

Abstract
Three different classes of thiol-oxidoreductases that facilitate the formation of protein disulfide bonds have been identified. They are the Ero1 and SOX/ALR family members in eukaryotic cells, and the DsbB family members in prokaryotic cells. These enzymes transfer oxidizing potential to the proteins PDI or DsbA, which are responsible for directly introducing disulfide bonds into substrate proteins during oxidative protein folding in eukaryotes and prokaryotes, respectively. A comparison of the recent X-ray crystal structure of Ero1 with the previously solved structure of the SOX/ALR family member Erv2 reveals that, despite a lack of primary sequence homology between Ero1 and Erv2, the core catalytic domains of these two proteins share a remarkable structural similarity. Our search of the DsbB protein sequence for features found in the Ero1 and Erv2 structures leads us to propose that, in a fascinating example of structural convergence, the catalytic core of this integral membrane protein may resemble the soluble catalytic domain of Ero1 and Erv2. Our analysis of DsbB also identified two new groups of DsbB proteins that, based on sequence homology, may also possess a catalytic core similar in structure to the catalytic domains of Ero1 and Erv2.
Keywords: DsbB, Ero1, Erv2, disulfide, structure
Extracellular proteins and exoplasmic domains of membrane proteins often contain disulfide bonds, which stabilize native protein structures relative to their unfolded states. The pathways for disulfide bond formation in the lumen of the endoplasmic reticulum (ER) in eukaryotic cells or the periplasmic space of prokaryotic cells are similar in general outline. In both cases, disulfide bonds are introduced into folding proteins by transfer from a disulfide bond carrier that is a member of the thioredoxin family of proteins. In eukaryotic cells the major disulfide bond carrier is protein disulfide isomerase (PDI), a soluble protein of the ER lumen, whereas in bacteria the major disulfide bond carrier is the soluble periplasmic protein DsbA. The disulfide carrier proteins, in turn, receive disulfide bonds from a second class of protein thiol-oxidoreductases that couple intracellular biochemical reactions that can provide oxidizing potential to the formation of protein disulfide bonds (for recent reviews, see Collet and Bardwell 2002; Fassio and Sitia 2002; Sevier and Kaiser 2002; Thorpe et al. 2002; Kadokura et al. 2003).
Three different classes of protein thiol-oxidoreductases are known to facilitate the oxidation of PDI or DsbA; these include members of the Ero1 and SOX/ALR families in eukaryotic cells and the DsbB family in prokaryotic cells. Ero1, a flavoprotein bound to the lumenal surface of the ER membrane, uses oxygen as a terminal electron acceptor for the generation of a disulfide bond within itself, which can then be transferred to PDI (Frand and Kaiser 1998, 1999; Pollard et al. 1998; Cabibbo et al. 2000; Tu et al. 2000; Mezghrani et al. 2001; Tu and Weissman 2002). Enzymes of the SOX/ALR family are also flavoenzymes that generate disulfide bonds using oxygen as an electron acceptor (Hoober et al. 1999; Lee et al. 2000; Gerber et al. 2001; Lisowsky et al. 2001; Sevier et al. 2001). Members of the SOX/ALR family operate in a variety of different cellular compartments. For example, in yeast cells a SOX/ALR protein known as Erv2 participates in disulfide bond formation in the ER (Sevier et al. 2001), whereas in metazoan cells a family member known as quiescin, which contains both a flavoenzyme domain and a thioredoxin-like domain, is thought to have a role in the formation of disulfide bonds in the extracellular matrix (Hoober et al. 1999; Coppock et al. 2000). The prokaryotic protein DsbB, which generates disulfide bonds for transfer to DsbA (Guilhot et al. 1995; Kishigami et al. 1995a,b), is an integral membrane protein with four transmembrane segments (Jander et al. 1994). Rather than using a flavin cofactor to mediate the reaction with oxygen, DsbB uses a membrane-bound quinone as an electron acceptor (Bader et al. 1999, 2000). The quinone (ubiquinone in aerobic conditions or menaquinone in anaerobic conditions) in turn acts as a mobile electron carrier for electron transfer to electron transport chains in the cytoplasmic membrane, ultimately transferring electrons to molecular oxygen or an anaerobic acceptor, such as nitrate or fumarate (Kobayashi et al. 1997; Bader et al. 1999, 2000; Kobayashi and Ito 1999).
We recently obtained by X-ray crystallography the structure of an enzymatically active portion of Ero1 (Gross et al. 2004), allowing a comparison to the previously solved structure of the SOX/ALR family member Erv2 (Gross et al. 2002). Despite the fact that Erv2 and Ero1 do not share obvious sequence similarity, the core catalytic domains of the two proteins display a remarkable structural similarity. Both proteins hold the FAD cofactor within a bundle of four anti-parallel α-helices. The redox active isoalloxazine moiety of the FAD is close to a pair of cysteines localized to a tight turn between two of the α-helices. Presumably electrons are transferred directly from the cysteine pair of Erv2 or Ero1 (with the concomitant formation of a disulfide bond) to the isoalloxazine ring via a charge-transfer intermediate. Except for some of the residues in proximity to the isoalloxazine ring, which are described below, there is no detectable sequence similarity between the corresponding α-helices in Erv2 and Ero1. Although there is not yet a structure for DsbB, we were struck by the fact that DsbB also has the potential to form a bundle of antiparallel α-helices, in this case comprising four hydrophobic transmembrane segments. We therefore sought to examine the DsbB sequence for features that may indicate structural resemblance to Erv2 and Ero1. Our sequence analysis of DsbB unexpectedly revealed two uncharacterized groups of DsbB-like proteins that were also analyzed for structural features.
Results and Discussion
Four-helix bundle
To compare the helical structure of DsbB to that of Erv2 and Ero1 it is useful to develop a standard description of the four α-helices in the conserved structure. We will designate the two antiparallel α-helices that carry the active site cysteine pair on the short connecting loop between them as helices A and B (in Fig. 1 ▶ helices A and B are labeled yellow and green, respectively). To complete the bundle of four antiparallel helices, the α-helix with its C terminus oriented towards the end of the bundle with the cysteine pair is designated helix C (Fig. 1 ▶, labeled red) and the α-helix oriented with its N terminus towards the cysteine pair is designated helix D (Fig. 1 ▶, labeled blue). Note that in the structures of Erv2 and Ero1 these structurally defined helices occur in a different order in the primary sequence: for Erv2 the order of helices is C-A-B-D, whereas for Ero1 the order is C-D-A-B.
Figure 1.
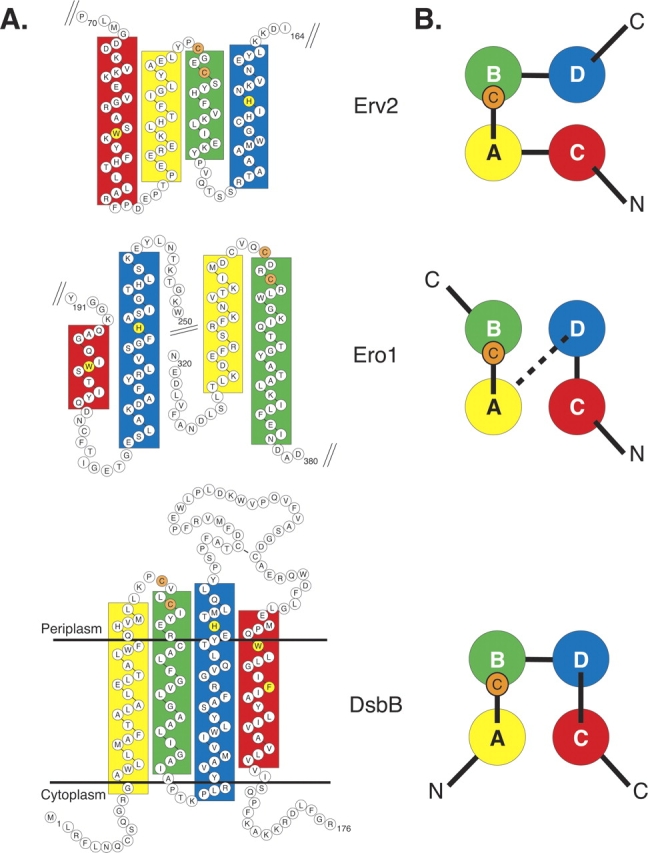
Primary sequence and secondary structure of the four-helix core of Erv2, Ero1, and DsbB. (A) Boxes highlight residues forming α-helices in the three-dimensional structures of Erv2 (PDB files 1JR8 and 1JRA) and Ero1 (1RP4 and 1RQ1) or predicted to form α-helices in DsbB by the PSIPRED program (Jones 1999; McGuffin et al. 2000). The helices of Erv2 and Ero1 are colored according to their position in the three-dimensional structure. The helices of DsbB are colored on the basis of conserved features, including the Cys-x-x-Cys active site (marked with solid orange circles) and the relative orientation of the helices across the membrane. Conserved histidine and aromatic amino acid residues in the transmembrane helices are highlighted in yellow. The transmembrane domains of DsbB are positioned in the membrane based on the predictions outlined previously (for example, see Bardwell et al. 1993; Jander et al. 1994). (B) End-on view of the four-helix bundle of Erv2, Ero1, and DsbB represented as seen in the structures of Erv2 and Ero1, or as postulated for DsbB. An orange circle with the letter C marks the location of the Cys-x-x-Cys active site residues. The dotted line between Ero1 helices D and A represents the 92 residue segment connecting the two helices, which includes three α-helices that are not part of the core domain.
Using previous predictions of the DsbB transmembrane spans (for example, see Bardwell et al. 1993; Jander et al. 1994) and the secondary structure prediction program PSIPRED (Jones 1999; McGuffin et al. 2000), we modeled the potential secondary structure and topology of Escherichia coli K12 DsbB, the first identified and best characterized of the DsbB homologs. Our predictions place the active site cysteine pair (Cys41 and Cys44) on a tight turn connecting two hydrophobic α-helical transmembrane spans (Fig. 1 ▶). Notably, the position of this cysteine pair relative to the predicted α-helices is strikingly similar to the position of the active site Cys-x-x-Cys motif at the end of helix B in the Erv2 and Ero1 structures (Fig. 1 ▶, orange circles). Therefore we propose the first two transmembrane helices of DsbB correspond to helices A and B in the Erv2 and Ero1 structures. Based on their orientation across the membrane, the third and fourth helices of DsbB are predicted to correspond to helices D and C, respectively, of Erv2 and Ero1. Thus, the four helices in DsbB are predicted to occur in order A-B-D-C, which is different from the order of helices in either the Erv2 or Ero1 proteins (Fig. 1 ▶).
Cofactor binding
The coordination of FAD by the four helices of Erv2 and Ero1 differs from the mode of FAD binding in other known flavoprotein structures, both in the conformation of the FAD molecule itself and in the precise manner in which the FAD interacts with planar amino acid side chains in the protein. FAD binds between helices C and D of Erv2 or Ero1, and is oriented with the flavin isoalloxazine ring proximal to the Cys-x-x-Cys active site present on helix B (Fig. 2 ▶; Gross et al. 2002, 2004). The orientation of the FAD backbone positions the planar flavin isoalloxazine and adenine rings in a roughly parallel arrangement. A conserved tryptophan on helix C packs against the isoalloxazine ring system, which functions as the electron acceptor in FAD, and is in a position to hydrogen bond with the hydrocarbon tail, while a conserved histidine from helix D occupies the space between the typtophan and adenine rings of the flavin, forming a hydrogen bond with the AMP phosphate of the flavin (Fig. 2 ▶). The planar stacking of the tryptophan and histidine side chains between the FAD rings results in a structure reminiscent of base stacking in polynucleotides.
Figure 2.

Conserved histidine and tryptophan residues bind the cofactor through planar stacking. Ball-and-stick format reveals the conserved geometry of the FAD cofactor (yellow), histidine (blue), tryptophan (red), and the Cys-x-x-Cys active site (orange) in the flavin binding sites of (A) Erv2 and (B) Ero1. The four core α-helices are colored to match the diagrams in Fig. 1 ▶. (C) An ero1-1 strain was transformed with the control plasmid pRS315, with ERO1-myc, or with ERO1-myc containing Trp200 or His231 mutations. Strains were incubated at 30°C for 2 d on media with the indicated amounts of dithiothreitol (DTT). The ability to confer resistance to the reducing agent DTT was used to evaluate the relative oxidoreductase activity of the His or Trp mutants.
It appears that the tryptophan and histidine residues in Ero1 aid in the binding and orientation of the FAD molecule since amino acid substitutions in either Trp200 or His231, or the nearby residue Gly229, result in decreased Ero1 function (Fig. 2 ▶; ero1-1 and ero1-2 mutants described in Frand and Kaiser 1998; Pollard et al. 1998). Not surprisingly, themost dramatic loss of oxidoreductase activity is exhibited when the residue closest to the FAD isoalloxazine ring, Trp200, is replaced (Fig. 2 ▶). More flexibility appears to be tolerated at the His231 position in Ero1, as proteins containing either alanine or glutamine at this position retain wild-type activity (Fig. 2 ▶). This flexibility is also evident among the Ero1 homologs: Ero1 proteins from Cryptosporidium parvum, Encephalitozoon cuniculi, Plasmodium falciparum, Plasmodium yoelii, and Yarrowia lipolytica each contain a glutamine in place of histidine.
Like FAD, the redox-active part of quinone adopts a planar ring structure. To explore the possibility that amino acids with a planar ring structure in the transmembrane helices of DsbB play a role in the interaction of DsbB with its quinone cofactor, we acquired 112 DsbB-like sequences from the nonredundant database at NCBI and searched them for conserved amino acids (Table 1). A multiple alignment of the DsbB homologs reveals few highly conserved amino acids. The only completely conserved residues are two cysteines, Cys41 and Cys44, and an arginine, Arg48, which is predicted to localize within the second transmembrane domain (helix B), one helical turn below Cys44, and has previously been implicated in the interaction of DsbB with quinone (Kadokura et al. 2000). Unexpectedly, after the protein sequence encoding the second transmembrane domain, the multiple alignment of the DsbB proteins diverges into three distinct clusters: (group I) proteins with an E. coli K12 DsbB topology including four transmembrane domains and a conserved cysteine pair present within an amino acid loop connecting transmembrane domains three and four; (group II) proteins containing five transmembrane domains ending with a periplasmic domain containing two conserved cysteines; and (group III) proteins that have five transmembrane spans and a C-terminal periplasmic tail, but lack a conserved cysteine pair beyond the cysteines on helix B. The group III sequences appear to contain two subtypes: proteins with an extensive C-terminal periplasmic domain of −300 amino acids (from Campylobacter jejuni, Helicobacter pylori, and Corynebacterium diptheriae), and proteins with a short −5 amino acid C terminus (from Bordetella parapertussi and Bordetella pertussis). Strikingly, among the most conserved amino acids shared between all the DsbB proteins are amino acids with a planar ring structure predicted to lie within the third and fourth membrane spanning helices (highlighted in Fig. 3 ▶). The sequence conservation between the first four transmembrane domains of the group I, II, and III proteins, including the Cys-x-x-Cys pair between the first two transmembrane spans as well as planar residues on transmembrane domains three and four, suggests the core domain in all the identified DsbB proteins is formed by the first four transmembrane helices: A-B-D-C.
Table 1.
TTDsbB homologs

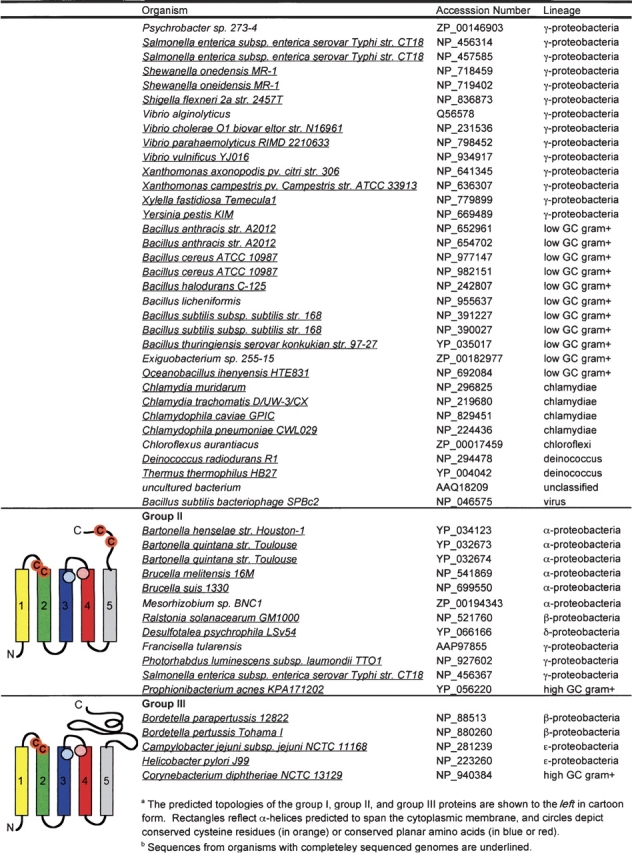
Figure 3.
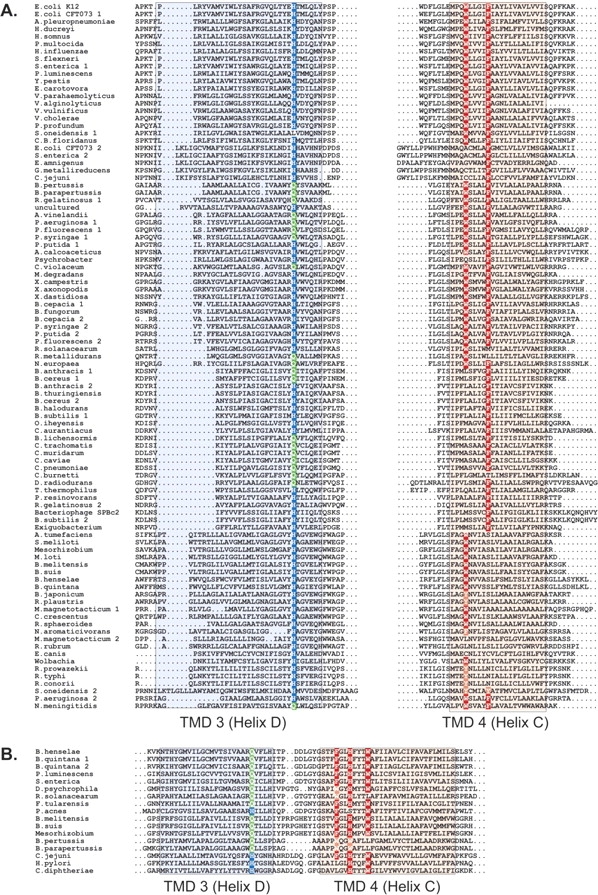
The DsbB homologs contain conserved histidine and aromatic amino acid residues in the third and fourth transmembrane domains. Sequence alignment of third and fourth transmembrane domains of the DsbB sequences from (A) group I and (B) groups II and III. Colored boxes shade the residues predicted to form α-helices. Highly conserved histidine and aromatic amino acid residues are highlighted in dark blue and red, respectively. The conserved glutamine, often present instead of histidine, in helix D is highlighted in green.
The well-conserved amino acids with a planar ring structure on the fourth transmembrane domain, helix C, include a tryptophan and phenylalanine in the group I proteins, and a cluster of residues including a phenylalanine, histidine and tryptophan in the group II and III proteins (Fig. 3 ▶). The most highly conserved of these residues is His91 from the third transmembrane domain of DsbB, helix D, which is present as a histidine or glutamine in all but one of the 112 analyzed sequences (Fig. 3 ▶). As described above, an identical amino acid variation is observed at the His231 position in helix D of Ero1. The predicted positions of the conserved amino acids in DsbB on helices D and C raise the possibility that these residues participate in the binding of the quinone cofactor in a manner analogous to the stacking interactions that bind flavin to Erv2 and Ero1. Interestingly, a photoactivatable ubiquinone derivative can be cross-linked to a peptide containing His91 when purified DsbB is illuminated with UV light in the presence of this quinone analog, also suggesting His91 is located near the quinone binding site (Xie et al. 2002).
To test the importance of the three conserved planar amino acid residues within the predicted helices D and C (His91, Trp145, Phe150) in the function of DsbB, nine amino acid substitutions (H91A, H91W, H91Y, W145A, W145H, W145Y, F150A, F150Y, F150L) were made, introduced into a His6-Myc-tagged derivative of DsbB on a plasmid, and the mutant proteins expressed in a ΔdsbB strain. The functionality of the mutant proteins was investigated by determining the in vivo redox state of the mutant proteins themselves and their substrate, DsbA. To assess the oxidation status of DsbA and DsbB, cellular proteins were first treated with acid to inhibit thiol-disulfide reactivity and the free cysteines were alkylated with 4-acetamido-4′-maleimidylstilbene- 2,2′-disulfonic acid (AMS). This modification retards the mobility of the reduced proteins on gels. The plasmid coding for the wild-type DsbB maintained DsbA almost completely in the oxidized form in a ΔdsbB strain (Fig. 4A ▶). However, all of the plasmids coding for DsbB His91 mutants (H91A, H91W, H91Y) caused the accumulation of a band of an apparent molecular mass of 36 kDa (Fig. 4A ▶) in addition to the oxidized form of DsbA. This band, which disappeared when samples were treated with reductant (data not shown), was also recognized by antibody to DsbB (Fig. 4B ▶). Thus, it represents an intermediate in DsbA reoxidation by DsbB, the mixed-disulfide complex between DsbA and DsbB (Kadokura and Beckwith 2002; Grauschopf et al. 2003; Inaba et al. 2004). Accumulation of substantial amounts of the reaction intermediate in all of the mutants with an alteration at this residue indicates that His91 plays a particularly important role in the function of this enzyme.
Figure 4.

Characteristics of the dsbB mutants. HK320 (ΔdsbB) (Kadokura and Beckwith 2002) carrying a plasmid, pAM238 (empty vector), pHK517 (wild-type DsbB-His6-c-Myc; DsbB), or pHK517 derivatives with the indicated substitution mutations were grown at 30°C in M63 minimal Glc medium. Proteins from the exponential culture were alkylated with AMS, and separated by SDS-PAGE. Redox state of DsbA and DsbB in the mutants was visualized by western analysis with antibody to DsbA (A) or c-Myc (B), respectively. Open arrowheads, nonspecific bands; closed arrowheads, reduced DsbB. The positions of marker proteins are indicated on the left in kDa.
We previously reported that Arg48 of DsbB is important for the interaction of DsbB with quinone (Kadokura et al. 2000). The R48H mutant, which showed reduced affinity for ubiquinone in vitro, accumulated the DsbA– DsbB complex in vivo, which led us to propose that the interaction of DsbB with quinone is required for the resolution of the intermediate complex (Kadokura et al. 2000). All of the following findings are consistent with our proposal that His91 may participate in the interaction of DsbB with quinone. First, like the R48H mutant, His91 mutants accumulate the DsbA–DsbB complex. Secondly, the H91W and H91Y mutants, as well as the R48H mutant, accumulate a small fraction of DsbB in their reduced form aerobically (Fig. 4B ▶), suggesting inefficient transfer of electrons from DsbB to quinones. Anaerobically, H91A mutant also accumulated a small fraction of DsbB in its reduced form (data not shown). Finally, substitutions that increase the size of the side chain (H91W and H91Y) caused stronger defects than a substitution that decreases the size of the side chain (H91A), a property that is often observed with substitutions at an enzyme– substrate interface.
We were unable to detect specific defects with the Trp145 or Phe150 mutants by these assays. Since slight defects inmutants are often difficult to detect (Haebel et al. 2002), it could be that His91 acts as one of the primary sites for the interaction with quinone and Trp145 or Phe150 serves as a secondary site. Notably, not all substitutions at Ero1 residues His231 and Trp200 substantially altered the oxidoreductase activity of Ero1 in our assay despite the observed stacking of these two amino acids with the flavin ring in the X-ray structure (Fig. 2 ▶).
Binding of a quinone to DsbB most likely involves burying the isoprenoid chain of the quinone molecule within the membrane bilayer, leaving the redox-reactive quinone ring near the periplasmic membrane surface in a position favorable for withdrawal of electrons from the Cys-x-x-Cys active site. In both the Erv2 and Ero1 structures the second cysteine of the Cys-x-x-Cys active site is in contact with the isoalloxazine ring of FAD (Gross et al. 2002, 2004). A similar relationship between the active site cysteines and the quinone cofactor appears to hold true for DsbB, since a recent spectroscopic analysis of a charge transfer complex between DsbB and its ubiquinone cofactor demonstrated that the Cys44 thiol of the Cys41–Cys44 pair reacts directly with ubiquinone or menaquinone (Inaba et al. 2004; Takahashi et al. 2004). The transmembrane helices of DsbB containing Arg48 and His91 may form a pocket to hold the quinone ring in a proper orientation with respect to the Cys-x-x-Cys active site, in a structure analogous to the flavin binding site and Cys-x-x-Cys active site in Erv2 and Ero1. Whether Trp145 and Phe150 play a similar role is unclear.
Dynamic catalytic mechanism
In addition to the active site Cys-x-x-Cys motif described above, Erv2, Ero1, and E. coli K12 DsbB each contain a second pair of cysteine residues that is required for activity (Jander et al. 1994; Frand and Kaiser 2000; Gross et al. 2002). The spacing between the cysteines in the second pair differs among the enzymes: Erv2, Ero1, and DsbB contain Cys-x-Cys, Cys-x4-Cys, or Cys-x25-Cys motifs, respectively (Figs. 5 ▶, 6 ▶, pink cysteine pair). A common mechanism has been proposed for all three oxidoreductases wherein the cysteine pair arranged in a variable motif directly oxidizes the soluble thioredoxin-like partner protein (PDI for Erv2 and Ero1; DsbA for DsbB), and this second cysteine pair is reoxidized by the internal transfer of electrons to the conserved active site Cys-x-x-Cys pair (Guilhot et al. 1995; Kishigami and Ito 1996; Kobayashi and Ito 1999; Frand and Kaiser 2000; Gross et al. 2002; Inaba and Ito 2002; Kadokura and Beckwith 2002; Grauschopf et al. 2003).
Figure 5.

Structures of the catalytic cores of (A) Erv2 and (B) Ero1 show the similar relationship between the two pairs of cysteine residues. The four core helices of Erv2 and Ero1 are colored as in Fig. 1 ▶. Ball-and-stick representation shows the position of the flavin cofactor (yellow), the active site cysteine pair (orange), and the second conserved cysteine pair and its flanking sequence (pink). Note the lack of secondary structure in the pink segments surrounding the second cysteine pair.
Figure 6.

(A) Diagram of the proposed active site of the group I (left) and group II (right) DsbB proteins. The transmembrane helices of DsbB are drawn as colored circles. The Cys-x-x-Cys active site and the second conserved cysteine pair are represented as an orange or pink “S-S,” respectively, and a yellow ring labeled Q represents the quinone cofactor. (B) DsbB amino acid sequence connecting helices D and C in the group I proteins, or forming the C-terminal domain of the group II proteins. The E. coli and B. henselae DsbB proteins were arbitrarily chosen as representative of the group I and group II proteins, respectively. Cysteines are represented in pink text, while purple rectangles highlight residues predicted to form helices by the PSIPRED program (Jones 1999; McGuffin et al. 2000). The nonhighlighted sequence is predicted to adopt a conformation differing from canonical secondary structures (α-helix and β-sheet).
As described above, in both the Erv2 and Ero1 proteins, the active site Cys-x-x-Cys pair is situated close to the FAD cofactor, at the beginning of one of the four helices forming the structurally conserved four-helix bundle. However, the location of the second cysteine pair is quite different in the two proteins. In Erv2, the second pair of cysteines is located at the C terminus of the protein in a flexible tail (Gross et al. 2002), whereas in Ero1 the second pair of cysteines are part of a flexible loop lying across the surface of the protein (Gross et al. 2004). Despite these differences, the regions of Erv2 and Ero1 that contain the second cysteine pair have features in common. The second pair of cysteines in Erv2 and Ero1 are both located in a region of sequence that is devoid of significant α-helical or β-sheet content and can adopt at least two different conformations in the crystalline state (Gross et al. 2002, 2004). In one conformation, the second cysteine pair is close to the Cys-x-x-Cys active site, suggesting a direct disulfide transfer between the two pairs of cysteine residues. In the second conformation, the second cysteine pair is located further away from the protein core and the Cys-x-x-Cys cysteine pair. The apparent flexibility of the region of protein containing the second cysteine pair may explain how these cysteines gain access to the Cys-x-x-Cys active site as well as the soluble partner protein PDI. It is tempting to speculate that the lack of secondary structure in these flexible sections of protein common to both structures may facilitate the interaction with PDI.
The second cysteine pair in E. coli K12 DsbB, and the other group I proteins, is located within a loop of sequence between transmembrane helices D and C, reminiscent of the loop surrounding the second cysteine pair in the Ero1 protein (Fig. 5 ▶). Interestingly, the second cysteine pair in the newly identified group II DsbB proteins is positioned within a periplasmic C-terminal tail, similar to the location of the second cysteine pair in Erv2 (Fig. 5 ▶). Both regions of DsbB sequence containing the second pair of cysteines are predicted by PSIPRED (Jones 1999; McGuffin et al. 2000) to lack extensive secondary structure (Fig. 6 ▶). In experiments designed to study the role of the second pair of cysteines in E. coli DsbB, it was observed that when reduced DsbA and a mutant DsbB protein with Cys104 and Cys130 replaced with serine residues are mixed together in vitro, the disulfide transfer between the two proteins is greatly attenuated relative to the transfer seen between reduced DsbA and wild-type DsbB, suggesting steric hindrance of disulfide exchange between DsbA and the remaining Cys41–Cys44 active site disulfide pair (Regeimbal and Bardwell 2002; Grauschopf et al. 2003). An analysis of the opposite reaction, the disulfide transfer between oxidized DsbA and a Cys104–Cys130 mutant of DsbB, also points to the inaccessibility of the Cys-x-x-Cys pair to DsbA as the strong oxidant DsbA was unable to oxidize the Cys41–Cys44 pair (Regeimbal and Bardwell 2002). It is an attractive possibility that the peptide loop or tail in DsbB containing the second cysteine pair can adopt multiple conformations to facilitate shuttling of disulfide bonds from the otherwise inaccessible active site Cys-x-x-Cys pair to DsbA.
A unique group of DsbB homologs includes proteins with a five transmembrane domain topology and a lone Cys-x-x-Cys active site localized to the turn between helices A and B (see Table 1, group III). The group III proteins may utilize a different mechanism than the DsbB proteins from group I and II to oxidize substrate proteins. Group III DsbB proteins may have a cysteine-containing partner protein that mediates the transfer of oxidizing equivalents from the buried Cys-x-x-Cys pair to substrate proteins. The viral Erv2 homolog, E10R, utilizes the cysteines in a second viral protein A2.5L to transfer oxidizing equivalents from its lone Cys-x-x-Cys active site to the thioredoxin-like viral protein G4L (Senkevich et al. 2002). A function of the extensive C-terminal domain in the C. jejuni, H. pylori, and C. diptheriae group III proteins could be to facilitate the interaction with its partner. The relationship between the group III and the group I and II DsbB proteins parallels the relationship between two other redox-active microbial proteins Rhodobacter capsulatus CcdA and its functional homolog E. coli DsbD: CcdA contains a cysteine pair in a DsbD-like transmembrane core, but lacks the two redox-active cysteine pairs that are present in the periplasmic domains of DsbD. CcdA can transfer electrons to a limited set of DsbD’s substrates: CcmG/HelX but not DsbC (Katzen et al. 2002). Consistent with CcdA’s inability to oxidize DsbC, DsbC proteins have not been identified in organisms containing CcdA and lacking a DsbD protein (Katzen et al. 2002). Interestingly, the two organisms exclusively encoding a group III DsbB homolog (H. pylori and C. diphtheriae) appear to lack a DsbA homolog, suggesting that the group III DsbB proteins transfer oxidizing equivalents to a yet uncharacterized protein. Considering the functional and mechanistic similarities between the enzymes that catalyze the oxidation of PDI or DsbA and the distinct enzymatic mechanism and unique substrates that may be used by the group III proteins, the group III proteins may merit a different name to distinguish them from the classical DsbB proteins.
Conclusions
Over the past few years, biochemical and genetic studies have uncovered striking parallels between the prokaryotic and eukaryotic cellular disulfide bond formation path-ways. Considering the functional and mechanistic similarities between the thiol-oxidoreductases that catalyze the formation of disulfide bonds, it was puzzling that these proteins do not display the type of sequence similarities shared by the disulfide carrier proteins, which are all members of the thioredoxin superfamily. However, the recent elucidation of the crystal structures of Erv2 and Ero1 revealed a remarkable conservation of the active sites of these enzymes at the structural level, leading us to speculate that the prokaryotic enzyme DsbB may share the same core structure.
Examination of the DsbB sequence revealed several key features conserved with Erv2 and Ero1. These include: (1) four predicted anti-parallel α-helices; (2) a conserved Cys-x-x-Cys pair located at the end of one of the four helices, with the second cysteine in close proximity to the redox cofactor; (3) conserved amino acid residues with a planar ring structure that may interact with the cofactor ring; and (4) a second essential cysteine pair located in a region lacking significant α-helical or β-sheet content. The proposed relationship between DsbB and the eukaryotic thiol-oxidoreductases may represent a remarkable example of convergent evolution in which a membrane-embedded helical bundle in DsbB forms a site to hold the hydrophobic quinone cofactor in proximity to a Cys-x-x-Cys active site with a similar geometry as the binding site for the hydrophilic cofactor FAD formed by the hydrophilic helical bundles in Erv2 and Ero1.
In light of the structural convergence and functional parallels between DsbB, Erv2, and Ero1, it is interesting to speculate about additional implied similarities between the three proteins. The two different conformations observed in X-ray crystallography for the region of Erv2 or Ero1 containing the second essential cysteine pair suggest a highly dynamic protein segment. The loop of sequence between helices C and D, or the periplasmic tail, with the second cysteine pair of DsbB may have similar properties. An appealing possibility is that the flexible domain has evolved to direct the flow of disulfides along specific pathways. The second cysteine pair on a flexible peptide may serve as a substrate selectivity filter in two ways: (1) by partially occluding the Cys-x-x-Cys pair, keeping the redox active cysteine pair from nonselectively oxidizing small molecules or proteins that would otherwise have access the active site, and (2) by specifically relaying disulfides from the partially buried Cys-x-x-Cys active site directly to a thioredoxin-like disulfide carrier protein. Perhaps the unstructured nature of these flexible regions facilitates an association with the thioredoxin-like partner proteins by mimicking unfolded nascent polypeptide chains, which are known to be substrates for PDI and DsbA.
Materials and methods
DsbB sequences
To identify homologs of DsbB, the nonredundant (nr) database at NCBI was searched with the E. coli K12 DsbB protein sequence (accession no. P30018) using the PSI-BLAST algorithm (http://www.ncbi.nih.gov/BLAST/) (Altschul et al. 1997). Searching was terminated after five iterations when no additional sequences were returned above the default inclusion threshold of 0.005. Five of the nine sequences with e values closest to the threshold (1×10−9 to 2×10−5) did not contain a Cys-x-x-Cys motif and were not included in our final dataset. When sequence data was available from multiple strains, one set of sequence data was chosen to be representative of that organism. An exception was made for E. coli where data from E. coli K12 and E. coli CFT073 are both included in our dataset. In order to obtain additional homologs, the most divergent sequence from the PSI-BLAST dataset, a hypothetical protein from Bartonella quintana (accession no. YP_032674), was used to search the nr database using the PSI-BLAST algorithm for nine iterations, which returned six new sequences. One of the six sequences, a Legionella pneumophila protein sequence, does not represent a completely translated open reading frame and was not used in our alignments. A search of the nr database with the most divergent of the six new sequences, a putative membrane protein from Bordetella parapertussis (accession no. NP_883513), failed to identify any new protein sequences outside our compiled dataset. The accession numbers for the complete DsbB homologs dataset are displayed in Table 1.
Secondary structure predictions of the DsbB homologs were obtained using the program PSIPRED version 2.4 (http://bioinf.cs.ucl.ac.uk/psipred/) (Jones 1999; McGuffin et al. 2000). The PSIPRED program was chosen based on its ability to accurately predict, within a few amino acids, the four α-helices determined by X-ray crystallography to form the catalytic core of Ero1 and Erv2 (Gross et al. 2002). Notably, additional secondary structure prediction programs including JPRED (Cuff and Barton 2000), PHD (Rost and Sander 1993, 1994), POR-TER (Pollastri and McLysaght 2004), PROF (Ouali and King 2000), SOPMA (Geourjon and Deleage 1994), SSpro (Pollastri et al. 2002), and Target99 (Karplus et al. 1998) also placed the active site cysteines of DsbB, Cys41 and Cys44, at the N-terminal end of an α-helix predicted by other methods to form the second transmembrane domain (see Bardwell et al. 1993; Jander et al. 1994). The secondary structure output between the programs did vary in the predicted lengths for the DsbB α-helices, especially the third and forth transmembrane spanning helices.
Ero1 Trp200 and His231 mutants
Amino acid replacements were created by site-directed mutagenesis with the QuikChange site-directed mutagenesis kit (Stratagene) using ERO1-myc in a LEU2-marked, CEN plasmid as a template. The mutated plasmids were verified by sequencing. Plasmids containing wild-type or mutant ERO1- myc were transformed into the yeast strain CKY598 (MATa GAL2 ura2-52 leu2-3,112 ero1-1). After overnight growth in synthetic minimal media (SMM) minus leucine with 2% (w/v) glucose, strains were plated onto rich YPD plates containing the indicated amounts of dithiothreitol (DTT). Strains were grown for 2 d at the semipermissive temperature of 30°C.
DsbB mutants
The mutant DsbB expression plasmids were constructed by introducing substitution mutations into the DsbB-His6-c-Myc gene of pHK517 (pAM238 carrying DsbB-His6-c-Myc) (Kadokura and Beckwith 2002) using QuikChange site-directed mutagenesis (Stratagene). In the Results and Discussion section, we call the DsbB-His6-c-Myc polypeptide simply DsbB. To analyze the in vivo redox state of proteins, the free cysteine residues of proteins were acid trapped and alkylated with AMS (4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid) as described (Kadokura et al. 2000). The alkylated proteins were separated by SDS-PAGE, and subjected to immuno-blotting with anti-Myc (Santa Cruz Biotechnology, Inc.) and anti-DsbA (Bardwell et al. 1993).
Acknowledgments
We thank lab members, especially Markus Eser and Seung-Hyun Cho for helpful discussions. J.B. is an American Cancer Society Professor. This work was supported by NIH grants GM41883 (J.B.) and GM46941 (C.A.K).
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051355705.
References
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader, M., Muse, W., Ballou, D.P., Gassner, C., and Bardwell, J.C. 1999. Oxidative protein folding is driven by the electron transport system. Cell 98 217–227. [DOI] [PubMed] [Google Scholar]
- Bader, M.W., Xie, T., Yu, C.A., and Bardwell, J.C. 2000. Disulfide bonds are generated by quinone reduction. J. Biol. Chem. 275 26082–26088. [DOI] [PubMed] [Google Scholar]
- Bardwell, J.C., Lee, J.O., Jander, G., Martin, N., Belin, D., and Beckwith, J. 1993. A pathway for disulfide bond formation in vivo. Proc. Natl. Acad. Sci. 90 1038–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabibbo, A., Pagani, M., Fabbri, M., Rocchi, M., Farmery, M.R., Bulleid, N.J., and Sitia, R. 2000. ERO1-L, a human protein that favors disulfide bond formation in the endoplasmic reticulum. J. Biol. Chem. 275 4827–4833. [DOI] [PubMed] [Google Scholar]
- Collet, J.F. and Bardwell, J.C. 2002. Oxidative protein folding in bacteria. Mol. Microbiol. 44 1–8. [DOI] [PubMed] [Google Scholar]
- Coppock, D., Kopman, C., Gudas, J., and Cina-Poppe, D.A. 2000. Regulation of the quiescence-induced genes: quiescin Q6, decorin, and ribosomal protein S29. Biochem. Biophys. Res. Commun. 269 604–610. [DOI] [PubMed] [Google Scholar]
- Cuff, J.A. and Barton, G.J. 2000. Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins 40 502–511. [DOI] [PubMed] [Google Scholar]
- Fassio, A. and Sitia, R. 2002. Formation, isomerisation and reduction of disulphide bonds during protein quality control in the endoplasmic reticulum. Histochem. Cell Biol. 117 151–157. [DOI] [PubMed] [Google Scholar]
- Frand, A.R. and Kaiser, C.A. 1998. The ERO1 gene of yeast is required for oxidation of protein dithiols in the endoplasmic reticulum. Mol. Cell 1 161–170. [DOI] [PubMed] [Google Scholar]
- ———. 1999. Ero1p oxidizes protein disulfide isomerase in a pathway for disulfide bond formation in the endoplasmic reticulum. Mol. Cell 4 469–477. [DOI] [PubMed] [Google Scholar]
- ———. 2000. Two pairs of conserved cysteines are required for the oxidative activity of Ero1p in protein disulfide bond formation in the endoplasmic reticulum. Mol. Biol. Cell 11 2833–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geourjon, C. and Deleage, G. 1994. SOPM: A self-optimized method for protein secondary structure prediction. Protein Eng. 7 157–164. [DOI] [PubMed] [Google Scholar]
- Gerber, J., Muhlenhoff, U., Hofhaus, G., Lill, R., and Lisowsky, T. 2001. Yeast ERV2p is the first microsomal FAD-linked sulfhydryl oxidase of the Erv1p/Alrp protein family. J. Biol. Chem. 276 23486–23491. [DOI] [PubMed] [Google Scholar]
- Grauschopf, U., Fritz, A., and Glockshuber, R. 2003. Mechanism of the electron transfer catalyst DsbB from Escherichia coli. EMBO J. 22 3503–3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross, E., Sevier, C.S., Vala, A., Kaiser, C.A., and Fass, D. 2002. A new FAD-binding fold and intersubunit disulfide shuttle in the thiol oxidase Erv2p. Nat. Struct. Biol. 9 61–67. [DOI] [PubMed] [Google Scholar]
- Gross, E., Kastner, D.B., Kaiser, C.A., and Fass, D. 2004. Structure of Ero1p, source of disulfide bonds for oxidative protein folding in the cell. Cell 117 601–610. [DOI] [PubMed] [Google Scholar]
- Guilhot, C., Jander, G., Martin, N.L., and Beckwith, J. 1995. Evidence that the pathway of disulfide bond formation in Escherichia coli involves interactions between the cysteines of DsbB and DsbA. Proc. Natl. Acad. Sci. 92 9895–9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haebel, P.W., Goldstone, D., Katzen, F., Beckwith, J., and Metcalf, P. 2002. The disulfide bond isomerase DsbC is activated by an immunoglobulin- fold thiol oxidoreductase: Crystal structure of the DsbC–DsbDα complex. EMBO J. 21 4774–4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoober, K.L., Glynn, N.M., Burnside, J., Coppock, D.L., and Thorpe, C. 1999. Homology between egg white sulfhydryl oxidase and quiescin Q6 defines a new class of flavin-linked sulfhydryl oxidases. J. Biol. Chem. 274 31759–31762. [DOI] [PubMed] [Google Scholar]
- Inaba, K. and Ito, K. 2002. Paradoxical redox properties of DsbB and DsbA in the protein disulfide-introducing reaction cascade. EMBO J. 21 2646–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba, K., Takahashi, Y.H., Fujieda, N., Kano, K., Miyoshi, H., and Ito, K. 2004. DsbB elicits a red-shift of bound ubiquinone during the catalysis of DsbA oxidation. J. Biol. Chem. 279 6761–6768. [DOI] [PubMed] [Google Scholar]
- Jander, G., Martin, N.L., and Beckwith, J. 1994. Two cysteines in each periplasmic domain of the membrane protein DsbB are required for its function in protein disulfide bond formation. EMBO J. 13 5121–5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, D.T. 1999. Protein secondary structure prediction based on position- specific scoring matrices. J. Mol. Biol. 292 195–202. [DOI] [PubMed] [Google Scholar]
- Kadokura, H. and Beckwith, J. 2002. Four cysteines of the membrane protein DsbB act in concert to oxidize its substrate DsbA. EMBO J. 21 2354–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadokura, H., Bader, M., Tian, H., Bardwell, J.C., and Beckwith, J. 2000. Roles of a conserved arginine residue of DsbB in linking protein disulfide- bond-formation pathway to the respiratory chain of Escherichia coli. Proc. Natl. Acad. Sci. 97 10884–10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadokura, H., Katzen, F., and Beckwith, J. 2003. Protein disulfide bond formation in prokaryotes. Annu. Rev. Biochem. 72 111–135. [DOI] [PubMed] [Google Scholar]
- Karplus, K., Barrett, C., and Hughey, R. 1998. Hidden Markov models for detecting remote protein homologies. Bioinformatics 14 846–856. [DOI] [PubMed] [Google Scholar]
- Katzen, F., Deshmukh, M., Daldal, F., and Beckwith, J. 2002. Evolutionary domain fusion expanded the substrate specificity of the transmembrane electron transporter DsbD. EMBO J. 21 3960–3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishigami, S. and Ito, K. 1996. Roles of cysteine residues of DsbB in its activity to reoxidize DsbA, the protein disulphide bond catalyst of Escherichia coli. Genes Cells 1 201–208. [DOI] [PubMed] [Google Scholar]
- Kishigami, S., Akiyama, Y., and Ito, K. 1995a. Redox states of DsbA in the periplasm of Escherichia coli. FEBS Lett. 364 55–58. [DOI] [PubMed] [Google Scholar]
- Kishigami, S., Kanaya, E., Kikuchi, M., and Ito, K. 1995b. DsbA–DsbB interaction through their active site cysteines. Evidence from an odd cysteine mutant of DsbA. J. Biol. Chem. 270 17072–17074. [DOI] [PubMed] [Google Scholar]
- Kobayashi, T. and Ito, K. 1999. Respiratory chain strongly oxidizes the CXXC motif of DsbB in the Escherichia coli disulfide bond formation pathway. EMBO J. 18 1192–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, T., Kishigami, S., Sone, M., Inokuchi, H., Mogi, T., and Ito, K. 1997. Respiratory chain is required to maintain oxidized states of the DsbA–DsbB disulfide bond formation system in aerobically growing Escherichia coli cells. Proc. Natl. Acad. Sci. 94 11857–11862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J., Hofhaus, G., and Lisowsky, T. 2000. Erv1p from Saccharomyces cerevisiae is a FAD-linked sulfhydryl oxidase. FEBS Lett. 477 62–66. [DOI] [PubMed] [Google Scholar]
- Lisowsky, T., Lee, J.E., Polimeno, L., Francavilla, A., and Hofhaus, G. 2001. Mammalian augmenter of liver regeneration protein is a sulfhydryl oxidase. Dig. Liver Dis. 33 173–180. [DOI] [PubMed] [Google Scholar]
- McGuffin, L.J., Bryson, K., and Jones, D.T. 2000. The PSIPRED protein structure prediction server. Bioinformatics 16 404–405. [DOI] [PubMed] [Google Scholar]
- Mezghrani, A., Fassio, A., Benham, A., Simmen, T., Braakman, I., and Sitia, R. 2001. Manipulation of oxidative protein folding and PDI redox state in mammalian cells. EMBO J. 20 6288–6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouali, M. and King, R.D. 2000. Cascaded multiple classifiers for secondary structure prediction. Protein Sci. 9 1162–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard, M.G., Travers, K.J., and Weissman, J.S. 1998. Ero1p: A novel and ubiquitous protein with an essential role in oxidative protein folding in the endoplasmic reticulum. Mol. Cell 1 171–182. [DOI] [PubMed] [Google Scholar]
- Pollastri, G. and McLysaght, A. 2004. Porter: A new, accurate server for protein secondary structure prediction. Bioinformatics e-pub Dec 7. [DOI] [PubMed]
- Pollastri, G., Przybylski, D., Rost, B., and Baldi, P. 2002. Improving the prediction of protein secondary structure in three and eight classes using recurrent neural networks and profiles. Proteins 47 228–235. [DOI] [PubMed] [Google Scholar]
- Regeimbal, J. and Bardwell, J.C. 2002. DsbB catalyzes disulfide bond formation de novo. J. Biol. Chem. 277 32706–32713. [DOI] [PubMed] [Google Scholar]
- Rost, B. and Sander, C. 1993. Prediction of protein secondary structure at better than 70% accuracy. J. Mol. Biol. 232 584–599. [DOI] [PubMed] [Google Scholar]
- ———. 1994. Combining evolutionary information and neural networks to predict protein secondary structure. Proteins 19 55–72. [DOI] [PubMed] [Google Scholar]
- Senkevich, T.G., White, C.L., Koonin, E.V., and Moss, B. 2002. Complete pathway for protein disulfide bond formation encoded by poxviruses. Proc. Natl. Acad. Sci. 99 6667–6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevier, C.S. and Kaiser, C.A. 2002. Formation and transfer of disulphide bonds in living cells. Nat. Rev. Mol. Cell Biol. 3 836–847. [DOI] [PubMed] [Google Scholar]
- Sevier, C.S., Cuozzo, J.W., Vala, A., Aslund, F., and Kaiser, C.A. 2001. A flavoprotein oxidase defines a new endoplasmic reticulum pathway for biosynthetic disulphide bond formation. Nat. Cell Biol. 3 874–882. [DOI] [PubMed] [Google Scholar]
- Takahashi, Y., Inaba, K., and Ito, K. 2004. Characterization of the menaquinone dependent disulfide bond formation pathway of Escherichia coli. J. Biol. Chem. 279 47057–47065. [DOI] [PubMed] [Google Scholar]
- Thorpe, C., Hoober, K.L., Raje, S., Glynn, N.M., Burnside, J., Turi, G.K., and Coppock, D.L. 2002. Sulfhydryl oxidases: Emerging catalysts of protein disulfide bond formation in eukaryotes. Arch. Biochem. Biophys. 405 1–12. [DOI] [PubMed] [Google Scholar]
- Tu, B.P. and Weissman, J.S. 2002. The FAD- and O(2)-dependent reaction cycle of Ero1-mediated oxidative protein folding in the endoplasmic reticulum. Mol. Cell 10 983–994. [DOI] [PubMed] [Google Scholar]
- Tu, B.P., Ho-Schleyer, S.C., Travers, K.J., and Weissman, J.S. 2000. Biochemical basis of oxidative protein folding in the endoplasmic reticulum. Science 290 1571–1574. [DOI] [PubMed] [Google Scholar]
- Xie, T., Yu, L., Bader, M.W., Bardwell, J.C., and Yu, C.A. 2002. Identification of the ubiquinone-binding domain in the disulfide catalyst disulfide bond protein B. J. Biol. Chem. 277 1649–1652. [DOI] [PubMed] [Google Scholar]