

Abstract
Here, we report a novel protein sequence descriptor-based remote homology identification method, able to infer fold relationships without the explicit knowledge of structure. In a first phase, we have individually benchmarked 13 different descriptor types in fold identification experiments in a highly diverse set of protein sequences. The relevant descriptors were related to the fold class membership by using simple similarity measures in the descriptor spaces, such as the cosine angle. Our results revealed that the three best-performing sets of descriptors were the sequence-alignment-based descriptor using PSI-BLAST e-values, the descriptors based on the alignment of secondary structural elements (SSEA), and the descriptors based on the occurrence of PROSITE functional motifs. In a second phase, the three top-performing descriptors were combined to obtain a final method with improved performance, which we named DescFold. Class membership was predicted by Support Vector Machine (SVM) learning. In comparison with the individual PSI-BLAST-based descriptor, the rate of remote homology identification increased from 33.7% to 46.3%. We found out that the composite set of descriptors was able to identify the true remote homolog for nearly every sixth sequence at the 95% confidence level, or some 10% more than a single PSI-BLAST search. We have benchmarked the DescFold method against several other state-of-the-art fold recognition algorithms for the 172 LiveBench-8 targets, and we concluded that it was able to add value to the existing techniques by providing a confident hit for at least 10% of the sequences not identifiable by the previously known methods.
Keywords: remote homology, sequence descriptor, secondary structure, sequence alignment, sequence motif, support vector machine
In the post-genomic era, the exponential increase in the number of genome sequences came as a result of several hundreds of genome projects and made the functional annotation of gene translation products an overwhelming task. The most common way of inferring the biological function of a new gene is based on evaluating its sequence similarity with proteins of known function. Classical sequence comparison algorithms such as FASTA (Pearson and Lipman 1988), BLAST (Altschul et al. 1990), or Smith-Waterman dynamic programming (Smith and Waterman 1981) were developed to compute these similarities. However, an increasing number of proteins with weak sequence similarity were found to share similar or related biological functions and to adopt similar three-dimensional (3D) folds, referred to as remote homologs (Koppensteiner et al. 2000). To deal with such proteins, some profile-based sequence similarity searching methods like PSI-BLAST (Altschul et al. 1997) and Hidden Markov Models (HMM) (Sonnhammer et al. 1997) have been used, and they resulted in a marked improvement to put in evidence remote homology. Nevertheless, in the case of related sequences situated within the twilight zone (i.e., sequence identity ≤20%) (Rost 1999), the similarity-based methods were performing poorly. Significant efforts have been therefore deployed to develop more sensitive and powerful remote homology detection techniques. These efforts were based on the argument that protein structural diversity is much lower than the diversity of protein sequences (Chothia 1992; Alexandrov and Go 1994). Indeed, the protein structural manifold is highly degenerate because protein folds are objects embedded in the real, physical three-dimensional space. Using 3D-lattice simulations, Lindgard and Bohr were able to reproduce a very limited number of possible packings for protein structures, characterized by distinct magic numbers of secondary structural elements, also observed in real protein structures solved by X-ray crystallography (Lindgard and Bohr 1996).
During the last decade, a variety of fold recognition methods have been developed to push remote homology identification beyond the level of sequence-based similarity searches. The overall good performances of these techniques have been widely addressed in a series of Critical Assessment of Techniques for Protein Structure Prediction (CASP) experiments (Levitt 1997; Murzin 1999; Sippl et al. 2001) as well as in real-time comparison of structure prediction servers (e.g., LiveBench) (Bujnicki et al. 2001). By combining different types of structural and sequence information, a number of automatic methods have been developed and used to enhance automatic structure-based functional annotation of whole genomes (e.g., GenTHREADER [McGuffin and Jones 2003], INBGU [Fischer 2000], FFAS03 [Rychlewski et al. 2000], ORFeus [Ginalski et al. 2003], 3D-PSSM [Kelley et al. 2000], and FUGUE [Shi et al. 2001]). The basic operation in the underlying algorithms consists in comparing the amino acid sequence of a new protein with the 3D amino acid profiles of proteins with solved structure to measure the compatibility between sequence and structure (Bowie et al. 1991; Godzik et al. 1992; Jones et al. 1992; Bryant 1996; Panchenko et al. 2000). Some frequently used structural characteristics are mean force field (Sippl 1995), structure-seeded profile obtained by structural alignment (Kelley et al. 2000), observed secondary structure (Fischer 2000), and solvation energy (McGuffin and Jones 2003), to cite a few. However, the variety of methods can be grouped in two main classes: (1) structure-seeded profile-based and (2) profile–profile alignment-based. For instance, the 3D-PSSM and FUGUE methods are probably the two best-known structure-seeded profile-based fold recognition algorithms. Both methods rely on the PSI-BLAST search algorithm, but available structural information is extensively used to generate the structure-seeded profiles as well as to obtain the solvation potential and the structure-environment-related amino acid substitution matrix. In contrast, the ORFeus and FFAS algorithms belong to the class of profile–profile alignment-based methods. These methods have recently shown to be quite powerful in remote homology identification as well as in creating accurate sequence alignments. ORFeus uses the alignment of two profiles based on the PSI-BLAST-derived sequence information from the family of homologous proteins, complemented by the predicted secondary structure. Without using the predicted secondary structure information, the good performance of FFAS03 has been also demonstrated in the LiveBench series of experiments.
Because some fold-recognition methods often require the knowledge of the 3D structure of one of the two compared proteins, they could be effectively applied only for finding remote homologs of proteins with already solved 3D structure. Nevertheless, in many cases it is highly desirable to address the structural or functional relationship of two sequences in the absence of any explicit structural information. This problem attracted our attention, and we present here our efforts to develop a novel sequence descriptor-based remote homology identification technique. Since the landmark work of Anfinsen (1973), it was generally recognized that the native structure of a given protein is uniquely determined by that protein’s amino acid sequence. Therefore we expected that a relevant description of the primary structure would correlate with the structural/fold class of a given sequence.
The study we present in this paper consists of two main parts: First, the performance of 13 different types of sequence descriptors was assessed for structural/fold-type classification. Next, the top three best-performing descriptors were combined to obtain a novel remote homology identification method, which we called DescFold. A structurally diverse data set was compiled, and the SVM algorithm was applied to relate the sequence representations with the corresponding structural/fold type.
Results and Discussion
Remote homology identification rates using individual descriptors
In the present study, different protein sequence descriptors were assessed in their ability to retrieve remote homology relationships among dissimilar but structurally related protein sequences. During the first round of the evaluation, we benchmarked the performance in remote homology identification of 13 descriptors. The descriptors were classified in three general classes—global, nonlocal, and local—depending on the type of information they are capturing in a given protein sequence. Initially, the performance was quantified only in terms of the sensitivity by recording the number of correctly assigned remote homology relationships within 445 test proteins taken from a reference data set. The details about the exact nature of the different types of sequence descriptors and how the reference data set was compiled are outlined in Materials and Methods. The rates of remote homology identification based on these descriptors are listed in Table 1.
Table 1.
Sensitivity of remote homology identification based on different sequence descriptors
| Descriptor class | Sequence descriptor | Sensitivitya |
| Global | PSI-BLASTb | 123/445 = 27.6% |
| FASTAc | 30/445 = 6.7% | |
| SSEA | 95/445 = 21.3% | |
| AAC | 32/445 = 7.1% | |
| DPC | 17/445 = 3.8% | |
| Nonlocal | ACCT_AA | 20/445 = 4.5% |
| ACCT_SS | 55/445 = 12.4% | |
| CTD_AA | 14/445 = 3.1% | |
| CTD_SS | 33/445 = 7.4% | |
| Triplet | 12/445 = 2.7% | |
| Local | Motif_SCOPd | 94/445 = 21.1% |
| Motif_CATHe | 69/445 = 15.5% | |
| ISITES_CATHf | 17/445 = 3.8% |
a The sensitivity was defined as the percentage of correctly assigned remote homologous protein pairs.
b The modified e-value from PSI-BLAST searching was taken as the descriptor for the similarity between two evaluated sequences.
c The opt alignment score from the FASTA alignment was used as the descriptor.
d The term MOTIF_SCOP_SIM, described in equation 9, was used to measure the similarity.
e The descriptor was based on MOTIF_CATH_SIM.
f The descriptor was based on ISITES_CATH_SIM.
Of the 13 descriptors, five were global sequence-based descriptors with a sensitivity ranging from 3.8% to 27.6%. The top performing one was the PSI-BLAST-based descriptor type, allowing successful remote homology identification for every third protein sequence. We adopted this descriptor as a reference because the algorithm is an integral part of several state-of-the-art fold-recognition methods (Kelley et al. 2000; Shi et al. 2001; McGuffin and Jones 2003). Throughout our work we tried to investigate how much we would be able to improve on it. A first conclusion was that with an identification rate of 20% higher the profile-based alignment descriptor (PSI-BLAST) performed better than the pairwise alignment descriptor (FASTA) (cf. Table 1). Furthermore, even in the case in which the cutoff for e-values was set to 0.01 to filter out the easily identifiable homology pairs, the PSI-BLAST-based descriptor retained its good performance. However, in this case the obtained confidence levels tended to decrease.
Coding based on the predicted secondary structure with an identification rate of nearly every fourth sequence was a second top-performing descriptor. In the present study, three descriptors based on the predicted secondary structure from PSIPRED were assessed. In our work, the SSEA descriptor performed better than the two other nonlocal descriptors CTD_SS and ACCT_SS relying on the predicted secondary structure, which is in line with the results of McGuffin and Jones (2002). As a successful example of application of the SSEA descriptor, the remote homology between 1cem and 1qazA was confidently identified, although the sequence identity between them is only 11%. The secondary structure prediction accuracy from PSIPRED was 83% and 81% for the two proteins, respectively (Fig. 1 ▶). However, we would like to emphasize that the secondary structure on its own is not sufficient to address reliably the problem of structural relationship (McGuffin and Jones 2002), as the same secondary structure topology may correspond to different folds.
Figure 1.

Graph illustrating two remote homologs (1cem and 1qazA) successfully detected by the SSEA descriptor. With the predicted secondary structure from PSI-PRED, all the FSSP607 sequences were converted into the format of predicted secondary structure elements as described in the dedicated paragraph of Materials and Methods. Taking 1cem (1,4-β-D-glucan-glucanohydrolase catalytic domain; FSSP family: 126.4.1.1.2.1) (Alzari et al. 1996) as the query sequence against all other proteins in FSSP607, the remote homolog 1qazA (Alginate lyase A1-III from Sphin-gomonas Species; Chain: A; FSSP family index: 126.4.3.2.1.1) (Yoon et al. 1999) was selected as the top hit with an SSEA score of 0.7041. The identity between the two sequences was only 11%. (A) The SSEA alignment between 1cem and 1qazA. The identical secondary structure elements in the same alignment position were displayed in bold type. (B) The Cα superimposed models of 1cem (cyan) and 1qazA (purple). The superimposition was carried out by using CE algorithm (Shindyalov and Bourne 1998). With the 233 aligned residues, the root mean square distance (RMSD) for the superimposed structures is 4.3 Å, and the Z-score is 5.6, implying that they should share higher than just fold-level remote homology.
The poor performances of the class of nonlocal descriptors were somewhat unexpected in our work, as we speculated that this type of coding should be able to capture some information related to the nonlocal nature of the protein folding phenomenon. However, there appeared to be no convincing evidence in the literature that nonlocal descriptors can be useful in remote homology detection, either.
Farther on in our work we obtained indications that a protein fold seems to be essentially determined by the occurrence of particular clusters of amino acids in its sequence. Data published in the literature support this speculation (Mirny and Shakhnovich 2001). On one hand, in the current theory of protein folding, the existence and importance of folding nuclei are generally accepted. On the other hand, observations seem to indicate that precise functional sites are more often conserved than the rest of the sequence. In our work we used the PROSITE database, as it is one of the most widely used and comprehensive sequence motif databases. The PROSITE motifs are defined as regular expressions (“patterns”), derived from analyses of sequences of known function. To illustrate the traceable correlation between folds and sequences motifs, even for pairs of proteins sharing low sequence identity, we performed a preliminary statistical investigation. Taking the CATH fold hierarchy as an example, we recorded the frequencies of significant matches to PROSITE motifs. The results are presented in Figure 2 ▶. We found that the top 10 folds ranked by the number of significant matches to PROSITE motifs (i.e., SFM > 1.0) were the Rossman fold, the TIM barrel, the α–β plaits, the jelly-rolls-type fold, the immunoglobulin-like fold, the four-helix bundle, the arc repressor mutant fold type, the OBfold, the two-layer sandwich, and glycosyltransferase, respectively. Interestingly enough, seven out of these top 10 folds belong to the 10 superfolds defined by the authors of CATH (Orengo et al. 1999).
Figure 2.

Frequency of occurrence of PROSITE motifs within the folds in the CATH database. Each column represents the accumulation of the number of folds with an increase of the number of functionally important motifs (i.e., SFM ≥ 1.0). Out of the 813 folds included in the current CATH database, 550 (67.6%) folds were found to contain at least one important PROSITE motif. About 505 (61.8%) of the CATH folds have a number of important motifs in the range 1–4, while the maximum number of important motifs occurring within one CATH fold was found to be 105, occurring in the Rossman-type fold.
The importance of precise clusters of amino acids in determining the fold type was confirmed by our results obtained with the class of purely local descriptors. To determine to what extent these clusters could have a structural origin, we extended our analysis of functional motifs to include the I-sites database of folding initiation sites. Puzzlingly enough, the descriptors relying on the occurrence of predefined functional motifs performed much better than the ones using the I-sites library. The motif descriptor based on the SCOP (Murzin et al. 1995) protein structure classification allowed a successful remote homology identification for nearly every fifth protein sequence, with a rate higher than the one derived by using the CATH (Orengo et al. 1997) classification. As an illustrative example, in Figure 3 ▶ we have provided the 3D models for a pair of protein remote homologs (1dd8A and 1afwA), identified using motif-based descriptors.
Figure 3.
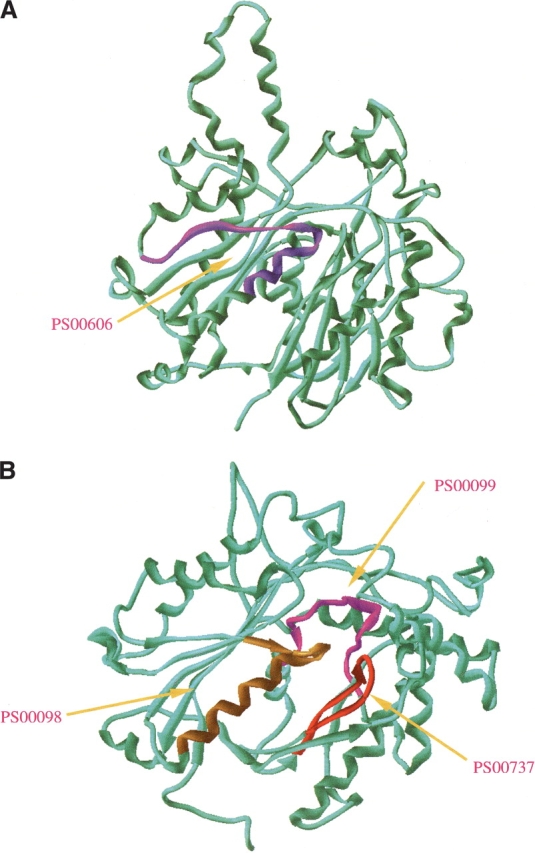
Ribbon representation of two remote homologs (1dd8A and 1afwA), confidently recognized by using the PROSITE motif-based descriptors. (A) 3D model of 1dd8A (β-ketoacyl [acyl carrier protein] synthase I from Escherichia coli; Chain: A; FSSP family index: 103.1.1.1.1.1) (Olsen et al. 1999); (B) 3D model of 1afwA (thiolase from Saccharomyces cerevisiae; Chain: A; FSSP family index: 103.1.1.1.2.1) (Mathieu et al. 1997). Although the sequence identity for the two proteins was only 12%, they were found to share high structural similarity by using CE (the RMSD for 229 structural aligned residues is 3.1 Å and the Z-score is 5.0). Taking the 1dd8A as the query sequence against the other proteins in FSSP607, the retrieved top hit was 1afwA with a score (i.e., MOTIF_SCOP_SIM) of 8.599. The PROSITE motifs contributing in this recognition were PS00606 (entry name: B_KETOACYL_SYNTHASE; pattern: G-x(4)-[LIVMFTAP]-x(2)-[AGC]-C-[STA](2)-[STAG]-x(3)-[LIVMF]), PS00098 (entry name: THIOLASE_1; pattern: [LIVM]-[NST]-x(2)-C-[SAGLI]-[ST]-[SAG]-[LIVMFYNS]-x-[STAG]-[LIVM]-x(6)-[LIVM]), PS00737 (entry name: THIOLASE_2; pattern: N-x(2)-G-G-x-[LIVM]-[SA]-x-G-H-P-x-[GA]-x-[ST]-G), and PS00099 (entry name: THIOLASE_3; pattern: [AG]-[LIVMA]-[STAGCLIVM]-[STAG]-[LIVMA]-C-x-[AG]-x-[AG]-x-[AG]-x-[SAG]). The localization of these motifs in the 3D models was marked with different colors in the two proteins. Interestingly enough, a remote homology was recognized by the MOTIF_SCOP_SIM similarity function due to the high SFM scores for these motifs within the SCOP tillage-like fold, although none of the motifs was found to co-occur in the two proteins.
These results emphasized the fact that the DescFold method depends strongly on the relationships between folds and functional motifs, and therefore it was not surprising to realize that its performance was influenced by classification schemes in the protein structural databases being used to generate the motif-based descriptors. For example, the SCOP-1.63.95 database used in the present work included 8720 sequences classified into 765 different folds, while CATH-v2.5.95 contained 7532 domains grouped into 813 folds. The better performance observed with the SCOP database may imply that the FSSP database is more similar to the SCOP than the CATH hierarchy. An illustration of this has been found in one remote homology pair that we have investigated. For instance, the remote homology relationship between 1uby (farnesyl pyrophosphate synthase, FSSP index: 113.13.1.2.1.1) and 1ezfA (squalene synthase, FSSP index: 113.13.1.3.1.1) was recognized by the motif-based descriptors generated from the SCOP classification, since both of them were assigned to adopt the SCOP fold of the terpenoid synthase (SCOP: a.128). In difference, these two proteins were classified into two different folds (farnesyl diphosphate synthase, CATH: 1.10.600; and 5-epi-aristolochene synthase, CATH: 1.10.615) when using the CATH database, and therefore, the CATH-based descriptors failed to identify their remote homology.
An advantage of using some descriptors based on functional motifs is the increase of the identification rate of true remote homologs by lowering the number of false-positive protein analogs, or proteins of different origin that evolved to adopt a similar fold. However, as also mentioned in the literature (Salwinski and Eisenberg 2001), this type of descriptor is certainly weakened by the fact that they rely on the a priori knowledge of a relatively small number of functional motifs, stored in the PROSITE database. This limitation might be overcome by using large, automatically generated motif libraries (e.g., eMOTIF; Huang and Brutlag 2001).
In summary, during the first evaluation round, three types of descriptors performed significantly better than all the 13 descriptors under scrutiny—the PSI-BLAST-based descriptor, the secondary structure element alignment (SSEA) descriptor, and the functional motif-based descriptor using the SCOP structural classification (MOTIF_SCOP). We further compared the frequency of consensual fold assignments by the pairwise examination of the top hits derived with these descriptors. The results of this survey are presented in Table 2. The number of consensual top hits identified by these three descriptors was generally low. For example, the PSI-BLAST- and the SSEA-based descriptors jointly identified 36 similar remote homologs out of the 445 test proteins, but among them only 10 were identical. This suggested that the top three descriptors were uncorrelated and captured different aspects of the information contained in a protein sequence. A combination of these three descriptors could be therefore envisaged with the expectation of an improved performance in remote homology identification experiments.
Table 2.
Comparison of the consensus among the top three best-performing descriptors
| PSI-BLASTa | SSEA | Motifb | |
| PSI-BLAST | — | 36 (10) | 36 (15) |
| SSEA | — | 30 (5) | |
| Motif | — |
The value outside the parentheses denotes the total number of proteins where the remote homologs could be correctly recognized by both methods, while the value inside the parentheses denotes the number of proteins where identical remote homologs could be retrieved by both methods.
a Based on the modified e-value from PSI-BLAST searching (i.e., PSIBLAST_SIM).
b Based on the PROSITE motif-fold correlation in the SCOP database (i.e., MOTIF_SCOP_SIM).
Performance of data mining
Prior to the second round of the evaluation, the original reference data set was partitioned in two unrelated training and test sets. The details about how these sets were built are discussed in Materials and Methods. The partitioning was motivated by statistical arguments and by some memory limitations inherent to the SVM implementation we have used in our work. We proceeded consequently with the evaluation of the fold-recognition sensitivity of the top three best-performing descriptor classes using SVM learning. In Table 3 we present the results obtained by using a radial basis kernel function, which were slightly better than the ones based on the linear and polynomial kernels (data not shown). The PSI-BLAST descriptors alone reached a 33.7% identification rate, which is nearly 6% higher compared to the case in which we used a simple assignment based on the PSIBLAST_SIM similarity measure (cf. Table 1). When the two other best descriptors were added, the identification rate rose to 46.3%, allowing a fold assignment for almost every second sequence in the test data set. The incorporation of the other 13 descriptors did not result in any significant improvement. Therefore, these three best descriptors were used as the input of the final version of our remote homology identification technique, termed as DescFold. Compared with the individual performance of the 13 different descriptors, the increased fold identification rate for DescFold was in the range 18% to 43%.
Table 3.
Sensitivity of remote homology identification using SVM learning
| Descriptors included | Sensitivity |
| PSI-BLASTa | 150/445 = 33.7% |
| PSI-BLAST + Motifb | 171/445 = 38.4% |
| PSI-BLAST + SSEA | 196/445 = 44.0% |
| PSI-BLAST + SSEA + Motif | 206/445 = 46.3% |
a The PSI-BLAST-searching-based descriptor, including four parameters in this SVM model.
b The PROSITE-motifs-based descriptors, containing MOTIF_SCOP_SIM and MOTIF_CATH_SIM.
The simple benchmarking presented above has not provided any information on the confidence level for an SVM-generated remote homology assignment score. However, in blind fold-identification experiments, an indication of the probability that a given fold assignment is correct is of central importance. Therefore, we tried to evaluate the reliability of our method by investigating how the error per query increased with the cumulated number of true-positive remote homologs identified. The results obtained are presented in Figure 4 ▶, in a way similar to the Receiver Operator Characteristic (ROC) analysis (Gribskov and Robinson 1996). The detailed way of carrying out the calculations can be found in Materials and Methods. Figure 4 ▶ illustrates the fact that the recovery of true positives was increasing with the incorporation of different types of descriptors in the SVM model. In the context of practical applications, such as blind genome annotation, the most important region of Figure 4 ▶ corresponds to the 95% confidence assignment (i.e., <5% error per query). When using the top three descriptors together (PSI-BLAST, SSEA, and PROSITE motifs) within the SVM learning procedure, we correctly identified 73 remote homologs out of a total of 445. This amounted to 16.4% coverage at a high-confidence level of 95%, which is nearly 10% higher than the coverage obtained by an SVM model based uniquely on the PSI-BLAST scores. The performance of our method should be evaluated in view of the success rates of well-known fold-recognition techniques such as 3D-PSSM, which achieved a similar 18% coverage at the 95% confidence level (Kelley et al. 2000).
Figure 4.

Graph illustrating the detection of remote homologies when applying SVM learning to the combination of top-three best-performing sequence descriptors including the PSI-BLAST-based descriptors (four parameters), the SSEA, and the PROSITE motif-based descriptor (two parameters).
Support vector machine learning has been already used for the purposes of remote homology identification. As reported in the literature (Liao and Noble 2002; Ben Hur and Brutlag 2003; Hou et al. 2004), the similarity of two sequences was evaluated by a kernel function, which can be described itself in terms of the feature vectors. Using such a similarity measure, the SVM learning algorithm was previously applied in one-against-all or winner-takes-all multiple-classes prediction experiments (Liao and Noble 2002; Hou et al. 2004). However, in order to keep enough representative sequences in each structural family, the approach required lower cutoff e-values (e.g., 1e-20) when preparing the training data set. Therefore, SVM models derived through such a scheme will probably experience loss of performance when applied to sets of diverse sequences with very low pairwise identities.
In our study, the SVM learning was carried out in a completely new way, due to the novel sequence similarity evaluation based on descriptors-encoded protein sequences. In our scheme of combining different descriptors, SVM was used to predict if a pair of proteins shared a similar structure or not, i.e., SVM was trained to distinguish between two classes—homology pair or nonhomology pair. Therefore, the number of required proteins per family was not limited and our method could be benchmarked with sets of highly dissimilar remotely homologous protein sequences. To our knowledge, this is the first report of using SVM in such a way.
An essential drawback of our method is the use of different structural, sequence, or motif databases, such as SCOP, CATH, PROSITE, and I-sites. Therefore, by its very nature it is knowledge-based with the disadvantage that it should not be able to extrapolate far away from the currently available protein structural data upon which it is built. A second drawback of the technique is that in its current version it cannot provide an alignment between the query and the best matching sequence, due to the sequence descriptor-based strategy being applied. Although sequence alignments could be derived with several algorithms (Smith and Waterman 1981; Pearson and Lipman 1988), we found these methods not to be very reliable in the context of our work. The sequence alignment of distant remote homologs is currently an open field of research (Contreras-Moreira et al. 2003; Marti-Renom et al. 2004). Paradoxically, the descriptor-based strategy used to encode protein sequences, which is at the origin of the main disadvantage of our method, turns out to be its principal strength. Indeed, in many situations it is desirable to infer structural or functional relationships for two sequences in the absence of any explicit structural information.
Finally, our work provided a rule-of-thumb technique to carry out structure-based functional annotation. Indeed, it indicates that PSI-BLAST searching, followed by analysis of the predicted secondary structure, and functional motif matching should be performed as the necessary and sufficient steps to obtain preliminary indications about a protein’s structural and functional class membership.
Comparison with state-of-the-art fold recognition methods
It was certainly interesting to benchmark our remote homology identification technique DescFold with other state-of-the-art fold-recognition methods, especially on a data set of truly novel structures. As a continuous benchmarking program, LiveBench experiment has been setting up a good testing environment. Every week new PDB proteins are submitted to the participating fold-recognition servers. The corresponding results are collected and evaluated. So far, nine rounds of LiveBench experiments have been carried out, and the detailed results are publicly available (Bujnicki et al. 2001). Here, we have selected the targets (172 proteins) that entered the eighth round of LiveBench (Live-Bench-8) as the reference test set used to compare the performance of DescFold and its peer fold-recognition methods.
We compiled a reference fold library by using the sequences included in the SCOP database in its version 1.63, sharing <40% identity, and containing 5226 entries. We labeled it as SCOP-1.63.40. The secondary structures for these sequences were predicted with PSIPRED, rather than assigning them based on the known 3D structures. The DescFold algorithm was executed on an SGI Octane2 workstation, with a typical processing time of 15 min per query sequence. Our method suggested a list of 20 top hits per query, ranked according to the corresponding confidence levels. The LiveBench-8 targets were represented by proteins deposited in the PDB database in the period starting on August 1, 2003, and ending on December 31 of the same year. We verified that these targets were not included in our fold library SCOP-1.63.40. In the analysis of the results we considered two hits as similar, provided that the Z-score obtained by applying the CE structural alignment algorithm, was >4.2. Our new method DescFold was therefore able to identify the folds for 86 out of the 172 LiveBench-8 targets. Regarding the receiver operator characteristics (ROC), our method was able to correctly identify the folds for 37, 70, and 75 targets with less than 1, 5, and 10 false positives, respectively (cf. Table 4).
Table 4.
Comparison of receiver operator characteristics (≤10 false positives) for different fold-recognition methods based on all LiveBench8 targets
| Correct predictionsa | |||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | Totalb | |
| ORFeusc | 83 | 89 | 93 | 93 | 93 | 93 | 93 | 93 | 93 | 93 | 100 |
| FFAS03c | 74 | 81 | 83 | 83 | 83 | 83 | 85 | 87 | 88 | 88 | 98 |
| FUGUE2.0c | 79 | 79 | 81 | 81 | 82 | 83 | 83 | 83 | 83 | 85 | 90 |
| 3D-PSSMc | 43 | 52 | 62 | 62 | 62 | 63 | 66 | 66 | 72 | 78 | 91 |
| DescFoldd | 37 | 56 | 58 | 65 | 70 | 70 | 72 | 73 | 73 | 75 | 86 |
| DescFolde | 37 | 56 | 58 | 62 | 66 | 66 | 67 | 69 | 69 | 71 | 75 |
a 1–10: number of correct predictions with higher reliability than the 1–10 false prediction.
b Total number of correct predictions.
c The results for ORFeus, FFAS03, FUGUE2.0, and 3D-PSSM are cited from http://bioinfo.pl/Meta/results.pl?B=LiveBench&V=8.
d The performance was evaluated based on the number of correctly assigned folds.
e The performance was assessed by the quality of predicted 3D models based on the PSI-BLAST alignment.
Although more than 20 fold-recognition severs participated in the LiveBench-8 experiment, we compared our DescFold algorithm with four other popular fold-recognition methods, that is, 3D-PSSM (Kelley et al. 2000), FUGUE2.0 (Shi et al. 2001), ORFeus (Ginalski et al. 2003), and FFAS03 (Rychlewski et al. 2000). As revealed in Table 4, the performance of DescFold against the LiveBench-8 targets was reasonable and comparable to the performances of the four other fold-recognition methods. Considering the performance within <10 false positives, the DescFold was able to confidently identify 44% targets, which is 1%, 5%, 7%, and 10% less than the identification rates of 3D-PSSM, FUGUE2.0, FFAS03, and ORFeus, respectively. It is interesting to notice that this performance was somewhat closer to that of a typical structure-seeded profile-based fold-recognition method (3D-PSSM), and was weaker compared to the state-of-the-art profile–profile alignment-based fold-recognition methods such as ORFeus and FFAS03. This is probably due to the fact that profile–profile alignment is able to catch some sequence evolutionary relationships that DescFold is currently missing. Therefore, we expect that the incorporation of descriptors accounting for evolutionary relationship will lead to an improvement in the performance of our method.
In quantifying the prediction accuracy of our method, we have only considered the number of the correctly assigned folds, whereas the LiveBench assessment was mainly based on the quality of the predicted 3D models. It was our goal to compare our method with some state-of-the-art fold-recognition methods on an equal basis. For this we have evaluated the quality of the 3D model generated from an alignment of the query and target remote homologous sequences obtained with PSI-BLAST with the default settings. More precisely, the profile for the target sequence was saved at the fourth iteration of the PSI-BLAST searching against the nonredundant (NR) protein database and then used as a query against the SCOP-1.63.40 sequences for another iteration to obtain the alignment between the target and the chosen template. Furthermore, the corresponding 3D model for the target sequence was built up by using the AL2TS Web server (http://predictioncenter.llnl.gov/local/al2ts/al2ts.html). Finally, the quality of the predicted 3D model was evaluated by using MaxSub (Siew et al. 2000), an official evaluation method in the LiveBench experiments. MaxSub returns values between 0.0 and 1.0, where 1.0 indicates the identity of two structures. A value above 0.0 indicates usually a nonrandom structural similarity, that is, an acceptable model. As expected, the general performance based on the quality of predicted model was slightly lower than that based on the correctly assigned fold. Regarding the prediction within three false positives, as shown in Table 4, the accuracy of our method based on the two different measures was the same. This implies that the PSI-BLAST alignment was reasonably good for these easy targets, having not too weak sequence similarity. At higher levels of false positives, the number of the targets with an acceptable model quality was less than the targets with correctly assigned folds. This indicated that the predicted 3D models for some of the targets were not valid due to the poor alignment, although their folds were correctly assigned. Considering the number of correct predictions with <10 false positives, our method was able to generate acceptable models for 71 targets, while the folds of 75 of them were confidently identified. These results showed that our method was able to build reasonably good 3D models by using some classical sequence alignment methods (e.g., PSI-BLAST). However, to take full advantage of our current method, some more powerful sequence alignment techniques would be required.
It was well established that the different fold-recognition algorithms could outperform each other depending on the particular case, probably because each method is exploiting a different aspect of protein similarity to identify distant homologs. Clear evidence for this was found in a previous work of ours where we investigated the possible remote homology relationships in the glycosyltransferase (GTF) family (Zhang et al. 2003). We observed a new example for this when we assessed the performance of DescFold and the four other fold-recognition methods against the set of the LiveBench-8 targets. In the benchmarking we considered only the correct predictions with reliability >10 false predictions. The values in Table 5 represent the percentage of consensual hits, defined as the total number of sequences where a pair of methods, appearing in a row and a column, were able to provide a correct prediction divided by the total number of correct predictions generated by the single method appearing in the row. Therefore, a high consensus in the generated hits is observed for those pairs of methods for which the respective values in the lower and upper diagonals in Table 5 are nearly equal. We considered percentages instead of absolute numbers of jointly identified hits because the different methods were able to identify confidently different numbers of hits. We found out that in the LiveBench-8 experiment ORFeus, FFAS03, FUGUE2.0, and 3D-PSSM provided largely consensual answers. For instance, >90% of targets identified by 3D-PSSM were also recognized by the other three methods, while the strongest discrepancy was observed for the ORFeus/3D-PSSM pair. A slightly higher consensus was observed between ORFeus and FFAS03, probably due to the similar profile–profile matching algorithms they use. Interestingly, DescFold showed a noticeably lower consensus with the other four methods, probably due to the novelty of the algorithm it is based on. Although DescFold showed a somewhat lower rate in remote homology identification, it was able to provide a confident hit for 15%, 15%, 10%, and 20% of the sequences not identifiable by ORFeus, FFAS03, FUGUE2.0, and 3D-PSSM, respectively. Therefore, we believe that our method could be a competitive part and add value to a jury-like consensus-based fold identification system, such as the structure prediction meta-server (Lundstrom et al. 2001).
Table 5.
Comparison of the consensus among different fold-recognition methods
| ORFeus | FFAS03 | FUGUE2.0 | 3D-PSSM | DescFold | |
| ORFeus | — | 91% | 86% | 76% | 70% |
| FFAS03 | 95% | — | 89% | 80% | 73% |
| FUGUE2.0 | 94% | 92% | — | 85% | 78% |
| 3D-PSSM | 91% | 90% | 92% | — | 73% |
| DescFold | 87% | 85% | 88% | 76% | — |
The comparison was carried out for the correct predictions with reliability higher than 10 false predictions. The values represent the percentage of consensual hits, defined as the total number of sequences where a pair of methods, appearing in a row and a column, was able to provide a correct prediction divided by the total number of correct predictions generated by the single method, appearing in the row. The detailed fold-recognition results for ORFeus, FFAS03, FUGUE2.0, and 3D-PSSM were downloaded from the Web site of LiveBench-8 for this comparison.
Conclusions
The present work was dedicated to the development of a sequence descriptor-based remote homology identification approach, because most of the known proteins lack an experimentally determined structure. The rates of remote homology identification for 13 different descriptors were benchmarked with 445 remote homologs from FSSP. Our results indicated that the PSI-BLAST-based descriptor, the predicted secondary structure descriptor, and the PROSITE motif-based descriptors are the top three most informative descriptors. These three top-performing descriptors have been incorporated into a novel method for remote homology identification, which we call DescFold, relying on a support vector machine-learning algorithm. Compared with the single PSI-BLAST-based descriptor, the rate of remote homology identification was increased from 33.7% to 46.3%. Similarly, on the 95% confidence level of detection, our new method assigned 16.7% remote homologs, or nearly 10% more than the single PSI-BLAST-based descriptor. The method was benchmarked against four other state-of-the-art fold-recognition techniques by using a test set of some truly novel protein structures. Our method demonstrated a reasonable accuracy and was found to be complementary to the existing techniques.
Materials and methods
Data sets
Databases
In the present study, we used the FSSP database (Holm and Sander 1996) to assess the performance of the different descriptors for remote homology identification. First, a representative set of 3548 proteins was downloaded from ftp://ftp.ebi.ac.uk/pub/databases/fssp/TABLE1.html, corresponding to the version of FSSP dated 30/10/2002. We named this data set FSSP3548 in which pairwise sequence identity was expressly chosen to be <25%. Among the 13 different types of descriptors investigated in the current study, a minimum sequence length for a protein sequence was required for the proper computation of three of the descriptors. To be able to derive the triplet descriptor, for example, the sequences should be longer than 36 residues. Therefore, we removed the sequences of <100 residues from the reference data set. On the other hand, sequences longer than 500 residues were found to often code for multidomain proteins. To avoid addressing the remote homology relationship for those multidomain proteins, those sequences with >500 residues were also removed. Thus, the data set size was reduced to 2290 (FSSP2290) protein structures. Furthermore, this condensed data set was filtered using PSI-BLAST with an e-value cutoff of 0.01, complemented by a 20% cutoff for the pairwise identity. More details about how to calculate a PSI-BLAST e-value between two sequences appear below in this section in a dedicated paragraph. Finally, we obtained a highly diverse sequence data set, containing some 607 proteins (FSSP607), and covering 175 different folds.
To perform the PSI-BLAST searching, the NCBI nonredundant (NR) sequence database, containing 1,329,756 sequences, was downloaded from ftp://ncbi.nlm.nih.gov/blast/ in its version dated 10/02/2003. We obtained the PROSITE release 18.5 from http:// www.expasy.org/prosite/ (Hofmann et al. 1999). It contained 1647 entries, 1327 of which were patterns. In our study, these patterns were used as a library of motifs. The I-sites library 13.6.1, containing 331 motifs, was downloaded from http://isites.bio.rpi.edu/bystrc/pub/Isites. The sequences from the SCOP database of domains (version 1.63) sharing <95% pairwise identity were downloaded from http://astral.standford.edu/ (Murzin et al. 1995; Brenner et al. 2000). A representative set of protein domains (CATH-v2.5.95) was obtained from CATH by clustering the sequences at a level of 95% sequence identity. These were downloaded from ftp://ftp.biochem.ucl.ac.uk/pub/cathdata/v2.5 (Orengo et al. 1997). The two representative sequence sets, SCOP-1.63.95 and CATH-v2.5.95, were used in the calculation of the compatibility (SFM) of functional and structural (I-sites) motifs with the different folds defined in CATH and SCOP.
Training and test data sets
An initial data set of 607 highly diverse, but structurally related protein sequences was compiled starting with the original FSSP database (Holm and Sander 1996). Within the FSSP607 data set, we postulated that remote homology exists for two protein sequences if they share the same first two indices in the FSSP family hierarchy. In this way, 445 proteins were selected as “test” proteins, each having at least one matching remote homolog in the FSSP607 data set. Overall, the number of unique remote homologs for the 607 proteins was 162.
During the first round of the evaluation, for each set of descriptors we compared each “test” protein against all other proteins in FSSP607. At this point a splitting in training and test data sets was not required, as no learning procedure was actually applied. Instead, tentative remote homology relationships were identified based on the pairwise sequence similarities between the “test” protein and all other members of the FSSP607 data set. The protein with the highest similarity score hit (i.e., the top hit) was assigned as being the closest remote homolog.
A typical partitioning of the original FSSP607 data set in training-set and test-set was carried out during the second round of the evaluation, when the top three descriptors were assessed in their success rate of remote homology identification by using support vector machine (SVM) learning. The SVM algorithm was trained to discriminate a given protein pair as a pair of remote homologs out of a total of 607 × (606/2) different protein pairs, split in five distinct sets of equal sizes. In order to predict if a given protein pair was a pair of remote homologs, we labeled as “test” the set to which this precise pair belongs. The four remaining sets were labeled as “training,” and SVM models were developed for each of them. Consequently, each of the four models was applied to the “test” data set, and an average value taken among the four models was finally provided as an outcome. This partitioning was motivated by statistical arguments and by the memory limitation inherent to the SVM implementation we have used in our work.
Descriptors
Global sequence descriptors
Our first choice of descriptors to be applied in remote homology identification was oriented toward the scores generated by string-matching algorithms, relying on facts from the literature indicating that the PSI-BLAST algorithm on its own was quite successful in identifying the fold type.
PSI-BLAST-based descriptor.
PSI-BLAST searching for two sequences A and B belonging to the FSSP3548 extended data set was executed as follows. First, a sequence A was compared against the NR sequence database by PSI-BLAST for three iterations in order to generate a PSSM profile. The e-value for including sequences in the score matrix model was set to 0.01. Based on the score matrix model built in this first search, we further used PSI-BLAST to compare the sequence A to the FSSP3548 data set for one round. The e-value E(A,B) corresponding to the match between A and B was adjusted to a fixed size of the data set (i.e., 10,000). A similarity score was finally evaluated according to the following equation:
 |
(1) |
In our work we adopted a maximum cutoff of 100 for the E(A,B) value. As the E(A,B) score for any pair of sequences within the nonredundant FSSP607 data set was set to be larger than 0.01 by construction, the respective PSIBLAST_SIM(A,B) similarity score within this same data set ranges between 0 and 1.
FASTA-based descriptor.
We used the pairwise alignment opt score derived by FASTA (Pearson and Lipman 1988) to evaluate the similarity between two sequences. We further designate it by FASTA_SIM(A,B).
Secondary structure element alignment-based descriptor.
String matching is largely applied to protein sequences represented by the 20-letter alphabet corresponding to natural amino acids. Reduced representations of protein sequences are possible, however, where amino acids are clustered in a smaller number of classes following some predefined chemical or structural equivalence relationships. It is also possible to represent sequences by using the most probable secondary structure to which every amino acid in a chain might belong. Secondary structure prediction has reached a level of maturity where the best performing methods could attain levels of 80% (Jones 1999) of correct secondary structure assignment. String matching algorithms could be applied therefore to the reduced representations of protein sequences.
To perform a secondary structure element alignment (SSEA), a secondary structure prediction for the two evaluated sequences A and B was carried out by PSIPRED (Jones 1999). Second, the predicted structural strings were shortened such that a single character “H” represented a helix element, the single character “E” represented a strand element and the single character “C” represented a coil element. The initial and final coil elements were ignored. A β-strand would equal three or more consecutive Es, and an α-helix would equal five or more consecutive Hs. All other secondary structure elements were taken as coils. For example, the secondary structure string CCCCCCHHHHHHHCCCCEEEEE EECCCCCCCHHHHHHCCCCC would have been shortened to HCECH, the length of each element being retained for the scoring of SSEA. The two shortened strings, corresponding to sequences A and B, were pairwisely aligned by using a modified dynamic programming algorithm (Needleman and Wunsch 1970) with a scoring scheme adapted from Przytycka et al. (1999). The alignment score SSEA_SIM(A,B), ranging from 0 to 1, was used as the descriptor of the similarity between every two evaluated sequences.
Amino acid composition-based descriptors.
Amino acid composition, denoted further by AAC, is the simplest descriptor of this type. It is computed as the frequency of occurrence of the natural amino acids in a given protein sequence, leading to the embedding of such sequences in a 20-dimensional feature space. In this high-dimensional space, the similarity between two vectors corresponding to two sequences A and B was assessed by using the cosine measure:
 |
(2) |
where XA and XB denote the two feature vectors (i.e., AACs) encoding the sequences A and B. We named the similarity score AAC_SIM(A,B) to denote the similarity for two protein sequences A and B coded by the AACs descriptors.
Dipeptide composition-based descriptors.
In a similar manner, a given protein sequence can be easily represented by the frequency of occurrence of the vicinal dipeptides or DPC descriptors. This leads to an embedding of protein sequences in a feature space of dimension N = 400. In such high-dimensional spaces the data were often found to be unevenly distributed, an event often referred to as “the curse of dimensionality.” When applying the Singular Value Decomposition (SVD) technique (Xie et al. 2000; Grigorov et al. 2003), the dimensionality of the original data set could be reduced by discarding the small singular values, mainly representing the noise. This allows for a more uniform distribution of the data set in the space of reduced dimensionality, thus avoiding some numerical problems in the computation of the neighborhoods and increasing the signal-to-noise ratio within the data set. In the current study, the protein similarity between two sequences was computed by simple statistical measures (e.g., cosine angle), which should not suffer seriously from the curse of dimensionality. For example, when encoding the FSSP607 data set by DPC descriptors, we were able to decrease space dimensionality to 30, without significant information loss. The similarity for two sequences encoded by the DPC descriptors was again evaluated by the cosine angle appearing in equation 2, but applied to the corresponding vectors in the low-dimensional space. For the other three descriptors (Autocross-Covariance Transform-based descriptor, Secondary structure ACCT-based descriptor, and Triplet descriptor), the dimensionality was also decreased to 30 by using SVD. The performances of the four types of descriptors spanning the high-dimensional spaces were found to be generally low independent of whether the original data set or the data set of reduced dimension was used. Therefore, the main effect of the dimension reduction was to save some computational time.
Nonlocal descriptors
It can be argued that protein folding is a nonlocal phenomenon, as typical force fields used to simulate protein folding are not based uniquely on the pointwise properties of the amino acids ordered along a protein’s primary structure. We expected that string matching and raw statistics based on the occurrence of amino acids, peptides, or more complicated structural motifs in protein sequences can capture only a part of the information lying in the basis of such a complicated phenomenon as protein folding. Therefore, we included in our study several sets of nonlocal descriptors gathering information from topologically distant regions on the amino acid sequence string under scrutiny.
Autocross-covariance transform-based descriptors.
We used some autocorrelation relations over protein sequences in our attempt to describe better the nonlocal nature of protein folding. To this end a set of autocross-covariance transform-based descriptors (Lapinsh et al. 2002) (ACCT_AA) was calculated as follows:
 |
(3) |
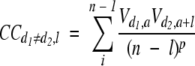 |
(4) |
where d varies from 1 to D, while l varies from 1 to L. D is the number of amino acid indices; L is the maximum lag; n is the total number of amino acids in the sequence; V is a specific property related to every particular amino acid; a is the amino acid position in the sequence; and p is the degree of normalization of the ACCT term. This type of description leads to the embedding of protein sequences in a vector space of dimension LD2. In our work, p was set to 1 and L was set to 30. Three different indices of amino acids were considered (i.e., D = , based on the lipophilic, steric, and electronic properties. The V values corresponding to every amino acid are available from the literature (Sandberg et al. 1998). The dimensionality of the ACCT data matrix was reduced to 30 by SVD, and the similarity between any pair of protein sequences was evaluated again by using the cosine measure. We denoted the 30 resulting descriptors as ACCT_AA.
Secondary structure ACCT-based descriptor.
The predicted secondary structure for a given protein sequence was also used to derive an ACCT-type of descriptor (i.e., ACCT_SS). In contrast to the previous paragraph, we substituted the amino acid physico-chemical properties at positions d1, d2, and d3 on the protein chain by the corresponding predicted three basic types of secondary structure states. For any di, dj ∈ d1, d2, d3), we defined Vdi,aVdj,a + 1 appearing in equations 3 and 4 as:
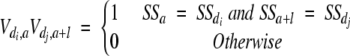 |
(5) |
In any other aspect the computational procedure was kept identical to the one described in the previous paragraph. We denoted the 30 resulting descriptors as ACCT_SS.
CTD-based descriptors.
The composition (C) transition (T) distribution (D) descriptors, labeled below CTD_AA, provided another way to incorporate nonlocal effects in our analysis by taking into account some basic structural and physicochemical properties of the natural amino acids, such as their normalized van der Waals volumes, hydrophobicities, and polarities. Using these descriptors, the 20 amino acids can be grouped into a limited number of classes. The coding of a given protein sequence by the CTD_AA descriptors is evaluated as follows: C represents the number of amino acids sharing a particular property divided by the total number of amino acids in the sequence; T characterizes the frequency with which an amino acid of a particular property is followed by amino acids falling in a class with a different property; and D measures the chain length within which the first, 25%, 50%, 75%, and 100% of the amino acids sharing a similar property are located, respectively. In most studies, amino acids are divided into three classes depending on the considered property (Murphy et al. 2000). In our study, the 20 amino acids are grouped into three classes (LASGVTIPMC, EKRDNQH, and FYW), which basically reflect the differences in the hydrophobicities and side-chain volumes (Dubchak et al. 1995; Cai et al. 2003). This representation led to an embedding of protein sequences in a vector space of dimension 21. The similarity between any two protein sequences encoded by these descriptors could be evaluated by using equation 2.
Secondary structure CTD-based descriptors.
Similar to CTD_AA, the sequence can be described by CTD with predicted secondary structure (i.e., CTD_SS). Instead of using the classification based on the physicochemical properties, the amino acids were classified into three predicted secondary structure states generated from PSIPRED (Jones 1999). Thus, a 21-dimensional vector for each sequence was available for measuring the similarity between two evaluated sequences.
Triplet descriptors.
Finally, an interesting nonlocal set of descriptors could be derived by analogy to that largely used in the pharmaceutical industry representation of small bioactive molecules, termed triangular fingerprints (Grigorov et al. 2003). In our study we constructed a list of triangular fingerprints to encode protein sequences through the following steps. First, the usual 20-letter amino acid alphabet was reduced by applying a structure-based classification of the natural amino acids into four classes (A, LVIMC; B, AGSTP; C, FYW; D, EDNQKRH) (Murphy et al. 2000). In this representation every protein sequence was represented by a four-letter alphabet. Next, an exhaustive list of triangular descriptors was generated, by counting all triplets of amino acids types separated by 4, 8, and 12 amino acids, respectively. Finally, triangles were ordered lexicographically, which allowed us to unambiguously encode an arbitrary protein sequence by the frequency of occurrence in the sequence of all triangular descriptors from the reference list. In our work we used a list of 576 triangular descriptors, further reduced to 30 dimensions by SVD. The similarity between every two sequences encoded by this method was evaluated by applying the cosine similarity measure, appearing in equation 2.
Local descriptors
We already stated that protein folding could be driven to a large extent by nonlocal forces. Nevertheless, the idea of the existence of folding initiation clusters in protein sequences has been now largely accepted (Mirny and Shakhnovich 2001). Although general local sequence descriptors are expected to lead to poor performance for remote homology identification, we speculated that some carefully chosen local description schemes, known to correlate with protein structure, are worth being evaluated in our study. To this end, we chose two types of encodings: one based on the 1327 PROSITE functional motifs (Hofmann et al. 1999), the other based on the I-sites library of 331 folding initiation sites (Bystroff and Baker 1998). Using these two motif libraries, we encoded a given protein sequence as a vector of real numbers, each number indicating the probability of occurrence of a given functional or structural motif in a protein sequence.
Motif-based descriptors.
Amino acid motifs were found to be characteristic of well-defined protein functional classes and were compiled in several databases, the most well known being the PROSITE database. In our work we used 1327 patterns as the representation basis for protein sequences. We encoded a given protein sequence as a binary vector, indicating by 0 and 1 the presence or absence of every one of the 1327 motifs. In order to provide a measure of the similarity based on the PROSITE motifs in two evaluated sequences, the statistical analysis for the motif-fold compatibility (Salwinski and Eisenberg 2001) was calculated based on two structural classification databases (i.e., SCOP and CATH). Certainly, we could also have used the whole FSSP data set for deriving the correlation between motifs and folds. However, since our test set was based on FSSP database, the FSSP607 sequences would be directly involved in the construction of the similarity score SFM. To avoid the statistically unreliable situation of drawing conclusions about a method’s performance based on predictions made within the training set, we used the sequences from the SCOP and CATH classifications to build our model. By doing this, we were able to provide statistically reliable predictions by applying the derived model to the FSSP607 data set.
Taking the analysis on SCOP as the example, the correlation between motif presence and protein fold in SCOP can be evaluated by calculating the log-odds score, SFM, defined as:
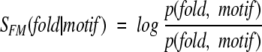 |
(6) |
where p(motif) and p(fold) are individual probabilities of finding a particular sequence motif and a particular fold in the SCOP domains (i.e., SCOP-1.63.95), and p(fold, motif) is the corresponding joint probability. Furthermore, the motif-based compatibility for the query sequence in the specified folds can be assigned as:
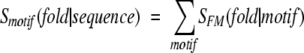 |
(7) |
where SFM_SCOP was calculated from equation 6 and summation was performed over all motifs found in the evaluated sequence and fulfilling the following criteria:
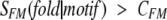 |
(8) |
where CFM is an adjustable parameter, with 0.5 being a good choice for it. For the evaluated sequence, the potential fold (PTF) should be identified as the fold where Smotif(fold |sequence) achieves the maximum. Then, the motif-based similarity between two sequences A and B in the context of SCOP fold space is assigned as:
 |
(9) |
In a similar manner, the motif-based similarity in the context of CATH fold space [i.e., MOTIF_CATH_SIM(A,B)] can be also calculated.
I-sites-based descriptor.
The Rosetta protein structure prediction server, relying on the I-sites library of folding initiation structural motifs, was successful in several rounds of the CASP challenge. We therefore downloaded the I-sites library as a representation basis for encoding protein sequences. In a way similar to the derivation of functional motif-based similarities, we derived a structural-motif-based similarity score, which we termed ISITES_CATH_SIM(A,B), aided by the CATH database.
Data mining methods
Measures of performance of remote homology identification
The performance of the 13 different descriptors in identifying remote homologs was quantified in terms of sensitivity and reliability. We defined the sensitivity as the percentage of correctly assigned remote homologous protein pairs in a database of a given size. This quantity is frequently termed the rate of remote homology identification. However, as reported in the literature, the sensitivity is not sufficient to measure the performance (McGuffin and Jones 2003). Therefore, in our work we used a second quantity, termed the reliability. It is described as the slope of the curve characterizing the change in the error rate of a query process versus its sensitivity, that is, the identification ratio of true positives (cf. Fig. 4 ▶). In practice, we evaluated the reliability of our method by investigating how the error per query increases with the cumulated number of true-positive remote homologs identified. Our analysis was carried out in a way similar to the one appearing in the work of Kelley et al. (2000), sharing features of ROC plots. It consisted essentially in compiling statistics on the observed number of true and false positives for several different queries. As Figure 4 ▶ illustrates, the recovery of true positives was increasing with the incorporation of different types of descriptors in the SVM model. For practical purposes, the most important region of Figure 4 ▶ corresponds to a 95% confidence assignment (i.e., <5% error rates per query). Throughout our work, we provided the sensitivity at the 95% confidence level by computing the ratio of true positives with an estimated error per query of 0.05.
All the evaluation methods addressed above are focused on the hit with highest similarity score or “top hit” for each query sequence. The above so-called “one-to-many” definition of coverage was previously described by Muller et al. (1999), which was subsequently used to benchmark 3D-PSSM and GenTHREADER. In the real world, the performance of “one-to-many” experiments is very important, since we are only looking for a single high-confidence match for every query sequence (Muller et al. 1999).
Support vector machine learning
The SVM is a machine-learning algorithm for two classes of classification with the goal to find a rule that best maps each member of training set to the correct classification (Cai et al. 2003; Dobson and Doig 2003). In linearly separable cases, SVM constructs a hyperplane that separates two different groups of feature vectors in the training set with a maximum margin. The orientation of a test sample relative to the hyperplane gives the predicted score, and hence the predicted class can be derived. In our work, the SVM was used to distinguish the remote homology pairs and nonhomology pairs combining the top three descriptors (i.e., PSI-BLAST term, motif term, and SSEA score). The FSSP607 set can be divided into 183,921 pairwise comparisons [C6072 = (607 × 606)/2 = 183,921; N.B. the pair (A, B) is the same as (B, A)], in which 2847 are remote homologs (i.e., they share at least the same first two indices in the FSSP family hierarchy). In addition to the PSIBLAST_SIM(A,B), the alignment score for the evaluated sequences A and B is also used. Due to the way the PSI-BLAST searching is carried out, the A → B query is different from the B →A one. Therefore, the PSIBLAST_SIM(B,A) similarity score and the corresponding alignment score were also included in the PSI-BLAST descriptor, composed of four parameters in total. For the motif term associated to the pair (A, B), both the MOTIF_SCOP_SIM(A,B) and the MOTIF_CATH_SIM(A,B) scores were used.
The data set of 183,921 protein pairs was divided into five subsets of equal size. In order to predict if a given protein pair was, indeed, a pair of remote homologs, we labeled as “test” the set to which this precise pair belongs. The four remaining sets were labeled as “training” and SVM models were developed for each of them. The class label for homologous and nonhomologous pairs were set to +1 and -1, respectively. The ratio of homologous pairs to nonhomologous pairs was ~1:64 in the training set. Presenting the data in this ratio causes the SVM to predict invariably every pair as nonhomologous. The best balance in the training set was found for a ratio of 1:2.5. Each training group was balanced by discarding a random selection of the nonhomologous pairs prior to training. The training resulted in four separate SVM models, the predicted score being obtained as an average value over the scores from the four different SVM models.
The implementation of the SVM algorithm we used in our work was SVM-Light (Joachims 1999; http://svmlight.joachimms.org). The applied kernel functions were the linear, the polynomial, and the radial basis function. Other than varying the kernel function, the algorithm was run with the default settings.
Electronic supplemental material
The Supplemental Material contains one table showing the lists of PDB codes for the FSSP607 data set.
Acknowledgments
We are thankful to Prof. Dr. Heribert Watzke for the many discussions on fundamental aspects of the concept of structure in molecular science.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041035505.
Supplemental material: see www.proteinscience.org
References
- Alexandrov, N.N. and Go, N. 1994. Biological meaning, statistical significance, and classification of local spatial similarities in nonhomologous proteins. Protein Sci. 3 866–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215 403–410. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzari, P.M., Souchon, H., and Dominguez, R. 1996. The crystal structure of endoglucanase CelA, a family 8 glycosyl hydrolase from Clostridium thermocellum. Structure 4 265–275. [DOI] [PubMed] [Google Scholar]
- Anfinsen, C.B. 1973. Principles that govern the folding of protein chains. Science 181 223–230. [DOI] [PubMed] [Google Scholar]
- Ben Hur, A. and Brutlag, D. 2003. Remote homology detection: A motif based approach. Bioinformatics 19 (Suppl. 1) I26–I33. [DOI] [PubMed] [Google Scholar]
- Bowie, J.U., Luthy, R., and Eisenberg, D. 1991. A method to identify protein sequences that fold into a known three-dimensional structure. Science 253 164–170. [DOI] [PubMed] [Google Scholar]
- Brenner, S.E., Koehl, P., and Levitt, M. 2000. The ASTRAL compendium for protein structure and sequence analysis. Nucleic Acids Res. 28 254–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant, S.H. 1996. Evaluation of threading specificity and accuracy. Proteins 26 172–185. [DOI] [PubMed] [Google Scholar]
- Bujnicki, J.M., Elofsson, A., Fischer, D., and Rychlewski, L. 2001. LiveBench-1: Continuous benchmarking of protein structure prediction servers. Protein Sci. 10 352–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bystroff, C. and Baker, D. 1998. Prediction of local structure in proteins using a library of sequence-structure motifs. J. Mol. Biol. 281 565–577. [DOI] [PubMed] [Google Scholar]
- Cai, C.Z., Han, L.Y., Ji, Z.L., Chen, X., and Chen, Y.Z. 2003. SVM-Prot: Web-based support vector machine software for functional classification of a protein from its primary sequence. Nucleic Acids Res. 31 3692–3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chothia, C. 1992. Proteins. One thousand families for the molecular biologist. Nature 357 543–544. [DOI] [PubMed] [Google Scholar]
- Contreras-Moreira, B., Fitzjohn, P.W., and Bates, P.A. 2003. In silico protein recombination: Enhancing template and sequence alignment selection for comparative protein modelling. J. Mol. Biol. 328 593–608. [DOI] [PubMed] [Google Scholar]
- Dobson, P.D. and Doig, A.J. 2003. Distinguishing enzyme structures from non-enzymes without alignments. J. Mol. Biol. 330 771–783. [DOI] [PubMed] [Google Scholar]
- Dubchak, I., Muchnik, I., Holbrook, S.R., and Kim, S.H. 1995. Prediction of protein folding class using global description of amino acid sequence. Proc. Natl. Acad. Sci. 92 8700–8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, D. 2000. Hybrid fold recognition: Combining sequence derived properties with evolutionary information. Pac. Symp. Biocomput. 119–130. [PubMed]
- Ginalski, K., Pas, J., Wyrwicz, L.S., von Grotthuss, M., Bujnicki, J.M., and Rychlewski, L. 2003. ORFeus: Detection of distant homology using sequence profiles and predicted secondary structure. Nucleic Acids Res. 31 3804–3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godzik, A., Kolinski, A., and Skolnick, J. 1992. Topology fingerprint approach to the inverse protein folding problem. J. Mol. Biol. 227 227–238. [DOI] [PubMed] [Google Scholar]
- Gribskov, M. and Robinson, N.L. 1996. Use of receiver operating characteristic (ROC) analysis to evaluate sequence matching. Comput. Chem. 20 25–33. [DOI] [PubMed] [Google Scholar]
- Grigorov, M.G., Schlichtherle-Cerny, H., Affolter, M., and Kochhar, S. 2003. Design of virtual libraries of umami-tasting molecules. J. Chem. Inf. Comput. Sci. 43 1248–1258. [DOI] [PubMed] [Google Scholar]
- Hofmann, K., Bucher, P., Falquet, L., and Bairoch, A. 1999. The PROSITE database, its status in 1999. Nucleic Acids Res. 27 215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1996. Mapping the protein universe. Science 273 595–603. [DOI] [PubMed] [Google Scholar]
- Hou, Y., Hsu, W., Lee, M.L., and Bystroff, C. 2004. Efficient remote homology detection using local structure. Bioinformatics 19 2294–2301. [DOI] [PubMed] [Google Scholar]
- Huang, J.Y. and Brutlag, D.L. 2001. The EMOTIF database. Nucleic Acids Res. 29 202–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joachims, T. 1999. Making large-scale SVM learning practical. In Advances in kernel methods—Support vector learning (eds. B. Scholkopf et al.), pp. 41–56. MIT Press, Cambridge, MA.
- Jones, D.T. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292 195–202. [DOI] [PubMed] [Google Scholar]
- Jones, D.T., Taylor, W.R., and Thornton, J.M. 1992. A new approach to protein fold recognition. Nature 358 86–89. [DOI] [PubMed] [Google Scholar]
- Kelley, L.A., MacCallum, R.M., and Sternberg, M.J. 2000. Enhanced genome annotation using structural profiles in the program 3D-PSSM. J. Mol. Biol. 299 499–520. [DOI] [PubMed] [Google Scholar]
- Koppensteiner, W.A., Lackner, P., Wiederstein, M., and Sippl, M.J. 2000. Characterization of novel proteins based on known protein structures. J. Mol. Biol. 296 1139–1152. [DOI] [PubMed] [Google Scholar]
- Lapinsh, M., Gutcaits, A., Prusis, P., Post, C., Lundstedt, T., and Wikberg, J.E. 2002. Classification of G-protein coupled receptors by alignment-independent extraction of principal chemical properties of primary amino acid sequences. Protein Sci. 11 795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt, M. 1997. Competitive assessment of protein fold recognition and alignment accuracy. Proteins Suppl. 1 92–104. [DOI] [PubMed] [Google Scholar]
- Liao, L. and Noble, W.S. 2003. Combining pairwise sequence similarity and support vector machines for detecting remote protein evolutionary and structural relationships. J. Comput. Biol. 10 857–868. [DOI] [PubMed] [Google Scholar]
- Lindgard, P.A. and Bohr, H. 1996. Magic numbers in protein structures. Phys. Rev. Lett. 77 779–782. [DOI] [PubMed] [Google Scholar]
- Lundstrom, J., Rychlewski, L., Bujnicki, J., and Elofsson, A. 2001. Pcons: A neural-network-based consensus predictor that improves fold recognition. Protein Sci. 10 2354–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti-Renom, M.A., Madhusudhan, M.S., and Sali, A. 2004. Alignment of protein sequences by their profiles. Protein Sci. 13 1071–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu, M., Modis, Y., Zeelen, J.P., Engel, C.K., Abagyan, R.A., Ahlberg, A., Rasmussen, B., Lamzin, V.S., Kunau, W.H., and Wierenga, R.K. 1997. The 1.8 Å crystal structure of the dimeric peroxisomal 3-ketoacyl-CoA thiolase of Saccharomyces cerevisiae: Implications for substrate binding and reaction mechanism. J. Mol. Biol. 273 714–728. [DOI] [PubMed] [Google Scholar]
- McGuffin, L.J. and Jones, D.T. 2002. Targeting novel folds for structural genomics. Proteins 48 44–52. [DOI] [PubMed] [Google Scholar]
- ———. 2003. Improvement of the GenTHREADER method for genomic fold recognition. Bioinformatics 19 874–881. [DOI] [PubMed] [Google Scholar]
- Mirny, L. and Shakhnovich, E. 2001. Protein folding theory: From lattice to allatom models. Annu. Rev. Biophys. Biomol. Struct. 30 361–396. [DOI] [PubMed] [Google Scholar]
- Muller, A., MacCallum, R.M., and Sternberg, M.J. 1999. Benchmarking PSI-BLAST in genome annotation. J. Mol. Biol. 293 1257–1271. [DOI] [PubMed] [Google Scholar]
- Murphy, L.R., Wallqvist, A., and Levy, R.M. 2000. Simplified amino acid alphabets for protein fold recognition and implications for folding. Protein Eng. 13 149–152. [DOI] [PubMed] [Google Scholar]
- Murzin, A.G. 1999. Structure classification-based assessment of CASP3 predictions for the fold recognition targets. Proteins 37 88–103. [DOI] [PubMed] [Google Scholar]
- Murzin, A.G., Brenner, S.E., Hubbard, T., and Chothia, C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247 536–540. [DOI] [PubMed] [Google Scholar]
- Needleman, S.B. and Wunsch, C.D. 1970. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 48 443–453. [DOI] [PubMed] [Google Scholar]
- Olsen, J.G., Kadziola, A., Wettstein-Knowles, P., Siggaard-Andersen, M., Lindquist, Y., and Larsen, S. 1999. The X-ray crystal structure of β-keto-acyl [acyl carrier protein] synthase I. FEBS Lett. 460 46–52. [DOI] [PubMed] [Google Scholar]
- Orengo, C.A., Michie, A.D., Jones, S., Jones, D.T., Swindells, M.B., and Thornton, J.M. 1997. CATH—A hierarchic classification of protein domain structures. Structure 5 1093–1108. [DOI] [PubMed] [Google Scholar]
- Orengo, C.A., Todd, A.E., and Thornton, J.M. 1999. From protein structure to function. Curr. Opin. Struct. Biol. 9 374–382. [DOI] [PubMed] [Google Scholar]
- Panchenko, A.R., Marchler-Bauer, A., and Bryant, S.H. 2000. Combination of threading potentials and sequence profiles improves fold recognition. J. Mol. Biol. 296 1319–1331. [DOI] [PubMed] [Google Scholar]
- Pearson, W.R. and Lipman, D.J. 1988. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. 85 2444–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przytycka, T., Aurora, R., and Rose, G.D. 1999. A protein taxonomy based on secondary structure. Nat. Struct. Biol. 6 672–682. [DOI] [PubMed] [Google Scholar]
- Rost, B. 1999. Twilight zone of protein sequence alignments. Protein Eng. 12 85–94. [DOI] [PubMed] [Google Scholar]
- Rychlewski, L., Jaroszewski, L., Li, W., and Godzik, A. 2000. Comparison of sequence profiles. Strategies for structural predictions using sequence information. Protein Sci. 9 232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salwinski, L. and Eisenberg, D. 2001. Motif-based fold assignment. Protein Sci. 10 2460–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg, M., Eriksson, L., Jonsson, J., Sjostrom, M., and Wold, S. 1998. New chemical descriptors relevant for the design of biologically active peptides. A multivariate characterization of 87 amino acids. J. Med. Chem. 41 2481–2491. [DOI] [PubMed] [Google Scholar]
- Shi, J., Blundell, T.L., and Mizuguchi, K. 2001. FUGUE: Sequence–structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol. 310 243–257. [DOI] [PubMed] [Google Scholar]
- Shindyalov, I.N. and Bourne, P.E. 1998. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 11 739–747. [DOI] [PubMed] [Google Scholar]
- Siew, N., Elofsson, A., Rychlewski, L., and Fischer, D. 2000. MaxSub: An automated measure for the assessment of protein structure prediction quality. Bioinformatics 16 776–785. [DOI] [PubMed] [Google Scholar]
- Sippl, M.J. 1995. Knowledge-based potentials for proteins. Curr. Opin. Struct. Biol. 5 229–235. [DOI] [PubMed] [Google Scholar]
- Sippl, M.J., Lackner, P., Domingues, F.S., Prlic, A., Malik, R., Andreeva, A., and Wiederstein, M. 2001. Assessment of the CASP4 fold recognition category. Proteins Suppl. 5 55–67. [DOI] [PubMed] [Google Scholar]
- Smith, T.F. and Waterman, M.S. 1981. Identification of common molecular subsequences. J. Mol. Biol. 147 195–197. [DOI] [PubMed] [Google Scholar]
- Sonnhammer, E.L., Eddy, S.R., and Durbin, R. 1997. Pfam: A comprehensive database of protein domain families based on seed alignments. Proteins 28 405–420. [DOI] [PubMed] [Google Scholar]
- Xie, D., Tropsha, A., and Schlick, T. 2000. An efficient projection protocol for chemical databases: Singular value decomposition combined with truncated-newton minimization. J. Chem. Inf. Comput. Sci. 40 167–177. [DOI] [PubMed] [Google Scholar]
- Yoon, H.J., Mikami, B., Hashimoto, W., and Murata, K. 1999. Crystal structure of alginate lyase A1-III from Sphingomonas species A1 at 1.78 Å resolution. J. Mol. Biol. 290 505–514. [DOI] [PubMed] [Google Scholar]
- Zhang, Z., Kochhar, S., and Grigorov, M. 2003. Exploring the sequence–structure protein landscape in the glycosyltransferase family. Protein Sci. 12 2291–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]