

Abstract
The unique β-hydroxyacyl-ACP dehydratase in Plasmodium falciparum, PfFabZ, is involved in fatty acid biosynthesis and catalyzes the dehydration of β-hydroxy fatty acids linked to acyl carrier protein. The structure was solved by single anomalous dispersion (SAD) phasing using a quick-soaking experiment with potassium iodide and refined to a resolution of 2.1 Å. The crystal structure represents the first structure of a Plasmodium β-hydroxyacyl-ACP dehydratase with broad substrate specificity. The asymmetric unit contains a hexamer that appears as a trimer of dimers. Each dimer shows the known “hot dog” fold that has been observed in only a few other protein structures. Each of the two independent active sites in the dimer is formed by equal contributions from both subunits. The active site is mainly hydrophobic and looks like an L-shaped tunnel. The catalytically important amino acids His 133 and Glu 147′ (from the other subunit), together with His98′, form the only hydrophilic site in this tunnel. The inner end of the active site tunnel is closed by the phenyl ring of Phe 169, which is located in a flexible, partly visible loop. In order to explain the acceptance of substrates longer than ~C-7, the phenyl ring must move away to open the tunnel. The present structure supports an enzymatic mechanism consisting of an elimination reaction catalyzed by His 133 and Glu147′. 3-decynoyl-N-acetylcysteamine, an inhibitor known to interact with the E. coli dehydratase/isomerase, turned out to interact covalently with PfFabZ. A first model of PfFabZ with this potent inhibitor is presented.
Keywords: Plasmodium falciparum, malaria, drug target, β-hydroxyacyl-ACP dehydratase, SAD phasing, 3-decynoyl-N-acetylcysteamine, fatty acid biosynthesis, crystal structure
Malaria is one of the greatest causes of illness and death in the world, and it is estimated that 500 million clinical episodes occur annually, claiming up to three million deaths per year (World Health Organization 1999). There are numerous factors that contribute to the persistence of malaria. Vector control is hampered by financial constraints and insecticide resistance. Treatment programs are limited by the poverty of most endemic areas, and despite enormous efforts an effective malaria vaccine is still not available. The anti-malarial treatment has depended on drugs developed decades ago, and the occurrence of resistance against almost all available drugs has largely contributed to the recent resurgence of malaria. The recent successful completion of the genome sequencing of Plasmodium falciparum, the causative agent for the most severe form of malaria, has been a milestone that provides a tremendous amount of information on a genetic level (Gardner et al. 2002).
A series of highly promising and so far unknown or only scarcely described metabolic pathways in P. falciparum were identified through the genome project, among which a complete type-II fatty acid biosynthesis system (FAS-II) could be detected (Gardner et al. 2002). The FAS-II pathway represents a particularly interesting drug target, as there are major differences between the structural organization of the plastid-associated enzymes found in plants and most microorganisms including Plasmodia, and the cytosolic enzymes of mammals and yeast (Rock and Cronan 1996). The latter carry a so-called FAS-I system that is built of a large homodimeric multi-functional protein, whereas the FAS-II system consists of discrete monofunctional enzymes for the same enzymatic reactions. Escherichia coli fatty acid biosynthesis has been studied extensively, and it serves as the paradigm for FAS-II systems. It is a sophisticated and highly versatile system, consisting of enzymatic steps that can be performed by several isoforms of the corresponding enzymes. This allows synthesis of saturated as well as unsaturated fatty acids. In contrast, P. falciparum harbors a simplified set of enzymes that are able to produce only saturated fatty acids. Subsequently, all enzymes taking part in the pathway seem to be unique.
The discovery of a series of compounds (e.g., thiolactomycin, diazaborines, isoniazid, triclosan) that can selectively inhibit FAS-I or FAS-II pathways served to validate FAS-II enzymes as drug targets (Rock and Cronan 1996; Campbell and Cronan 2001; Heath et al. 2001, 2002; Waller et al. 2003). Inhibition of P. falciparum with thiolactomycin was reported at an IC50 of 50 μM (Waller et al. 1998, 2003), and triclosan was found to inhibit P. falciparum growth with an IC50 of 1 μM (Perozzo et al. 2002). Furthermore, in vitro whole-cell studies exhibited high activity of triclosan against multidrug-resistant parasites, which was corroborated by in vivo efficacy studies in the P. berghei mouse model (Surolia and Surolia 2001). These findings clearly demonstrated the essential role of fatty acid biosynthesis, and it could be shown by us and others that P. falciparum FAS-II may comprise very attractive novel targets for the development of new and selective anti-malarials (McLeod et al. 2001; Surolia and Surolia 2001; Perozzo et al. 2002).
The third step in chain elongation during fatty acid biosynthesis in P. falciparum is carried out by the β-hydroxyacyl-ACP dehydratase (PfFabZ) and corresponds to the primary dehydratase (EcFabZ) participating in fatty acid biosynthesis of E. coli. A second dehydratase in E. coli (EcFabA) is involved in the dehydration and subsequent isomerization of β-hydroxydecanoyl- ACP to cis-3-decenoyl-ACP. 3-decynoyl-N-acetylcysteamine (3-decynoyl-NAC), an analog of the cis-3-decenoyl-ACP isomerization product, inhibits in vitro and in vivo the EcFabA dehydratase as well as the isomerase reaction. The inhibitor functions via triple bond isomerization to give a highly reactive 2,3-diene intermediate that covalently binds to the active-site histidine of EcFabA (Brock et al. 1967; Kass 1968; Rando and Bloch 1968; Endo et al. 1970; Morisaki and Bloch 1972). The covalent nature of 3-decynoyl-NAC binding to EcFabA has also been shown by a crystal structure (Leesong et al. 1996).
As deduced from studies performed on E. coli FabZ (Heath and Rock 1996), the primary dehydratases are only capable of performing the dehydration reaction. These enzymes are only distantly similar to EcFabA in the primary sequence, and there are no data published regarding 3-decynoyl-NAC inhibition of FabZ. Here we present the three-dimensional crystal structure of PfFabZ at 2.1 Å resolution as well as inhibition data of 3-decynoyl-NAC towards this enzyme.
Results
Structure solution
The crystal structure of P. falciparum β-hydroxyacyl-ACP dehydratase (PfFabZ) was solved with the single anomalous dispersion (SAD) method from a crystal that was soaked with 0.5 M potassium iodide for 30 sec, followed by extensive density modification and phase extension (see Materials and Methods). Iterative rounds of model building with MOLOC (Gerber and Muller 1995) and maximum-likelihood refinement with the CCP4 (Collaborative Computational Project 1994) program REFMAC 5.2 (Murshudov 1997) against a 2.1 Å data set measured at the Swiss Light Source (SLS), Villigen, Switzerland, resulted in a refined structure with very good R-factors and stereochemistry (Table 1). Since PfFabZ was expressed without the putative N-terminal signal- and translocation-sequence (amino acids 1–80), the crystallized protein consists of residues 81–230. Ninety-one percent of these amino acids could be modeled into the electron density maps, and the missing residues are at the N and C termini and in one or two flexible loops per subunit.
Table 1.
Crystallographic data and refinement statistics
| SLS native data set | In-house native data set | In-house KI derivative data set | |
| Space group | C2221 (No. 20) | C2221 (No. 20) | C2221 (No. 20) |
| Unit cell dimensions | |||
| a, b, c [Å] | 90.6, 127.5, 173.7 | 89.4, 127.4, 172.1 | 90.2, 129.0, 172.3 |
| α, β, γ [°] | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution range [Å] | 40.0–2.1 (2.15–2.1)a | 40.0–2.3 (2.33–2.3)a | 40.0–3.0 (3.14–3.0)a |
| No. of total reflections | 430,930 | 326,890 | 144,992b |
| No. of unique reflections | 59,472 | 44,448 | 38,133b |
| Rsymc | 0.10 (0.83)a | 0.081 (0.81)a | 0.073 (0.49)a |
| <I/σ (I)> | 10.9 (2.4)a | 12.1 (2.4)a | 15.6 (2.8)a |
| Completeness | 99.6% (100.0%)a | 99.8% (99.9%)a | 98.0% (84.6%)a |
| Phasing Powerd | — | — | 0.62e |
| RCullisf | — | — | 0.89e |
| Figure-of-Meritg | — | — | 0.27e |
| No. of refined atoms | — | — | |
| protein | 6401 | ||
| cacodylate | 30 | ||
| sulfate | 35 | ||
| chloride | 6 | ||
| water | 256 | ||
| R-factorh | 0.17 | — | — |
| Free R-factor | 0.22 | ||
| RMSD bond lengthsi | 0.013 Å | — | — |
| bond angles | 1.4° | ||
a Values in parentheses refer to the highest resolution shell.
b Friedel pairs not merged.
cRsym=∑h ∑i | Ii(h) − <I(h)>|/∑h ∑i Ii(h), where Ii(h) and <I(h)> are the ith and mean measurement of the intensity of reflection h, respectively.
d Phasing Power= ∑Fhcalc/∑| Fphobs − Fphcalc |, where Fhcalc is the calculated heavy atom structure factor amplitude, Fphobs and Fphcalc are the observed and calculated heavy atom derivative structure factor amplitudes, respectively.
e Acentric reflections only, because of SAD (see “Experimental Procedures”).
fRCullis = ∑| | Fphobs − Fpobs| − Fhcalc|/∑| Fphobs − Fpobs|, where Fphobs is the observed heavy atom derivative structure factor amplitude, Fpobs is the observed native structure factor amplitude, and Fhcalc is the calculated heavy atom structure factor amplitude.
g Figure-of-Merit = ∫P(α)*exp(iα) dα/∫P(α) dα, where P(α) is the probability of the phase α.
hR = ∑| Fpobs − Fpcalc|/∑Fpobs, where Fpobs and Fpcalc are the observed and calculated structure factor amplitudes, respectively.
i Root-mean-square-deviation from the parameter set for ideal stereochemistry (Engh and Huber 1991).
Structure of the hexamer
The asymmetric unit contains a hexamer of PfFabZ with local 32-point group symmetry (Fig. 1 ▶), indicating that the hexamer is a trimer of dimers (see Discussion). The six subunits are structurally very similar. After least-squares super-position with the CCP4 program LSQKAB, the overall RMSD values are 0.27 Å ± 0.06 Å for the positions of 127 common Cα atoms, and 0.16 ± 0.02 Å for the positions of 105 Cα atoms excluding the flexible termini and loop regions. The RMSD value for the positions of all 808 atoms from these 105 residues excluding the flexible regions is 0.38 ± 0.04 Å. Because of the high structural similarity, we will show and discuss only structural details of a selected dimer in the subsequent paragraphs.
Figure 1.

Schematic picture of the PfFabZ hexamer in the asymmetric unit. The dimer A/B is colored cyan/orange, the dimer C/D is red/yellow, and the dimer E/F is blue/magenta. The local threefold axis perpendicular of the paper plane is indicated with an open triangle; the local twofold axes in the plane of the paper are indicated by straight lines with open ellipses at their ends. The secondary structure assignment was done with the program DSSP (Kabsch and Sander 1983). For the six subunits, the following amino acid residues were visible in the electron density maps: A: 84–161, 166–229; B: 85–161, 170–199, 203–227; C: 86–161, 167–228; D: 84–160, 167–198, 205–227; E: 84–160, 166–199, 203–228; F: 83–161, 166–228. The crystallized protein consists of residues 81–230.
The total buried surface in the hexamer is ~14,900 Å2, calculated with the program DSSP (Kabsch and Sander 1983) as the difference between the sum of the six subunit accessible surfaces and the hexamer accessible surface. Of the total buried surface, ~2300 Å2 is buried at the contact surface of the two subunits in each dimer.
Structures of the subunit and the dimer
Each subunit consists of a six-stranded strongly curved anti-parallel β-sheet with a long α-helix spanning the concave side of the sheet, and a short α-helix perpendicular to the long α-helix lying on top of its C-terminal end. In the dimer, the two six-stranded β-sheets of the subunits form a continuous 12-stranded anti-parallel β-sheet with two long central α-helices lying on the concave side and two short α-helices perpendicular to the long α-helices on top of them (Fig. 2 ▶). Because of its appearance, this fold is called “hot dog” fold, first described for the structure of the E. coli β-hydroxydecanoyl thiol dehydratase, EcFabA (PDB entry 1MKA.pdb; Leesong et al. 1996), later observed as well in the structures of Pseudomonas sp. 4-hydroxybenzoyl- CoA thioesterase (PDB entry 1BVQ.pdb; Benning et al. 1998), Aeromonas caviae (R)-specific enoyl coenzyme A hydratase (PDB entry 1IQ6.pdb; Hisano et al. 2003), and recently Pseudomonas aeruginosa β-hydroxyacyl-ACP dehydratase, PaFabZ (PDB entry 1U1Z.pdb; Kimber et al. 2004). Super-positions of the EcFabA dimer and the PaFabZ dimer A/B onto the PfFabZ dimer A/B, calculated with the program SUPERIMPOSE (Diederichs 1995), are shown in Figure 3 ▶ together with an amino acid sequence alignment. The structures of the dimers of PaFabZ and PfFabZ are very similar, as can be expected for an amino acid sequence identity of 47%. Despite the low amino acid sequence identity of only 21%, there is also a striking overall structural similarity between EcFabA and PfFabZ, as was predicted by Leesong et al. (1996). In the structural superposition, the 12-stranded anti-parallel β-sheet appears to be very similar, whereas the two long central α-helices show different angles of their helical axes with respect to the β-sheet. The short N-terminal α-helices and the loops on top of the long α-helices are shifted with respect to each other.
Figure 2.
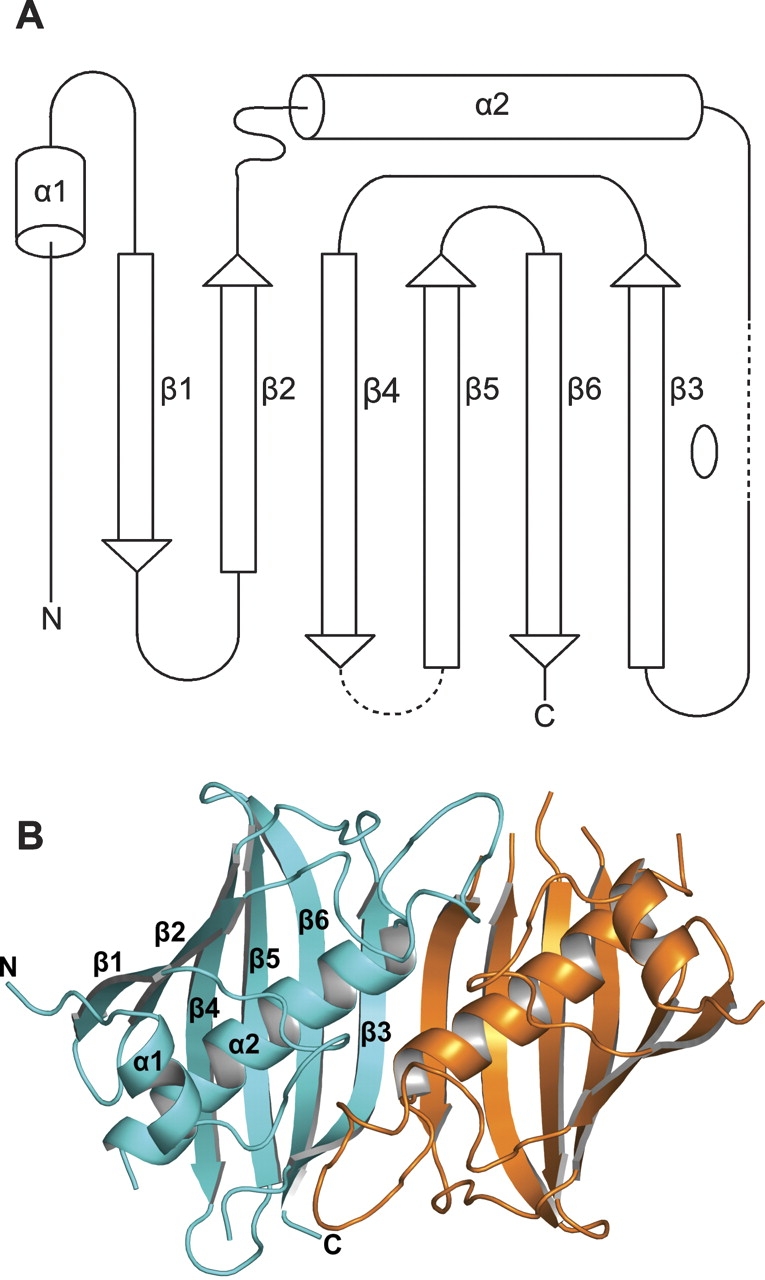

(A) Topology of the subunit fold. A long N-terminal stretch runs parallel to β1, and between β2 and α2 is a long coiled loop. The loops between α2 and β3 and between β4 and β5 are partially disordered and shown as dashed lines. The local twofold axis of the dimer is indicated by an open ellipse on the right side of β3. The secondary structure elements for the six subunits in the asymmetric unit, assigned with DSSP (Kabsch and Sander 1983), are as follows: α1, 89–95; β1, 107–112; β2, 116–122; α2, 142– 159 ± 1; β3, 170 ± 1–179; β4, 188–198 ± 1; β5, 205 ± 1–213; β6, 216–227 ± 1. (B) Schematic picture of the PfFabZ dimer formed by subunits A (cyan) and B (orange), viewed from the top of the “hot dog” fold. For subunit A, the secondary structure elements are labeled as in Fig. 2A. The partially disordered loop between β4 and β5 is only fully visible in subunit A. (C) Schematic picture of the PfFabZ dimer formed by subunits A (cyan) and B (orange), viewed along the “hot dog” fold.
Figure 3.
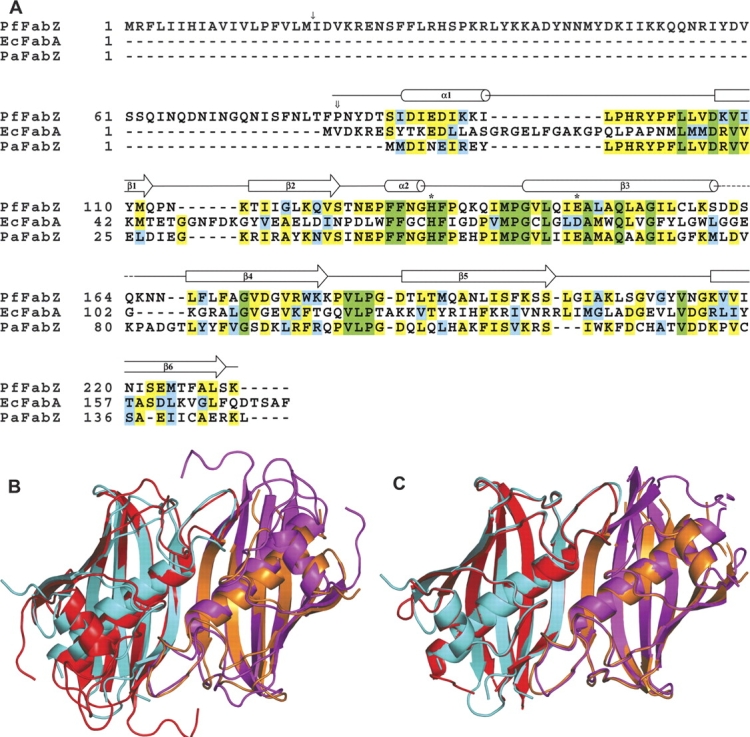
(A) Amino acid sequence alignment of PfFabZ with EcFabA and PaFabZ. Green indicates completely conserved residues, yellow indicates two or more highly conserved residues, and blue indicates at least one similar amino acid residue. The putative signal sequence cleavage site of PfFabZ located between Met18 and Ile19 is marked with a single arrow. The first residue of the expressed protein, Pro81, is marked with a double arrow. The catalytically important residues His133 and Glu147 are marked with asterisks. The secondary structure elements of PfFabZ, deduced from subunit A, are schematically drawn above the amino acid sequences. With reference to the 150 amino acids of the expressed PfFabZ protein, PaFabZ shows 47% sequence identity, and EcFabA shows 21% sequence identity. (B) Schematic picture of a superposition of a PfFabZ dimer (in cyan and orange) with the dimer of EcFabA (in red and magenta). (C) Schematic picture of a superposition of a PfFabZ dimer (in cyan and orange) with a dimer of PaFabZ (in red and magenta).
Our assignment of secondary structural elements for PfFabZ deviates in two locations from the one for EcFabA (Leesong et al. 1996): (1) In PfFabZ, the structurally equivalent part of β1 in EcFabA was not assigned to a β-strand, since only two residues make interstrand hydrogen bonds typical for a β-sheet. Thus, the first β-strand β1 in PfFabZ corresponds to the second β-strand β2 in EcFabA. (2) In PfFabZ, the structurally equivalent part of the short helix α2 in EcFabA was not assigned to a helix, since only 1–2 residues are in a 310-helix conformation. Thus, the long central α-helix α2 in PfFabZ corresponds to the long central α-helix α3 in EcFabA.
Structure of the active site
The catalytically important active site residues in EcFabA are His70 and Asp84′ (Leesong et al. 1996) (the prime indicates a residue from the other subunit in the dimer); in PaFabZ, the equivalent residues are His49 and Glu63′ (Kimber et al. 2004). In PfFabZ, the equivalent residues by both amino acid sequence alignment (Fig. 3A ▶) and structural superposition are His133 and Glu147′. The two active sites in the PfFabZ dimer are separated symmetrically about the twofold axis by ~18 Å (measured as the distance between the active site His133:Nɛ2 of the two subunits) and appear to be structurally completely independent of each other. Each active site is formed by equal contributions from the two subunits (Fig. 4 ▶). His133 is located in the coiled loop reaching over from one subunit, and Glu147′ is located in the first third of the long central helix α2′ from the other subunit. Together with His98′, they form the only hydrophilic site in an otherwise completely hydrophobic active site. The imidazole ring of His133 is stacked between the phenyl ring of Phe134 and the proline ring of Pro141. This stacking between the side chains of His133 and Phe134 is accomplished by a rather unusual cis-peptide bond between both residues, and has also been observed for the equivalent residues His70 and Phe71 in the EcFabA structure (Leesong et al. 1996), and for His49 and Phe50 in the PaFabZ structure (Kimber et al. 2004). A chloride anion has bound at each active site in hydrogen bonding distance to His133:Nɛ2 and the main chain nitrogen of Gly142 at the N terminus of α2. The identification of chloride was based on both the high electron density and the typical mixed hydrophobic and positively charged environment, including the N-terminal positive dipole moment of α2. The chloride binding is also indirectly confirmed by the observed binding of the iodides at the same sites from the quick soaking experiment with potassium iodide for phase determination (see Materials and Methods). Obviously, the hydrogen donor nitrogen atoms from His133 and Gly142 and the positive dipole moment at the N terminus of α2 create a local positive electrostatic field that appears to be attractive for halide atoms and might be important for the enzymatic mechanism (see Discussion). The sites in PaFabZ that correspond to the chloride binding sites in the PfFabZ structure have been modeled with water molecules exhibiting relatively low B-factors (Kimber et al. 2004), possibly indicating a partial occupation by chloride ions from the protein storage buffer in this structure, too. In contrast to the chloride binding, we believe that the loosely bound cacodylate at the active site of PfFabZ is an artifact from the crystallization buffer and has no physiological meaning. Each substrate binding pocket includes one water molecule that is hydrogen bonded to the main chain nitrogen of Gly142 and Val143, and to the carboxyl oxygen of Glu147′. In the EcFabA and PaFabZ structures, a water molecule is found at the very same position, and it was proposed to be the stored water of hydration (Leesong et al. 1996; Kimber et al. 2004).
Figure 4.

Stereo picture of the PfFabZ dimer active site shown in a ball-and-stick representation. The coloring is as follows: carbon atoms from subunit A, cyan; carbon atoms from subunit B, orange; oxygen atoms, red; nitrogen atoms, blue; sulfur atoms, yellow; chloride, green; arsenic and carbon from cacodylate in magenta and white, respectively. The catalytically important active-site residues His133 and Glu147′ and the stored water of hydration are labeled with H133, E147′, and WAT, respectively. Hydrogen bonds from the side chains of His133, Glu147′, chloride, cacodylate, and water are indicated as lines of small yellow spheres.
The active site of PfFabZ looks like an L-shaped tunnel with a diameter ranging from ~4–7 Å, a length of ~18 Å, and a 90° kink in the middle (Fig. 5A, B ▶). The presumed entrance of this tunnel is located at the interface of the two subunits in the dimer close to the contact of the two β-sheets. In contrast to the PaFabZ structure, where five of the six crystallographically independent entrances of the active sites are blocked by Tyr88, the entrances of the active sites in PfFabZ are open without steric hindrance from the equivalent residue Leu170. The catalytically important residues His133 and Glu147′ face the innermost arm of the L-shaped tunnel with their side chains. The tunnel is closed at its inner end by a “lid” formed by the phenyl ring of Phe169 in five of the six crystallographically independent active sites. Phe169 belongs to the highly flexible and partially disordered loop between α2 and β3 (see Fig. 2 ▶ for definition) and, due to its high flexibility, is poorly visible in the electron density maps of all subunits, with the exception of subunit B, where we could not see it in the electron density maps. Phe169 is not stabilized by crystal contacts nor by contacts to a neighboring dimer in the hexamer. As a result of the disordered Phe169 in subunit B, the corresponding active-site tunnel appears not to be closed and leads back to the surface with an “exit” after another ~11 Å, exhibiting an overall shape of a “U” (Fig. 5A, B ▶). Thus, without any structural rearrangements, the active site could accommodate a substrate molecule with a total length of less then ~18 Å with a closed Phe169 “lid”, whereas with an open Phe169 “lid”, the substrate could have a length of up to ~29 Å until it would stick out of the active-site tunnel through the “exit.”
Figure 5.

Surface representations of the L-shaped active site tunnels of PfFabZ. The dimer formed by subunitsA(cyan) and B (orange) is shownas aCα-trace. The active-site cavities were calculated with the program PASS (Brady and Stouten 2000). The active-site residues His133 and Glu147 and the Phe169 “lid” (not visible in subunit B) are shown in ball-and-stick representation and marked with labels. Atoms of the side chains are colored as follows: carbon atoms, white; nitrogen atoms, blue; oxygen atoms, red. (A)The active-site cavities viewed from the top are shown in dark gray surface representations, and the presumed entries to the active sites are marked with green arrows. The presumed exit of the active site is marked with a red arrow (only in subunit B). (B)View from the side without arrows. (C)The active-site cavities viewed from the top are shown in a gray semitransparent surface representation with the 3-decynoyl-NAC inhibitor modeled into the active site. The carbon atoms of the inhibitor are colored green. Note that the inhibitor fits in only one of the two observed active-site tunnels, depending on the presence or absence of the Phe 169 lid. In this model, Phe169 (of subunit A) has been rotated with respect to the observed position to avoid steric hindrance with the modeled inhibitor binding (see text). The presumed entries to the active sites are marked with green arrows, and the presumed exit of the active site is marked with a red arrow(only in subunit B). (D) View from the side without arrows.
PfFabZ inhibition by 3-decynoyl-NAC
Due to the striking overall structural similarity between EcFabA and PfFabZ, we tested 3-decynoyl-NAC (kindly provided by Dr. P. Valenti, University of Bologna) against P. falciparum FabZ. To date there are no data published describing 3-decynoyl-NAC to be an inhibitor of the corresponding primary dehydratase in E. coli (EcFabZ). Inhibition experiments clearly showed that PfFabZ was potently inhibited by 3-decynoyl-NAC in a time-dependent manner (see Fig. 6A ▶). Inhibition was accelerated when higher inhibitor concentrations were applied, which is indicative for mechanism-based inhibition mechanisms (Silverman 1988). The covalent nature of inhibitor binding to PfFabZ was confirmed by mass spectroscopy of the protein- inhibitor complex (m/z ratio: 17289, corresponding to one bound 3-decynoyl-NAC molecule per protein subunit).
Figure 6.

(A) PfFabZ inhibition by 3-decynoyl-NAC (shown in the inset). Total enzyme inhibition occurs within 30 min after applying a 1.5:1 molar ratio of inhibitor to protein subunit. The control reaction was performed under identical conditions but without β-hydroxybutyryl-CoA as substrate. (B) Proposed enzymatic mechanism of PfFabZ, adapted from Leesong et al. (1996) for EcFabA. The thioester carbonyl group is positively polarized by the hydrogen bond to the main chain nitrogen of Gly 142 and by the N-terminal positive dipole moment of helix α2 (indicated by an ellipse with plus and minus sign). The α-C-H bond is positively polarized by His 133 and the β-C-OH bond is positively polarized by Glu147′, thus lowering the activation energy(s) for dehydration.
Discussion
During the writing of this manuscript, the crystal structure of the Pseudomonas aeruginosa FabZ (PaFabZ) was published (Kimber et al. 2004). PaFabZ forms a hexamer similar to the one that we observe. Those authors reported that PaFabZ also runs as a hexamer on a gel filtration column, leading to their hypothesis that the hexameric state could have a physiological role in stabilizing the active site. In the case of PfFabZ, however, the oligomerization state is concentration-dependent, ranging from dimers at low protein concentrations to hexamers at high protein concentrations as deduced from gel filtration experiments. Moreover, native PAGE and Ferguson plot analysis (Hames 1981) showed that PfFabZ runs with the size of a dimer (data not shown). In addition, we have crystallized PfFabZ in a different crystal form with only one dimer in the asymmetric unit and no higher oligomerization state formed by crystallographic symmetry. The structures of the PfFabZ dimers are very similar in both crystal forms. It seems that for PfFabZ the hexameric form is not required for stabilizing the active-site structure and thus for enzymatic function. In contrast, the dimeric architecture plays an important role for enzyme activity as each active site is situated at the subunit interface and is formed by equal contributions of the two adjacent subunits, meaning that only the dimer can exhibit catalytic activity.
Based on the crystal structure of EcFabA in complex with the 3-decynoyl-NAC inhibitor (Leesong et al. 1996), we modeled the covalent binding of this inhibitor to the active-site His133 of a PfFabZ dimer (subunits A and B) with the program MOLOC. One of the two active-site tunnels is closed by the Phe169 “lid”; the other appears to be open with an “exit” to the outside (see Fig. 5A, B ▶). At the active site where the Phe169 lid was not visible, only slight structural adaptations of the inhibitor and the His133 side chain were necessary to model the binding of the inhibitor. In contrast, the 3-decynoyl moiety clashed with the phenyl ring of Phe169 while binding into the active site comprising the Phe169 lid. Allowing only minor structural adaptations of the surrounding residues of Phe169, we could remove this steric hindrance by rotating the phenyl ring of Phe169 around its X1 side chain torsion angle from energetically favorable +60° in the crystal structure to energetically unfavorable −103° in the model. Since Phe169 and its environment are parts of flexible loops, we assume that in solution, this lid will open upon substrate or inhibitor binding at a low energy cost only. Interestingly, the residue equivalent to Phe169 in the PaFabZ structure, Tyr87, has been observed in an open conformation similar to the modeled position of Phe169, supporting our hypothesis that the Phe169 lid can open relatively easily. In EcFabA, the residue equivalent to Phe169 is Gly104, which does not interfere with the binding of long fatty-acid substrates.
As 3-decynoyl-NAC was found to be a potent inhibitor of PfFabZ, we tried to determine crystal structures for the covalent enzyme-inhibitor complex. However, all soaking trials with 3-decynoyl-NAC into the present crystal form failed, despite an apparently good accessibility of the active sites as judged by the crystal packing. Although the Phe169-containing loop makes only a few hydrophobic interactions with the neighboring molecule in the dimer without having any other crystal contacts, it is obvious that the necessary structural changes of PfFabZ upon inhibitor binding are blocked in this crystal form. Cocrystallization trials of PfFabZ with 3-decynoyl- NAC resulted in a different crystal form diffracting to 2.8 Å, with one dimer in the asymmetric unit and no higher oligomers formed by crystallographic symmetry. We solved this crystal structure by molecular replacement using a PfFabZ dimer. Unfortunately, residues 97–102 from the loop between α1 and β1 and residues 130–137 from the spatially neighboring loop between β2 and α2 encompassing the active-site His133 and the covalently bonded inhibitor were disordered in both subunits. The overall structure looked very similar and no major conformational changes were detected compared to the free enzyme (data not shown).
In our model of the PfFabZ-inhibitor complex, the NAC moiety is located at the entrance of the active-site tunnel, and the 3-decynoyl moiety reaches deep into the active-site tunnel (Fig. 5C, D ▶). The NAC moiety mimics the acyl-carrier- protein (ACP) phosphopantetheine group that carries the intermediates during fatty acid synthesis, and the 3-decynoyl moiety mimics the long aliphatic chain of the substrates. Thus, we believe that the natural binding partner of PfFabZ during fatty acid synthesis, ACP, enters the active-site tunnel with its phosphopantetheine group through the same entrance where the NAC moiety of the inhibitor has been modeled. In this hypothesis, the long aliphatic chains of the substrates would stick deep into the hydrophobic active-site tunnel, like the modeled 3-decynoyl moiety of the inhibitor. PfFabZ has a broad substrate specificity with respect to chain length, using fatty acids from C-4 to C-14 (Surolia and Surolia 2001). Thus fatty acids comprising longer aliphatic chains than the one of the 3-decynoyl-NAC inhibitor will bind to the enzyme. Apparently, the observed active sites with the Phe169 lid could only bind short substrates (up to ~C-7) without any major structural rearrangements. For longer substrates, the Phe169 lid must open, presumably by a combination of a side chain rotation with a (transient) movement of the flanking α2-β3-loop and the neighboring C terminus.
Based on the structural similarity between EcFabA and PfFabZ that also extends to the active site (i.e., His133, Glu147′, catalytic water molecule), we can conclude that the catalytic mechanism for PfFabZ will be the same as that proposed for EcFabA (Leesong et al. 1996). An adapted enzymatic mechanism which takes into account the different acidic amino acid is shown in Figure 6B ▶. In this mechanism, the thioester carbonyl bond is positively polarized by the hydrogen bond to the main chain nitrogen of Gly142 and by the N-terminal positive dipole moment of helix α2. The α-C-H bond is positively polarized by the free electron pair of Nɛ2 in the imidazole ring of His133 and indirectly by the positive dipole moment of α-helix α2, and the β-C-OH bond is positively polarized by a hydrogen bond mediated through the carboxylic acid group of Glu147′. This local positive environment appears to be attractive for halide ions, as observed in this crystal structure and presumably other (partially) negatively charged ions or chemical moieties. The role of this halide binding site under physiological conditions is not known and may well be a crystallization artifact. However, such an “anion binding hole” could be important for the enzymatic mechanism through stabilization of a carbanion formed during the proposed substrate conversion mechanism.
Conclusion
The present study reports the three-dimensional crystal structure of PfFabZ at 2.1 Å resolution as well as inhibition data of 3-decynoyl-NAC towards this enzyme, providing a framework for the understanding of the reaction mechanism of this type of enzyme. Moreover, the essential role of PfFabZ in fatty acid biosynthesis makes it an interesting target for drug discovery, and the current structure means a promising advance in the development of new anti-malarials, as it will serve as a model for structure-based drug development. The identification of potential lead compounds with inhibitory activity against PfFabZ using computational structure-based database screening approaches is in progress.
Materials and methods
Cloning, expression, and purification of PfFabZ
A pffabZ sequence N-terminally truncated by 240 base pairs was PCR-amplified from a P. falciparum (3D7 strain) gametocyte cDNA pSPORT plasmid library (kindly provided by Dr. T. Templeton, Weill Medical College of Cornell University) using forward primer 5′-TCAATTTACATATG CCTAATTATGATACAAGTATTGATATAGAA and the reverse primer 5′-AAGAATTCTTATTTCGATAAGGCAAA CGTCATTTCTGA. NdeI and EcoRI sites are underlined. PCR conditions were: 1 × 94°C for 2 min; 35 × 94°C for 20 sec, 52°C for 10 sec, 48°C for 10 sec, 60°C for 2 min. After restriction digestion, the gene was ligated into the pET28b vector (Novagen) and transfected into DH5α E. coli. Correct clones were identified and verified by restriction digestion, PCR, and automated sequencing with T7 forward and reverse primers. The resulting clone was designed to express the enzyme without the putative N-terminal signal- and translocation-sequence, i.e., amino acids 1–80.
PfFabZ, consisting of residues 81–230, was expressed at <15°C using BL21(DE3) Codon+-RIL cells (Stratagene) and Terrific broth. Cell pellets were suspended in buffer A (20 mM Tris/HCL [pH 8.0], 500 mM NaCl) and disrupted using a French press, and the protein was isolated by metal chelate affinity chromatography. After washing with buffer B (20 mM Tris/HCL [pH 8.0], 500 mM NaCl, 150 mM imidazole) and elution with buffer C (20 mM Tris/HCL [pH 8.0], 500 mM NaCl, 500 mM imidazole), the His6-tag was removed by thrombin digestion. The protein was diluted fourfold and repurified by means of cationic exchange chromatography. All fractions containing the enzyme were collected, concentrated, and applied to a Superdex 75 gel filtration column (Amersham Biosciences) equilibrated with 10 mM HEPES (pH 7.0) and 100 mM NaCl.
Crystallization and data collection
PfFabZ was crystallized in apo form using the hanging drop vapor diffusion method. Two microliters of 5–12 mg/mL protein was mixed with 2 μL well solutions of crystal screens “I”, “II” , and “cryo” from Hampton Research, and equilibrated against the reservoir solution at 16°C. Initial crystals appeared in the cryoscreen condition No. 15 and were subsequently refined. The optimized conditions were 170 mM ammonium sulfate, 85 mM cacodylic acid buffered at pH 5.5–6.5, 21%–27% PEG 8000, and 15% glycerol. Thin crystal plates with irregular edges appeared within 4 wk and grew to their final sizes of ~0.7 × 0.2 × 0.05 mm3 for another 4 wk.
In-house data sets were collected as consecutive series of 0.5° rotation images on a MAR345 imaging plate detector using CuKα radiation produced by an Enraf-Nonius FR591 rotating anode generator. The synchrotron data set was collected at a wavelength of 1.0715 Å as a consecutive series of 0.5° rotation images on a MARCCD detector at the protein beam line X06SA at the Swiss Light Source (SLS), in Villigen, Switzerland. All data sets were processed with XDS (Kabsch 2001). Data set statistics are given in Table 1.
Structure determination
We solved the crystal structure of PfFabZ by soaking a crystal with 0.5 M KI for 30 sec (Dauter et al. 2002). The crystal diffracted to 3 Å resolution in-house, but the resulting KI derivative data set was not isomorphous to the in-house native data set (Table 1) with an overall isomorphous R-factor of 30%. Using the anomalous differences from the KI data set alone, we could clearly locate six iodide positions in the asymmetric unit with SHELXD (Uson et al. 2003). These six iodide positions were put into SHARP (De la Fortelle and Bricogne 1997) for single anomalous dispersion (SAD) heavy atom refinement and phasing, followed by solvent flattening with SOLOMON (Abrahams 1996). Despite a rather clear separation between protein and solvent regions, the resulting electron density map was not interpretable. In order to improve the phases, we tried various density modification schemes including noncrystallographic symmetry (NCS) averaging. Of those, only the following scheme resulted in a fully interpretable electron density map. The six iodide positions indicated a hexamer with local 32-point group symmetry. A starting protein mask for NCS averaging was created with the program MAMA from the Uppsala Software Factory (USF; Kleywegt et al. 2003) by uniting large spherical masks around the six iodide positions to occupy the expected protein volume. Using this mask and the centroid electron density map from SHARP, the six starting NCS operators derived from the iodide positions were optimized against the electron density correlation coefficient with the USF program IMP. The optimized operators together with the starting mask were used with the CCP4 program DM (Cowtan and Main 1998) for phase improvement by a combination of solvent flattening, histogram matching, and six-fold NCS averaging with refinement of the NCS mask and operators. The resulting electron density map was already partially interpretable, showing some features of β-sheets and right-handed α-helices. To further improve the phases, a skeletonized representation (Greer 1985) of this map was used to visually cut out a region encompassing the expected protein hexamer with the computer graphics and modeling program MOLOC. The program MAMA was used to create a protein mask from this cut-out skeleton, and the mask volume was optimized by maximizing the electron density correlation coefficient of the SHARP centroid electron density map for all six NCS operators within the mask with the program IMP. The so optimized mask had a diameter of ~70% of a full mask covering the expected volume of a hexamer, and thus will be called “core mask,” here. Despite the nonisomorphism, the SHARP phases and probability distributions at 3 Å resolution from the KI derivative data set were combined with the observed structure factor amplitudes at 2.3 Å resolution from the in-house native data set. This combined data set was used together with the core mask and the optimized NCS operators in a final DM run using solvent flattening, histogram matching, sixfold NCS averaging with mask and operator refinement, and phase extension from 3 Å to 2.3 Å in 200 resolution steps. The resulting electron density map was fully interpretable. The refined mask from DM covered the expected volume of the hexamer. Apparently, the combination of an optimized core mask with sixfold NCS averaging, including refinement of the mask and the NCS operators, together with phase extension was sufficient to overcome the initial nonisomorphism between the two data sets.
Model building and refinement
An almost complete model of the PfFabZ hexamer could be built into the starting electron density map with the computer graphics program MOLOC. Subsequent model building and refinement were done against the SLS data set at 2.1 Å resolution (Table 1). The initial model was improved by iterative rounds of model building with MOLOC and reciprocal space refinement with REFMAC 5.2, using TLS domains (Schomaker and Trueblood 1968) for the three dimers of the hexamer, a maximum-likelihood target, Engh and Huber ideal geometry restraints (Engh and Huber 1991), individual isotropic atomic B-factors, and NCS restraints between the six subunits, excluding the chain termini and flexible loop regions. The model was completed by adding 256 water molecules, seven sulfates, six chlorides, and six cacodylates. In the final round of refinement, the NCS restraints were removed, resulting in marginal parameter shifts only. Three larger electron density features on the local twofold axes close to the local threefold axis could not be interpreted. Interestingly, the six chlorides are in the same positions as the six iodides of the KI derivative, far from any crystal contact. Thus, the anisomorphism between the KI derivative crystal and the native crystal was either present before soaking with KI or was induced by the high concentration of 0.5 M KI (either by osmotic shock or by unspecific binding of iodides close to crystal contacts). The stereochemistry of the refined model was checked with the CCP4 programs PROCHECK (Laskowski et al. 1993) and ROTAMER (using the penultimate rotamer library; Lovell et al. 2000), and with the program WHAT_CHECK (Hooft et al. 1996). The final model has an R-factor of 0.17 and a Free-R-factor of 0.22 with very good stereochemistry (Table 1).
Inhibition assay
PfFabZ inhibition was determined using the enzyme-coupled spectrophotometric assay described below. The forward reaction, i.e., the dehydration of β-hydroxybutyryl- CoA to form crotonoyl-CoA, was monitored by following the subsequent transformation of crotonoyl-CoA into butanoyl-CoA by means of P. falciparum enoyl-ACP reductase (PfFabI) provided in the assay mixture and using NADH as cofactor. The standard reaction mixture volume of 1 mL contained 10 mM HEPES buffer (pH 7.4), 100 mM NaCl, 4 μg PfFabI, 200 μM NADH, and 200 μM β-hydroxybutyryl- CoA. For inhibition studies, a published protocol (Leesong et al. 1996) was modified. Briefly, 0.3 μL of a 3-decynoyl-NAC solution (100 mM) in acetone was added to 136 μL of an enzyme solution at 2.5 mg/mL (0.15 mM) to give a 1.5:1 molar ratio of inhibitor to protein subunit. The acetone concentration used in this assay (0.22%) did not affect enzyme activity. At several time intervals in the range of 0 to 90 min, 10 μL of the protein solution were removed and added to 990 μL of the reaction mixture to give a final amount of 25 μg PfFabZ in the assay. The reaction was monitored over 10 min by measuring the change in absorption at 340 nm due to oxidation of NADH.
Acknowledgments
We thank Clemens Schulze-Briese, Claude Pradervand, Takashi Tomizaki, and Armin Wagner from the protein beamline X06SA at the SLS, and Drs. P. Valenti and A. Bisi (University of Bologna) for the synthesis of 3-decynoyl-NAC. This work was supported by the Swiss Federal Institute of Technology, Zurich (TH-27/02-2) and the Novartis Foundation, formerly the Ciba-Geigy Jubilee Foundation. The coordinates of this crystal structure have been deposited with the Protein Data Bank with the entry code 1Z6B.pdb along with the observed structure factors.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051373005.
References
- Abrahams, J.P. 1996. Methods used in the structure determination of bovine mitochondrial F(1) ATPase. Acta Crystallogr. D 52 30–42. [DOI] [PubMed] [Google Scholar]
- Benning, M.M., Wesenberg, G., Liu, R., Taylor, K.L., Dunaway-Mariano, D., and Holden, H.M. 1998. The three-dimensional structure of 4-hydroxybenzoyl-CoA thioesterase from Pseudomonas sp. Strain CBS-3. J. Biol. Chem. 273 33572–33579. [DOI] [PubMed] [Google Scholar]
- Brady Jr., G.P. and Stouten, P.F. 2000. Fast prediction and visualization of protein binding pockets with PASS. J. Comput. Aid. Mol. Des. 14 383–401. [DOI] [PubMed] [Google Scholar]
- Brock, D.J., Kass, L.R., and Bloch, K. 1967. Beta-hydroxydecanoyl thioester dehydrase. II. Mode of action. J. Biol. Chem. 242 4432–4440. [PubMed] [Google Scholar]
- Campbell, J.W. and Cronan Jr., J.E. 2001. Bacterial fatty acid biosynthesis: Targets for antibacterial drug discovery. Annu. Rev. Microbiol. 55 305–332. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, N. 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D 50 760–763. [DOI] [PubMed] [Google Scholar]
- Cowtan, K. and Main, P. 1998. Miscellaneous algorithms for density modification. Acta Crystallogr. D 54 (Pt 4) 487–493. [DOI] [PubMed] [Google Scholar]
- Dauter, Z., Dauter, M., and Dodson, E. 2002. Jolly SAD. Acta Crystallogr. D 58 494–506. [DOI] [PubMed] [Google Scholar]
- De la Fortelle, E. and Bricogne, G. 1997. Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multi-wavelength anomalous diffraction methods. Methods Enzymol. 276 472–494. [DOI] [PubMed] [Google Scholar]
- Diederichs, K. 1995. Structural superposition of proteins with unknown alignment and detection of topological similarity using a six-dimensional search algorithm. Proteins 23 187–195. [DOI] [PubMed] [Google Scholar]
- Endo, K., Helmkamp Jr., G.M. and Bloch, K. 1970. Mode of inhibition of β-hydroxydecanoyl thioester dehydrase by 3-decynoyl-N-acetylcysteamine. J. Biol. Chem. 245 4293–4296. [PubMed] [Google Scholar]
- Engh, R.A. and Huber, R. 1991. Accurate bond and angle parameters for x-ray protein structure refinement. Acta Crystallogr. A 47 392–400. [Google Scholar]
- Gardner, M.J., Hall, N., Fung, E., White, O., Berriman, M., Hyman, R.W., Carlton, J.M., Pain, A., Nelson, K.E., Bowman, S., et al. 2002. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419 498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber, P.R. and Muller, K. 1995. MAB, a generally applicable molecular force field for structure modelling in medicinal chemistry. J. Comput. Aid. Mol. Des. 9 251–268. [DOI] [PubMed] [Google Scholar]
- Greer, J. 1985. Computer skeletonization and automatic electron density map analysis. Methods Enzymol. 115 206–224. [DOI] [PubMed] [Google Scholar]
- Hames, E.D. 1981. An introduction to polyacrylamide gel electrophoresis. In Gel electrophoresis of proteins: A practical approach (eds. E.D. Hames and D. Rickwood), pp. 4–92. IRL Press, Oxford.
- Heath, R.J. and Rock, C.O. 1996. Roles of the FabA and FabZ β-hydroxyacylacyl carrier protein dehydratases in Escherichia coli fatty acid biosynthesis. J. Biol. Chem. 271 27795–27801. [DOI] [PubMed] [Google Scholar]
- Heath, R.J., White, S.W., and Rock, C.O. 2001. Lipid biosynthesis as a target for antibacterial agents. Prog. Lipid Res. 40 467–497. [DOI] [PubMed] [Google Scholar]
- Heath, R.J., White, S.W., and Rock, C.O. 2002. Inhibitors of fatty acid synthesis as antimicrobial chemotherapeutics. Appl. Microbiol. Biotechnol. 58 695–703. [DOI] [PubMed] [Google Scholar]
- Hisano, T., Tsuge, T., Fukui, T., Iwata, T., Miki, K., and Doi, Y. 2003. Crystal structure of the (R)-specific enoyl-CoA hydratase from Aeromonas caviae involved in polyhydroxyalkanoate biosynthesis. J. Biol. Chem. 278 617–624. [DOI] [PubMed] [Google Scholar]
- Hooft, R.W., Vriend, G., Sander, C., and Abola, E.E. 1996. Errors in protein structures. Nature 381 272. [DOI] [PubMed] [Google Scholar]
- Kabsch, W. 2001. XDS. In Crystallography of biological macromolecules, 1st ed. (eds. M.G. Rossmann and E. Arnold), pp. 730–734. Kluwer Academic Publishers, Dordrecht, The Netherlands.
- Kabsch, W. and Sander, C. 1983. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22 2577–2637. [DOI] [PubMed] [Google Scholar]
- Kass, L.R. 1968. The antibacterial activity of 3-decynoyl-N-acetylcysteamine. Inhibition in vivo of β-hydroxydecanoyl thioester dehydrase. J. Biol. Chem. 243 3223–3228. [PubMed] [Google Scholar]
- Kimber, M.S., Martin, F., Lu, Y., Houston, S., Vedadi, M., Dharamsi, A., Fiebig, K.M., Schmid, M., and Rock, C.O. 2004. The structure of (3R)-hydroxyacyl-acylcarrier protein dehydratase (FabZ) from Pseudomonas aeruginosa. J. Biol. Chem. 2793 52593–52602. [DOI] [PubMed] [Google Scholar]
- Kleywegt, G.J., Henrick, K., Dodson, E.J., and van Aalten, D.M. 2003. Pound-wise but penny-foolish: How well do micromolecules fare in macromolecular refinement? Structure 11 1051–1059. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- Leesong, M., Henderson, B.S., Gillig, J.R., Schwab, J.M., and Smith, J.L. 1996. Structure of a dehydratase-isomerase from the bacterial pathway for biosynthesis of unsaturated fatty acids: Two catalytic activities in one active site. Structure 4 253–264. [DOI] [PubMed] [Google Scholar]
- Lovell, S.C., Word, J.M., Richardson, J.S., and Richardson, D.C. 2000. The penultimate rotamer library. Proteins 40 389–408. [PubMed] [Google Scholar]
- Matthews, B.W. 1968. Solvent content of protein crystals. J. Mol. Biol. 33 491–497. [DOI] [PubMed] [Google Scholar]
- McLeod, R., Muench, S.P., Rafferty, J.B., Kyle, D.E., Mui, E.J., Kirisits, M.J., Mack, D.G., Roberts, C.W., Samuel, B.U., Lyons, R.E., et al. 2001. Triclosan inhibits the growth of Plasmodium falciparum and Toxoplasma gondii by inhibition of the Apicomplexan FabI. Int. J. Parasitol. 31 109–113. [DOI] [PubMed] [Google Scholar]
- Morisaki, M. and Bloch, K. 1972. On the mode of interaction of β-hydroxydecanoyl thioester dehydrase with allenic acid derivatives. Biochemistry 11 309–314. [DOI] [PubMed] [Google Scholar]
- Murshudov, G.N. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 53 240–255. [DOI] [PubMed] [Google Scholar]
- Perozzo, R., Kuo, M., Sidhu, A.S., Valiyaveettil, J.T., Bittman, R., Jacobs Jr., W.R., Fidock, D.A., and Sacchettini, J.C. 2002. Structural elucidation of the specificity of the antibacterial agent triclosan for malarial enoyl acyl carrier protein reductase. J. Biol. Chem. 277 13106–13114. [DOI] [PubMed] [Google Scholar]
- Rando, R.R. and Bloch, K. 1968. Mechanism of action of β-hydroxydecanoyl thioester dehydrase. J. Biol. Chem. 243 5627–5634. [PubMed] [Google Scholar]
- Rock, C.O. and Cronan, J.E. 1996. Escherichia coli as a model for the regulation of dissociable (type II) fatty acid biosynthesis. Biochim. Biophys. Acta 1302 1–16. [DOI] [PubMed] [Google Scholar]
- Schomaker, V. and Trueblood, K.N. 1968. On the rigid-body motion of molecules in crystals. Acta Crystallogr. B 24 63–76. [Google Scholar]
- Silverman, R. 1988. Mechanism-based enzyme inactivation: Chemistry and enzymology. CRC Press, Boca Raton, FL.
- Surolia, N. and Surolia, A. 2001. Triclosan offers protection against blood stages of malaria by inhibiting enoyl-ACP reductase of Plasmodium falciparum. Nat. Med. 7 167–173. [DOI] [PubMed] [Google Scholar]
- Uson, I., Schmidt, B., von Bulow, R., Grimme, S., von Figura, K., Dauter, M., Rajashankar, K.R., Dauter, Z., and Sheldrick, G.M. 2003. Locating the anomalous scatterer substructures in halide and sulfur phasing. Acta Crystallogr. D 59 57–66. [DOI] [PubMed] [Google Scholar]
- Waller, R.F., Keeling, P.J., Donald, R.G., Striepen, B., Handman, E., Lang-Unnasch, N., Cowman, A.F., Besra, G.S., Roos, D.S., and McFadden, G.I. 1998. Nuclear-encoded proteins target to the plastid in Toxoplasma gondii and Plasmodium falciparum. Proc. Natl. Acad. Sci. 95 12352–12357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller, R.F., Ralph, S.A., Reed, M.B., Su, V., Douglas, J.D., Minnikin, D.E., Cowman, A.F., Besra, G.S., and McFadden, G.I. 2003. A type II pathway for fatty acid biosynthesis presents drug targets in Plasmodium falciparum. Antimicrob. Agents Chemother. 47 297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. 1999. Rolling back malaria. In The world health report 1999: Making a difference. (ed. WHO), pp. 49–63. World Health Organization, Geneva, Switzerland.