxylate oxygen of Arg-141 to the α-NH2 of Val-1. The 2.92 Å distance is from the same carboxylate oxygen of Arg-141 to the peptide bond N between Val-1 and Leu-2. The 2.40 Å distance is between the other carboxylate oxygen of Arg-141 to the ɛ-NH2 of Lys-127. In β–β the white lines from Val-98 are H-bonds between the peptide bond N and O of Val-98 to the phenolic OH of Tyr-145, whereas those between Val-1, His-2, Lys-144, and His-146 are distances. The 3.09 Å distance is between one carboxylate oxygen of His-146 and the ɛ-NH2 of Lys-144. The 5.10 Å distance is between the other carboxylate oxygen of His-146 and the α-NH2 of Val-1. The 6.08 Å distance is between the ɛ-NH2 of Lys-144 and the peptide bond O between Val-1 and His-2. The program Insight II (Accelrys) was used to construct these figures from the data of Shaanan (1983).
Effects on cooperativity by α-subunit N-terminal substitutions
Cooperative changes in interactions among Hb subunits are manifested by the sigmoidal shape of the O2-binding curve during rearrangement of the dimer pairs of the R and T states at the allosteric tetramer–dimer interface. It is the strength of these interactions in the R-state that is measured in Figures 4 ▶ and 5 ▶. How could the α-subunit substitutions affect cooperativity (Fig. 6 ▶)? During the normal R/T conformational transition that gives rise to cooperativity, changes occur in the degree of protonation of certain side chains, e.g., in the alkaline Bohr effect where Val-1(α) has an increased basicity (higher pKa) in the T-state compared to its pKa in the R-state. Since tetramer strength as well as basicity is affected in α2Aβ2 as described above, the α–α network shown in Figure 7A ▶ is likely weakened, also decreasing the efficiency of the overall cooperative interactions. However, the same substitutions on the β-subunit N terminus do not influence cooperativity perhaps because they are not exclusive interactions in this region but rather are supported by a network of others (see Fig. 7B ▶). Thus, the hypothesis described above to explain the differences in basicity of α2Aβ2 versus α2β2A and the effects on tetramer dissociation/association can also be extended to the differences found for each on cooperativity. Using digestion with specific carboxypeptidases to remove C-terminal residues of α- or β-subunits, Kilmartin et al. (1975) found that the C-terminal α-subunit interactions played a more important role than C-terminal β-interactions in maintaining cooperativity. This result is consistent with the conclusions made here. Since some aspects of cooperativity in Hb are still uncertain, any dissection of its various facets could conceivably lead to a better understanding of this important phenomenon.
Acetylation
Acetylation is among the most common types of protein modification, yet its function is incompletely understood. Acetylation of amino groups in proteins has received considerable attention especially as it relates to ɛ-NH2 acetylation of lysine residues in histones. Although it has been known for many years that ɛ-NH2 lysine acetylation is strongly correlated with DNA transcription (Allfrey et al. 1964), its mechanism is still not understood (Kouzarides 2000). Acetylation of the α-NH2 of N-terminal residues occurs during biosynthesis of eukaryotic proteins (Tsunasawa and Sakiyama 1984), although in mature proteins it also serves other functions (Caesar and Blomberg 2004).
Acetylation of α-amino groups occurs infrequently in human adult hemoglobins, although it is more common in other types of hemoglobin. N-terminal AcSer residues occur in several human embryonic hemoglobins and in many fish hemoglobins, but a specific function is not known. A few human Hb variants and a cat Hb component contain AcAla at their N-terminals. Some N-terminal Gly residues are subject to partial acetylation due to the relatively low catalytic efficiency of the acetyltransferase for this residue. Hence, the minor fetal hemoglobin HbF1 occurs at low concentrations (1%–2%) in human red cells. Its function, if any, is unknown, although we reported that this form of fetal Hb has a significantly increased dissociation constant (Kd=0.33 μM), which was close to that of HbA (Kd=0.68 μM) but much different from that of HbF itself (Kd=0.01 μM) (Dumoulin et al. 1997; Manning and Manning 2001). Hence, an additional effect of acetylation on HbF besides lowering the 2,3-DPG response (Bunn and Briehl 1970) is to promote a decrease in the strength of interactions between dimer pairs at the allosteric interface. This result is consistent with the results found here for α2AcSβ2 and α2β2AcS.
A possible explanation for this effect of N-terminal acetylation is provided in relation to the known structural features of the N-terminal regions of the α- and β-subunits. Since the tetramer strength of HbA is decreased about fivefold by the presence of AcSer at the N terminus of its β-subunit, it seems very likely that this occurs through weakening of β–β subunit interactions involving the N terminus/C terminus shown in Figure 7B ▶. These results reinforce the conclusions found for acetylated HbF1. It is possible that also in histones, lysine acetylation acts by disruption of nucleosomes through breakage of interactions between a histone tail protruding from a given nucleosome into an adjacent nucleosome to thereby loosen the assembly and enable efficient access by various transcription factors.
Possible physiological relevance of pH dependence of tetramer strength
Even though the extremes of pH employed in Figure 5 ▶ (from pH 6.5 to pH 9.0), may not be physiologically relevant for human hemoglobins, we would not have discovered the reason for the increased tetramer strength of HbF unless the studies had been extended into this range. This approach also permitted us to observe a reduced slope in the pH versus Ka plot (Fig. 5 ▶) for those hemoglobins substituted at Val-1(α) by either Ala or AcSer. This latter result suggests that Val-1(α) may normally play a role in liganded HbAin the uptake of protons involved in tetramer–dimer association/dissociation.
The hemoglobins shown in the plot of log Ka versus pH in Figure 5 ▶ are each of the αβ or the αγ type, with some having amino acid substitutions or acetyl groups on the N-terminal α-, β-, or γ-subunits. These types of tetramers undergo a reversible tetramer–dimer association/ dissociation equilibrium to varying extents, but the dimers do not further dissociate to monomers under physiological conditions. Based on the profiles in Figure 5 ▶, they can be broadly classified as being of three major types:
The αγ-type exemplified by HbF (dashed line at top) which has the strongest interactions at its N-/C-terminal network due largely to facile protonation of Gly-1(γ) and the presence of certain residues in the α-helix allowing efficient tetramer assembly at low Hb concentrations (Yagami et al. 2002). This type has the highest capacity to absorb protons per unit of tetramer assembled, at about two orders of magnitude more efficiently than the αβX-type.
The αβX-type (two middle lines in Fig. 5 ▶), which includes HbA and related tetramers with N-terminal β-substitutions, some of which have lost a single β–β interaction but otherwise have an intact β–β network. For each unit charge in pH, this type undergoes tetramer assembly about 1% as efficiently as the αγ-type.
The αXβ-type (two lowest lines in Fig. 5 ▶) where X represent a substitution leading to the loss of an important interaction in the α–α network. This type has the lowest capacity to absorb protons during tetramer assembly.
Even though each of these types is tetrameric at the Hb concentrations existing in the mature red cell, there may be a significant dimer population and a correspondingly low tetramer concentration for the weak tetramers in cells if overall Hb concentration is low. Thus, the ability of each of these three types to absorb protons as a function of the tetramer formation and the resultant different extents of O2-carrying capacity could have an influence on their relative extents of expression. Further studies are needed to explore this possibility.
Materials and methods
Mutagenesis, expression, and purification
These have been described above in the Results section.
Mass spectrometry
Reagents used for mass spectrometry were all of spectral or HPLC grade: methanol, OPTIMA grade (Fisher Scientific Co.), HPLC grade acetonitrile and water, and ACS grade formic acid 98% (Pierce Chemical Co.), sequencing grade trifluoroacetic acid (TFA), triethylamine (TEA), and phenylisothiocyanate (PITC) (Applied Biosystems), Teflon-bottled glacial acetic acid (Sigma Aldrich). Clear polypropylene microcentrifuge tubes were purchased from National Scientific Supply Co. Concentration values given here are related to hemoglobin tetramer concentration unless stated otherwise.
Sample preparation: Enzymatic cleavage of globin chains
One hundred picomoles hemoglobin were incubated in 100 mM ammonium bicarbonate buffer (>99%, Sigma-Aldrich) at pH 8.2, 5 mM calcium chloride (>99% Sigma-Aldrich), and 10% (v/v) acetonitrile for 5 min at 37°C. An aliquot of sequencing grade modified tosylphenylalanylchloroketone- treated trypsin (Roche Diagnostic Corp.), corresponding to an enzyme-to-hemoglobin molar ratio of 1:40, was added at time zero and the reaction mixture was placed in a shaker for 4 h at 37°C (Thermomixer, Eppendorf). A second aliquot of trypsin with an enzyme-to-hemoglobin molar ratio of 1:40 was added after 4 h and the reaction mixture was incubated at 37°C for another period of 4 h. The reaction was then stopped by addition of 25 μL of a mixture of methanol-acetic acid at 49:1 (v/v).
Derivatization of tryptic peptides
To differentiate between Val → AcSer and Val → Glu, tryptic peptides were derivatized by phenylisothiocyanate (Applied Biosystems) to their phenylthiocarbamyl derivatives. An aliquot of 5 μL of the trypsinized hemoglobin solutions, corresponding to 10 pmol hemoglobin, was completely dried in a 0.65-mL microcentrifuge vial using a rotary concentrator (Savant Instruments). The peptides were resuspended in 20 μL of methanol–water–triethylamine (2:2:1, v/v/v) and the solution evaporated again in a rotary concentrator for 15 min at 50°C. Finally, the peptides were resuspended in a mixture of methanol–water–triethylamine–phenylisothiocyanate (15:2:2:1, v/v/v/v) and the solution was incubated at room temperature for 20 min protected from light. Excess reagent was removed by evaporation in a rotary concentrator for 90 min at 50°C.
Measurement of themolecular mass of the intact globin chains
Average molecular masses of the intact globin chains α and β were measured in a Finnigan LCQ electrospray-ion trap mass spectrometer (ThermoElectron, Waltham, MA) equipped with an in-house constructed electrospray ionization source. Hemoglobin samples were diluted to a final concentration of 10 nM in water–methanol–acetic acid (24:75:1, v/v/v) and infused into the mass spectrometer at a constant flow rate of 0.5 μL-min−1 through a 50-μm I.D. fused silica capillary tapered at the spraying end. A potential of +2.8 kV was applied by means of a liquid junction to form the spray.
Desolvation of the protein ions was accomplished by maintaining the heated capillary at 150°C and applying a potential of −15 V between the heated capillary and the tube lens. Acquisition was performed with 3 microscans and the automatic gain control was set to 200 msec or a maximal number of counts in full scan mode of 5×106. One hundred scans were averaged prior to acquisition to produce a single full-scan spectrum in the m/z range of 500–2000. Individual sample spectra were externally calibrated using a spectrum of adult human hemoglobin acquired under the same conditions and subsequently processed by a deconvolution program developed at the Rockefeller University to transform the ion envelopes into single mass peaks.
Peptide maps
Peptide maps were obtained using two different mass spectrometers, both equipped with a MALDI ion source. To ensure maximal coverage of protein sequence, three matrix solutions were used in MALDI-TOF experiments. A saturated stock solution of recrystallized α-cyano-4-hydroxycinnamic acid (Sigma-Aldrich) was prepared in (1) water–acetonitrile (2:3, v/v) (herein referred to as 4CHCA-WA solution); (2) water– acetonitrile (2:3, v/v), 0.1% TFA (4CHCA-TWA); and (3) formic acid–water–isopropanol at 2:1:3 (v/v/v) (4CHCAFWI). The 4CHCA-WA and 4CHCA-TWA solutions were further diluted 2:1 (v/v) with the corresponding solvent mixture prior to its use. Individual aliquots of the 2-μM trypsinized hemoglobin solutions were diluted to 50 nM in the matrix solutions and aliquots of 0.5 μL were transferred onto a gold-coated sample plate. The droplets were allowed to air dry and the dried spots were washed three times with cold water or a cold aqueous solution of either 0.1% TFA or 0.1% formic acid, with excess liquid being removed by vacuum suction. The sample plate was inserted in the ionization source of a Voyager DE-STR MALDI-TOF mass spectrometer from PerSeptive Biosystems (Waltham, MA) equipped with delayed extraction, ion reflection and a 337-nm nitrogen laser pulsing at 20 Hz. Mass spectra from 500 laser shots were summed to produce a single, externally calibrated spectrum. Spectra collected were further processed using the programs M over z and Paws (Genomic Solutions).
A fourth matrix solution, DHB-TWA was prepared from a saturated matrix solution of recrystallized 2,5-dihydroxybenzoic acid in 0.1% TFA in water–acetonitrile–methanol (2:1:3, v/v/v) by a threefold dilution with the same solvent mixture. Aliquots of the trypsinized hemoglobin solutions were diluted to 50 mM in the DHB-TWA solution and 0.5-μL aliquots were transferred onto a transparent compact disk previously cleaned with methanol. The samples were allowed to air dry and were subsequently washed with cold 0.1% aqueous TFA solution. The compact disk was then inserted in a prototype QqTOF mass spectrometer (model Centaur, Sciex) equipped with an ionization source built at the Rockefeller University (Krutchinsky et al. 2000). Peptide ions were generated by 337- mm nitrogen laser pulses at 20 Hz, with each pulse yielding a full spectrum. Data collection time was under a minute for most samples. Data processing was performed using the program M over z.
Mass spectrometric fragmentation
Tryptic peptides with the expected amino acid substitutions and/or modifications were selected and fragmented on a Finnigan LCQ mass spectrometer equipped with in-house constructed ion source as described in step 1. Trypsinized hemoglobin solutions were diluted to 10 nM with water– methanol–acetic acid at 24:75:1 (v/v/v) and directly infused at 0.5 μL-min−1 into the mass spectrometer. Ions were formed by an applied potential of +2.8 k and desolvation was assisted by maintaining the heated capillary at 135°C and the use of a declustering potential of −57 V across the tube lens. One hundred spectra were averaged prior to acquisition using the following parameters: five microscans, automatic gain control set to 500 msec or a maximal number of counts of 5×107, isolation window of 4 m/z units, and a relative collision energy of 25% (Finnigan’s nomenclature).
Circular dichroism
These measurements were performed on liganded Hb using a Jasco 715 as described previously (Martin de Llano and Manning 1994) in the far ultraviolet region of the spectrum.
Tetramer–dimer dissociation constants (Kd)
These procedures have been described previously (Manning et al. 1996, 1999) and are summarized above in Results in conjunction with Figure 4 ▶. The concentration of each Hb was accurately determined by amino acid analysis for calculation of the final Kd or Ka value.
Oxygen affinity measurements
These determinations were done at a Hb concentration of 0.6–0.8 mM as tetramer in 50 mM bis-Tris Ac buffer at pH 7.5 as described previously (Manning 1981; Dumoulin et al. 1998) either in the absence or presence of a fivefold molar excess of 2,3-DPG over Hb, or 200 mM sodium chloride. To calculate the Hill plots, data points at each 5% O2-tension between 20%–70% O2 saturation were read from the sigmoidal curve on the HemOScan recorder chart. Since a small grid recorder paper was used, accurate data points could be obtained for the Hill plots.
Acknowledgments
We thank Dr. Michael Gilson of the University of Maryland Biotechnology Institute for helpful discussions, and Roger Avelino for typing the manuscript. We are grateful to Stephen DiCiaccio of the Chemical Engineering Department at Northeastern University for his very able assistance with the fermentation unit. This work was supported in part by NIH grants HL-18819 and HL-58512.
Abbreviations
MALDI, matrix-assisted laser desorption/ionization
ESI, electrospray ionization
TOF, time-of-flight
QqTOF, quadrupole –quadrupole time-of-flight.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041267405.
Footnotes
The term “tetramer strength” is synonymous with “tetramer stability” in this communication and indicates the facility with which the contacts between the dimer pairs are formed or are broken, as represented by the association constant (Ka) or the dissociation constant (Kd), of the tetramer–dimer equilibrium, respectively. However, “tetramer strength” is less ambiguous than “tetramer stability” since a decreased stability could suggest instability, e.g., arising from a substitution causing either protein denaturation or loss of heme, which is not the case with any of the substitutions described here.
References
- Allfrey, V.G., Faulkner, R., and Mirsky, A.E. 1964. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. 51 786–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atha, D.H. and Riggs, A. 1976. Tetramer–dimer dissociation in hemoglobin and the Bohr effect. J. Biol. Chem. 251 5537–5543. [PubMed] [Google Scholar]
- Beavis, R.C. and Chait, B.T. 1996. Matrix-assisted laser desorption ionization mass-spectrometry of proteins. Methods Enzymol. 270 519–551. [DOI] [PubMed] [Google Scholar]
- Bradshaw, R.A., Brickey, W.W., and Walker, K.W. 1998. N-terminal processing: The methionine aminopeptidase and N-acetyl transferase families. Trends Biochem. Sci. 23 263–267. [DOI] [PubMed] [Google Scholar]
- Bunn, H.F. and Briehl, R.W. 1970. The interaction of 2,3-diphosphoglycerate with various human hemoglobins. J. Clin. Invest. 49 1088–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caesar, R. and Blomberg, A. 2004. The stress-induced TfS1P requires Nat B-mediated acetylation to inhibit carboxypeptidase Y and to regulate the protein kinase A pathway. J. Biol. Chem. 279 38532–38543. [DOI] [PubMed] [Google Scholar]
- Chu, A.H. and Ackers, G.K. 1981. Mutual effects of protons, NaCl and oxygen on the dimer–tetramer assembly of human hemoglobin. The dimer Bohr effect. J. Biol. Chem. 256 1199–1205. [PubMed] [Google Scholar]
- Dumoulin, A., Manning, L.R., Jenkins, W.T.,Winslow, R.M., and Manning, J.M. 1997. Exchange of subunit interfaces between recombinant adult and fetal hemoglobins. Evidence for a functional inter-relationship among regions of the tetramer. J. Biol. Chem. 272 31326–31332. [DOI] [PubMed] [Google Scholar]
- Dumoulin, A., Padovan, J.C., Manning, L.R., Popowicz, A., Winslow, R.M., Chait, B.T., and Manning, J.M. 1998. The N-terminal sequence affects distant helix interactions in hemoglobin. Implications for mutant proteins from studies on recombinant hemoglobin felix. J. Biol. Chem. 273 35032–35038. [DOI] [PubMed] [Google Scholar]
- Garner, M.H., Bogardt Jr., R.A., and Gurd, F.R. 1975. Determination of the pK values for the α-amino groups of human hemoglobin. J. Biol. Chem. 250 4398–4404. [PubMed] [Google Scholar]
- He, Z. and Russell, J.E. 2001. Expression, purification, and characterization of human hemoglobins Gower-1 (ζ 2ɛ2), Gower 2 (α2ɛ2) and Portland-2 (ζ2β2) assembled in complex transgenic-knockout mice. Blood 97 1099–1105. [DOI] [PubMed] [Google Scholar]
- Huehns, E.R., Flynn, F.V., Butler, E.A., and Beaven, G.H. 1961. Two new hemoglobin variants in a very young human embryo. Nature 189 496–497. [DOI] [PubMed] [Google Scholar]
- Kalkum, M., Llyon, G.J., and Chait, B.T. 2003. Detection of secreted peptides by using hypothesis-driven multistage mass spectrometry. Proc. Natl. Acad. Sci. 100 2795–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilmartin, J.V., Hewitt, J.A., and Wootton, J.F. 1975. Alteration of functional properties associated with the change in quaternary structure in unliganded hemoglobin. J. Mol. Biol. 93 203–218. [DOI] [PubMed] [Google Scholar]
- Kouzarides, T. 2000. Acetylation: A regulatory modification to viral phosphorylation? EMBO J. 19 1176–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutchinsky, A.N., Zhang, W., and Chait, B.T. 2000. Rapidly switchable matrix-assisted laser desorption/ionization and electrospray quadrupole- time-of-flight mass spectrometry for protein identification. J. Am. Soc. Mass. Spectrom. 11 493–504. [DOI] [PubMed] [Google Scholar]
- Li, X., Himanen, J.-P., Martin de Llano, J.J., Padovan, J.C., Chait, B.T., and Manning, J.M. 1999. Mutational analysis of sickle haemoglobin (Hb) gelation. Biotechnol. Appl. Biochem. 29 165–184. [PubMed] [Google Scholar]
- Manning, J.M. 1981. Preparation of hemoglobin carbamylated at specific NH2-terminal residues. Methods Enzymol. 76 159–167. [DOI] [PubMed] [Google Scholar]
- Manning, L.R. and Manning, J.M. 2001. The acetylation state of human fetal hemoglobin modulates the strength of its subunit interactions: Long-range effects and implications for histone interactions in the nucleosome. Biochemistry 40 1635–1639. [DOI] [PubMed] [Google Scholar]
- Manning, L.R., Jenkins, W.T., Hess, J.R., Vandegriff, K., Winslow, R.M., and Manning, J.M. 1996. Subunit dissociations in natural and recombinant hemoglobins. Protein Sci. 5 775–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning, L.R., Dumoulin, A., Jenkins, W.T., Winslow, R.M., and Manning, J.M. 1999. Determining subunit dissociation constants in natural and recombinant proteins. Methods Enzymol. 306 113–129. [DOI] [PubMed] [Google Scholar]
- Martin de Llano, J.J. and Manning, J.M. 1994. Properties of a recombinant human hemoglobin double mutant: Sickle hemoglobin with Leu-88(β) at the primary aggregation site substituted by Ala. Protein Sci. 3 1206–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin de Llano, J.J., Schneewind, O., Stetler, G.L., and Manning, J.M. 1993. Recombinant human sickle hemoglobin expressed in yeast. Proc. Natl. Acad. Sci. 90 918–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1994. Purification and characterization of recombinant human sickle hemoglobin expressed in yeast. Methods Enzymol. 231 390–403. [DOI] [PubMed] [Google Scholar]
- Meister, A. 1965. Biochemistry of the amino acids, 2nd ed., Vol. I, p. 28. Academic Press, New York.
- Perutz, M. 1989. Mechanisms of cooperativity and allosteric regulation in proteins. Q. Rev. Biophys. 22 139–237. [DOI] [PubMed] [Google Scholar]
- Polevoda, B. and Sherman, F. 2003. N-terminal acetyltransferases and sequence requirements for N-terminal acetylation of eukaryotic proteins. J. Mol. Biol. 325 595–622. [DOI] [PubMed] [Google Scholar]
- Randhawa, Z.I., Jones, R.T., and Lie-Injo, L. 1984. Human hemoglobin Portland-2. J. Biol. Chem. 259 7325–7330. [PubMed] [Google Scholar]
- Rollema, H.S., Gros, G., and Bauer, C. 1980. pH changes accompanying the association of isolated α and β chains of human hemoglobins. J. Biol. Chem. 255 2756–2760. [PubMed] [Google Scholar]
- Root, R.W. 1931. The respiratory function of the blood of marine fishes. Biol. Bull. Mar. Biol. Lab. Woods Hole 61 427–456. [Google Scholar]
- Shaanan, B. 1983. Structure of human oxyhemoglobin at 2.1 Å resolution. J. Mol. Biol. 171 31–59. [DOI] [PubMed] [Google Scholar]
- Sheff, D.R. and Rubenstein, P.A. 1992. Amino-terminal processing of actions mutagenized at the Cys-1 residue. J. Biol. Chem. 267 2671–2678. [PubMed] [Google Scholar]
- Tsunasawa, S. and Sakiyama, F. 1984. Amino-terminal acetylation of proteins: An overview. Methods Enzymol. 106 165–170. [DOI] [PubMed] [Google Scholar]
- Urbancikova, M. and Hitchcock-De Gregori, S.E. 1994. Requirement of amino-terminal modification for striated muscle α-tropomyosin function. J. Biol. Chem. 269 24310–24315. [PubMed] [Google Scholar]
- Wagenbach, M., O’Rourke, K., Vitez, L., Wieczorek, A., Horrman, S., Durfee, S., Tedesco, J., and Stetler, G. 1991. Synthesis of wild-type and mutant human hemoglobins in Saccharomyces ceravisae. Biotechnology 9 57–61. [DOI] [PubMed] [Google Scholar]
- Yagami, T., Ballard, B.T., Padovan, J.C., Chait, B.T., Popowicz, A.M., and Manning, J.M. 2002. N-terminal contributions of the γ-subunit of fetal hemoglobins to its tetramer strength: Remote effects at subunit contacts. Protein Sci. 11 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, S.C., Grinnell, B.W., and Wold, F. 1989. Post-translational modifications of proteins: Some problems left to solve. Trends Biochem. Sci. 14 264–268. [DOI] [PubMed] [Google Scholar]