

Abstract
Using a recently developed program (SCOPmap) designed to automatically assign new protein structures to existing evolutionary-based classification schemes, we identify a evolutionarily conserved domain (EDD) common to three different folds: mannose transporter EIIA domain (EIIA-man), dihydroxyacetone kinase (Dak), and DegV. Several lines of evidence support unification of these three folds into a single superfamily: statistically significant sequence similarity detected by PSI-BLAST; “closed structural grouping” using DALI Z-scores (each protein inside a group finds all other group members with scores higher than those to proteins outside the group) that includes only these proteins sharing a unique α-helical hairpin at the C-terminus and excludes all other proteins with similar topology; similar domain fusions connect Dak and DegV, and genomic neighborhood organizations connect Dak and EIIA-man. Finally, both Dak and EIIA-man perform similar phosphotransfer reactions, suggesting a phosphotransferase activity for the DegV-like family of proteins, whose function other than lipid binding revealed in the crystal structure remains unknown.
Keywords: EDD domain, Dak1, Dak2, dihydroxyacetone kinase, DegV, mannose transporter EIIA, SCOPmap, homology detection, structure similarity, protein classification
Although expert manual analysis remains the gold standard in structural classification, the necessity for automatic methods that can reliably reproduce these results becomes increasingly apparent as biological databases continue to grow in size. We have recently developed a program (SCOPmap; Cheek et al. 2004) for automatically assigning domains in new protein structures to existing evolutionary-based fold classifications (i.e., SCOP superfamilies). To achieve this task SCOPmap uses various sequence-based and structure-based score thresholds that were trained with existing SCOP defined domains. Therefore, assignments are expected to exhibit evolutionary relevance reflective of manually curated SCOP superfamilies. Application of this method to existing classification schemes can also help recognize nontrivial evolutionary links between distant protein families that have been missed for various reasons, thus providing more standardized classifications.
SCOPmap assigns the N-terminal domain of dihydroxyacetone kinase (Dak, PDB ID 1oi2; Siebold et al. 2003a,b) to the mannose transporter IIA domain superfamily (EIIA-man, PDB ID 1pdo, Nunn et al. 1996). The structures of both Dak (Fig. 1A ▶, N-terminal yellow/cyan domain) and EIIA-man (Fig. 1B ▶) contain a common domain made up of a central parallel β-sheet of four strands (order 2134) with helices bounding either side. In contrast to this assignment, the SCOP database (Murzin et al. 1995) currently groups Dak (Fig. 1A ▶) with the tubulin GTPase domain (Fig. 1D ▶) at the superfamily level. These two folds share a similar topology across both Dak domains, assuming various inserted elements (Fig. 1A,D ▶, white N-terminal domain insertions and gray C-terminal domain insertions). Despite these general similarities, we were not able to find sequence support for a homologous relationship between Dak and tubulin, and their overall structural similarity is low (highest DaliLite Z score 4.6 between tubulin structure 1jff_B and Dak structure 1oi2_A).
Figure 1.

Structural similarities of EDD domain-containing families and tubulin GTPase domain.Structural models produced with MOLSCRIPT (Esnouf 1999) reveal similarities between (A) Dak representative structure (PDB ID 1un9) with N-terminal EDD domain, (B) EIIA-man structure (PDB ID 1pdo) of EDD domain swapped dimer, and (C) DegV representative structure (PDB ID 1pzx) with N-terminal EDD domain in comparison to (D) tubulin GTPase domain representative structure (PDB ID 1tub). The α-helices and β-strands of the EDD domain homologs and the N-terminal tubulin domain are colored blue and yellow, respectively. The α-helices and β-strands of the equivalent C-terminal domains are colored purple and green, respectively. Elements inserted into the EDD domain core fold are colored white while elements inserted into the C-terminal domain (or elements corresponding to the EIIA-man swapped dimer that replace the C-terminal domain) are colored gray. Bonds representations of bound ligands (Dihydroxyacetone in Dak and Palmitate in DegV) and family-conserved residues marking the active sites are colored according to atom type (gray for carbon, red for oxygen, blue for nitrogen, and pink for phosphate). The N-terminus and the C-terminus of each structure is labeled.
Dak functions to generate the glycolytic intermediate dihydroxyacetone phosphate using a phosphoryl donor from either an ATP molecule or a phosphoenolpyruvate (PEP)-dependent phosphotransferase cascade (Gutknecht et al. 2001). EIIA-man belongs to a similar phosphorelay protein cascade that constitutes the PEP: sugar phosphotransferase system (PTS) responsible for phosphorylation of sugar coupled with its translocation in bacteria. In the initial step of this cascade, enzyme I (EI) catalyzes phosphoryl transfer from PEP to histidine-containing phosphoryl carrier protein (HPr) (Hu and Saier 2002). HPr then serves as a phosphoryl donor to histidine or cysteine residues of enzyme II (EII) components, whose composition differ in various PTS. In the mannose PTS of Escherichia coli, EII consists of two soluble components that continue the phosphotransfer cascade (EIIA-man fused to EIIB) and two membrane-bound components that serve as the sugar transport machinery (EIIC and EIID) (Robillard et al. 1999). In the Dak PTS of E. coli, EII consists of an EIIA-man paralog (EIIA-dak) and two soluble subunits of Dak (Dak1 and Dak2) (Gutknecht et al. 2001).
To help resolve Dak classification, we decided to explore its evolutionary relationships using a combination of sequence- and structure-based methods. This report outlines the resulting support for an evolutionary link between the two SCOP folds represented by EIIA-man and Dak. Furthermore, our data establish a homologous relationship between these two families and members of a third SCOP fold, DegV-like proteins (PDB IDs 1pzx and 1mgp, Schulze-Gahmen et al. 2003). We propose to unite these three families (EIIA-Man, Dak, and DegV) into a single superfamily (EDD fold superfamily). In accordance with this classification, structural similarities between three families were previously noted (Schulze-Gahmen et al. 2003; Siebold et al. 2003a,b), although their homologous evolutionary relationships were not appreciated. Functional similarities between EIIA-man and Dak provide additional support for this classification and suggest a common activity for the DegV family of proteins, whose biological activity other than phospholipid binding revealed in crystal structures of two hypothetical proteins remains unknown.
Results and Discussion
SCOPmap identifies Dak homologs
SCOPmap recognized a link between Dak (PDB ID 1oi2) and EIIA-man (PDB ID 1pdo) based on a measure of structure-based conservation pattern similarity between these two protein families (conservation score = 0.20, based on DaliLite pairwise alignment with Z-score = 7.2). SCOP-map calculates this conservation score for a given pair of domains by evaluating whether the most highly conserved residue motifs in each domain reside in equivalent positions in the structure-based alignment. Such a score was developed to reflect the concept that homologous domains often accomplish various functional roles (e.g., substrate binding or catalysis) using similar active sites. Based on existing SCOP classifications, a pair of domains are likely homologs if they possess similar folds (DaliLite Z-score ≥ 5) and retain similar active site placement as defined by motifs (conservation score ≥ 0.1). The conservation score for Dak and EIIA-Man (0.20) falls well within this accepted range.
To further explore the relationship of the Dak family to existing SCOP classification, SCOPmap was modified as described in Methods to identify multiple homologs of the query structure that fall within established cutoffs. SCOP-map then indicated homology between Dak and an additional family of proteins (DegV) through superfamily level assignments. This link was established by a high degree of structural similarity between Dak queries (PDB IDs 1oi2 and 1un8) and a DegV-like protein, Thermotoga maritima hypothetical protein TM841 (PDB ID 1mgp; Fig. 1C ▶) (DaliLite Z-score for 1oi2–1mgp = 15.8; DaliLite Z-score for 1un8–1mgp = 15.5). SCOPmap cutoffs for superfamily assignments using such structural comparisons by DaliLite require a Z-score ≥ 14, regardless of the degree of sequence similarity. Importantly, the structural similarity of these two protein families extends beyond the N-terminal domain shared with EIIA-man and also encompasses the two C-terminal domains.
Sequence-based support for Dak classification
SCOPmap assignments for at least one query (1oi2) suggest that Dak, EIIA-man, and DegV structures should belong to the same superfamily. To help confirm this implied evolutionary link, we used sequences from each proposed EDD domain as queries in transitive PSI-BLAST searches. A representative EIIA-man sequence (gi| 22538040, range 2..133) identified the N-terminal domain of DegV (gi|26553729, detected in iteration 11 with E- value 0.004) while another representative EIIA-man sequence (gi|10957457, range 2..126) identified the N-terminal domain of Dak (gi| 15082277, detected in iteration 2 with E value 0.002). Additionally, sequence searches with the N-terminal domain of DegV (gi|28379075, range 1..152) find an EIIA-man-like transcription antiterminators (gi|48826184, range 579..663 with E-value 0.001 in iteration 2). The PSI-BLAST runs converge without identifying sequences from other families (false positives), and each of the PSI-BLAST hits encompasses a significant portion of the EDD domain (indicated with * in Fig. 2 ▶). Although representative sequences from Dak and DegV do not detect each other directly using these criteria, the two families display a transitive linkage through the EIIA-man family.
Figure 2.
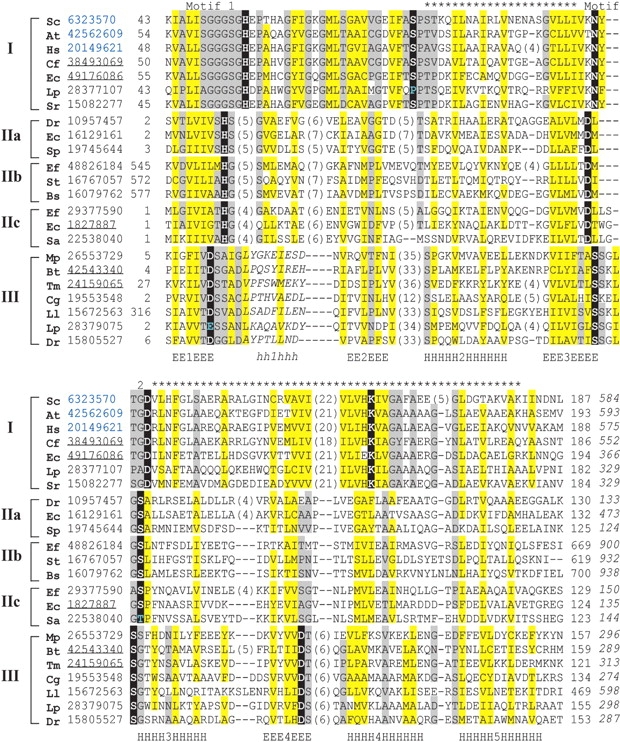
Multiple sequence alignment of EDD domain. Representative sequences of the three EDD domain containing families are labeled: (I) Dak, (II) EIIA-man, and (III) DegV; and grouped with subgroups (IIa, IIb, and IIc) according to Euclidian distance mapping shown in Figure 3 ▶. Each sequence is identified by the NCBI gene identification number (gi) colored according to superkingdom (black for bacteria and blue for eukaryote) and species name abbreviated as follows: Dr, Deinococcus radiodurans; Bt, Bacillus stearothermophilus; Tm, Thermotoga maritima; Cg, Corynebacterium glutamicum; Ll, Lactococcus lactis; Lp, Lactobacillus plantarum; Mp, Mycoplasma penetrans; Sa, Streptococcus agalactiae; Ef, Enterococcus faecalis; Ec, Escherichia coli; St, Salmonella typhimurium; Bs, Bacillus subtilis; Sp, Streptococcus pyogenes; Sr, Selenomonas ruminantium; Cf, Citrobacter freundii; Sc, Saccharomyces cerevisiae; At, Arabidopsis thaliana; and Hs, Homo sapiens. The sequence identifiers corresponding to known structures are underlined. Positions corresponding to structurally conserved secondary structural elements (E for β-strand and H for α-helix) and structurally unalignable elements (h for α-helix) are labeled and numbered (within the labels) below the sequences. The portion of the EDD domain that is detected between families with PSI-BLAST is labeled (*) above the alignment, as are the two motifs (1 and 2) detected with SCOPmap conservation scores. The first and last residue numbers of the shown sequences are indicated before and after each sequence, with the total length of the sequences following in italics. Some nonconserved residues in loops and inserts are omitted, and the number of omitted residues is shown in parentheses. Residues conserved among families are highlighted black, uncharged residues at mainly hydrophobic positions are highlighted in yellow, and conserved small residues are highlighted in gray.
The multiple sequence alignment illustrated in Figure 2 ▶ highlights the conservation patterns of the Dak (group I), the EIIA-man (group II), and the DegV (group III) superfamilies. Overall, each family displays similar hydrophobicity patterns (yellow highlights) corresponding to the secondary structural elements of the core fold. Highly conserved residues within each superfamily (black highlights) reside mainly in two loops: the loop connecting the first β-strand and α-helix and the loop connecting the third β-strand and α-helix (numbered consecutively along the core structural elements, as in Fig. 2 ▶). Although these loops often form the active sites of other Rossmann-type folds, the residues contributing to the active site of each EDD domain are similar. The motif-2 residues of each structure point toward their respective ligands: the conserved Asp in Dak hydrogen bonds to the covalently bound dihydroxyacetone, the conserved Asp of EIIA-man hydrogen bonds to the phosphoryl group acceptor histidine (His10, referred to as “ligand”), and the second conserved Ser in DegV hydrogen bonds to the bound palmitate. In fact a global superposition of each EDD domain reveals an overlap of each of these hydrogen-bonded sites, despite marked differences in the molecular properties of each ligand. The observed diversity between each EDD domain sequence probably reflects their adaptation to accommodate these different molecules.
To help visualize the evolutionary relationships between members of the EDD domain superfamily, we constructed a stereo plot of representative sequences mapped in Euclidian space according to distances (Fig. 3 ▶) (Grishin and Grishin 2002). In this three-dimensional plot, each symbol represents a sequence colored according to the three different families: Dak (black), EIIA (gray), and DegV (open circles); and the space between symbols reflects evolutionary distances. Those sequences that established links between families with PSI-BLAST are connected by arrows stemming from the queries, and the E-value of the initial PSI-BLAST hits are displayed. This mapping procedure clusters the EIIA family into three distinct subgroups that correspond to their COG classification (reflected by different symbols: circle for EIIA-dak, triangle for EIIA-man, and square for transcription antiterminator). Notably, the sequences from the different subgroups readily detect each other using our PSI-BLAST cutoffs (E-value 0.005, maximum 20 iterations). The Dak sequences form a tight cluster that reflects a high overall degree of sequence similarity between members. Accordingly, profiles built for these sequence queries with PSI-BLAST are not diverse enough to detect homologs from the other families. Sequences from the more diverse groups (EIIA-man and Dak) can establish evolutionary links to the other families.
Figure 3.

Euclidian distance mapping and evolutionary connections. The stereo plot represents the first three dimensions of a maximal scatter of points in multidimensional space. The data points correspond to the sequences shown in Figure 2 ▶, with the scatter approximating evolutionary distances. The symbols correspond to grouped sequences: black circles (Group I Dak); gray circles (Group IIA EIIA-dak); gray squares (Group IIB EIIA-transcription antiterminator); gray triangles (Group IIC EIIA-man); and open circles (Group III, DegV). The arrows stem from data points corresponding to query sequences that when used in PSI-BLAST establish evolutionary links across families to the data points representing sequence hits. Arrows are labeled with the PSI-BLAST E-values of initial hits across families.
Structural classification of Dak homologs
SCOP currently classifies EIIA-man, DegV, and Dak in different folds. While both the EIIA-man and the DegV folds remain independent, the Dak fold is grouped together with tubulin nucleotide binding domain (PDB ID: 1tub, range 1..245) in the same superfamily (Murzin et al. 1995). All of these structures display similar topologies in both their N-terminal domains and their C-terminal domains (with the exception of EIIA-man lacking the C-terminal domain). In fact by forming a Rossmann-type crossover connection, the three-layer αβα sandwich fold of the EDD domain appears quite frequently in the PDB database, and was even predicted prior to the crystal structure determination based on another Rossmann-type fold, flavodoxin (Markovic-Housley et al. 1994). To determine the degree of structural similarity between these folds and to help resolve existing classification schemes, we chose to compare each structure to a nonredundant (90% sequence identity) library of existing structures (as described in Methods). This library of 8653 representative structures contained two DegV representatives (1mgp and 1pzx), two Dak representatives (1oi2 and 1un8), and a single EIIA-man representative (1pdo).
As shown in Table 1, the top five hits (ordered by Z-score) for each EDD domain-containing query correspond to representative structures of the three families under consideration. The next best hits in each case are different for each query and belong to different SCOP folds: The EIIA-man query finds a structure belonging to the periplasmic binding protein-like I fold, the Dak query (1un8) finds a structure belonging to the isocitrate dehydrogenase, the Dak query (1oi2) finds a structure belonging to the chorismate mutase-like fold, and the two DegV queries find NifK structures belonging to the chelatase-like fold. Only one of these next best hits belongs to the tubulin family (chorismate mutase-like fold 1jff). Thus, each query structure finds all EDD domain-containing structures before finding any other structure, despite the common occurrence of this type of fold in the database. EIIA-man, Dak, and DegV structures form a “closed structural group” and are closer to each other than to any other known protein structure. This property of structural similarity has previously been used to delineate SHS2 domains (Anantharaman and Aravind 2004) and is suggestive of EDD domain monophyly.
Table 1.
Structural similarity search
| Hit | |||
| Query PDB | PDB | Family | DaliLite Z-score |
| 1pdo | 1pdo | EllA-Man | 29.5 |
| 1pzx | DegV-like | 9.8 | |
| 1un8 | Dak | 7.5 | |
| 1oi2 | Dak | 7.2 | |
| 1mgp | DegV-like | 6.8 | |
| 1dp4 | PBP-like | 6.4 | |
| 1oi2 | 1oi2 | Dak | 65.4 |
| 1un8 | Dak | 42.1 | |
| 1pzx | DegV-like | 16.1 | |
| 1mgp | DegV-like | 15.8 | |
| 1pdo | EllA-Man | 7.2 | |
| 1jff | Tubulin | 4.6 | |
| 1un8 | 1un8 | Dak | 64.1 |
| 1oi2 | Dak | 42.1 | |
| 1pzx | DegV-like | 16.7 | |
| 1mgp | DegV-like | 15.5 | |
| 1pdo | EllA-Man | 7.5 | |
| 1itw | IDH | 5.9 | |
| 1mgp | 1mgp | DegV-like | 50.3 |
| 1pzx | DegV-like | 31.8 | |
| 1oi2 | Dak | 15.2 | |
| 1un8 | Dak | 14.4 | |
| 1pdo | EllA-Man | 5.9 | |
| 1m1n | NifK | 3.6 | |
| 1pzx | 1pzx | DegV-like | 47 |
| 1mgp | DegV-like | 32.6 | |
| 1un8 | Dak | 16.5 | |
| 1oi2 | Dak | 15.1 | |
| 1pdo | EllA-Man | 7.1 | |
| 1mio | Nifk | 3.4 | |
Each structure hit from the PDB90 library is named according to SCOP database classification at the family level.
Fro each query structure, the top five hits are listed first, then the next-best hit is listed. Top five hits are the same for all queries and contain EDD domain, but the next best hits differ.
One structural feature that helps distinguish the EDD fold is the presence of a C-terminal “helical hairpin” as first described for EIIA-man (Nunn et al. 1996). The antiparallel configuration of these two helices helps establish the orientation of the Dak and DegV C-terminal domains, whose first strand directly follows the second helix of the hairpin. Interestingly, EIIA-man compensate for the loss of this C-terminal domain by forming a swapped dimer mediated by a strand C-terminal to the helical hairpin (Fig. 2B ▶, gray). Topologically, the tubulin structure also retains these two helices. However, the second helix of this fold falls perpendicular to the preceding helix, and appears to associate more closely with the C-terminal domain (Fig. 2D ▶, pink). This association is accompanied by a significant rotation of the C-terminal domain with respect to the other folds.
The evolutionary linking of EDD domain containing families justified by both sequence and structure methods leads to a more precise definition of the core fold, which includes an αβα sandwich with a four-stranded, parallel β-sheet (order 2134) surrounded by two α-helices (2 and 3) on one side and three on the other (1, 4, and 5). Each EDD domain family possesses various insertions with respect to the core fold. As previously discussed, the EIIA-man core β-sheet (Fig. 1B ▶) is extended by one β-strand positioned antiparallel to the rest (order 21345) that establishes the swapped dimer. An N-terminal extension of the Dak EDD domain (Fig. 1A ▶) also extends the core β-sheet by two β-strands in the opposite direction of the EIIA-man extension. Alternatively, the DegV EDD domain (Fig. 1C ▶) contains a small domain insertion (three-stranded antiparallel β-sheet with α-helix) that covers the lipid-bound active site.
Genomic neighborhood and domain organization of Dak homologs
The subset of EIIA-man sequences (EIIA-dak) that find the Dak N-terminal domain using PSI-BLAST function in the same PTS pathway as Dak. In bacteria these two proteins are often components of a single operon (Gutknecht et al. 2001) found in a tandem array and can be linked by genomic neighborhood analysis (string database score 0.95 of highest confidence) (von Mering et al. 2003). Such close proximity in bacterial genomes supports their emergence from a duplication event that includes the loss (or gain) of the Dak C-terminal domain, especially considering the PSI-BLAST-detected link between these two families.
The similarities between Dak and DegV extend beyond their EDD domains and include similar C-terminal domains that contribute to the active sites of each family. In contrast to the N-terminal EDD domain, this C-terminal domain (Fig. 1 ▶, green and purple) forms a distinctive fold with a topology that is not often found in the PDB. Both the ubiquitous presence of this domain and its unusual topology provide additional support for homology between these two families. Additionally, some members of both the DegV and the Dak families are fused to yet another domain (Dak2). This domain is responsible for binding ATP in the C. freundii Dak structure (1un9; Siebold et al. 2003a,b), and its presence in DegV may provide clues to its function.
Functional implications of Dak classification
While EIIA-man belongs to the PTS system responsible for coupling the import and phosphorylation of sugars in bacterial cells, Dak functions to produce the glycolytic intermediate dihydroxyacetone phosphate. The phosphoryl donor to EIIA-man is HPR from EI of the PTS system (Robillard and Broos 1999), and the phosphoryl donor for Dak is either ATP-bound to a fused Dak2 domain or the EIIA-dak PTS system (Gutknecht et al. 2001). Although these general biological functions differ, the molecular reactions EIIA-man and Dak use to carry out these functions share some common features. Each of the proteins uses a buried aspartic acid (Asp67 in EIIA-man and Asp119 in Dak EDD domain) to form hydrogen bonds with their phosphoryl group acceptor, an invariant histidine residue (His10) in EIIA-man (Nunn et al. 1996) and a Dihydroxyacetone molecule covalently bound to an invariant histidine (His270) in the Dak C-terminal domain (Siebold et al. 2003a,b). Each structure also contains a third spatially conserved serine residue near the phosphoryl acceptor (Ser72 in EIIA-man and Ser60 in Dak EDD domain) that may participate in catalysis. Accordingly, the presence of His10 and Ser72 are essential for EIIA-man activity (Stolz et al. 1993; Nunn et al. 1996).
Despite a preserved spatial location of these residues in the two structures (Fig. 1 ▶, residues in bonds representation), the side chains are contributed by nonidentical positions in the multiple sequence alignment (Fig. 2 ▶, black highlights). Such migration of active-site residues has been noted in many homologous folds (Todd et al. 1999; Kinch and Grishin 2002). Although the positions of these active-site residues are not invariant in the EDD folds, their high degree of conservation within the respective families supports their functional importance. SCOPmap motif conservation scores detected the presence of a common active site in Dak and EIIA-man, despite the noted migration of active-site residues. Two major motifs (Fig. 2 ▶) contribute to the conservation score. The first motif includes residues in the first β-strand and in the loop surrounding the EIIA-man phosphoryl acceptor histidine residue (His10). In both families this loop forms the active site and contains several conserved small residues. The invariant EIIA-man histidine aligns with an invariant Glycine in the Dak family, whose presence probably allows room for substrate binding. The second motif includes residues in the loop connecting the third strand and helix of the EDD fold. This loop includes the EIIA-man active-site residues (Asp67 and Ser72) and the Dak active-site residue (Asp119).
Although the function of DegV remains unknown, the active site of the protein can be assumed based on the presence of several invariant residues near the bound palmitate (Fig. 1C ▶). This active-site placement agrees with the established evolutionary link to other EDD domain-containing families. Similar to the active site Asp residues found in EIIA-man and Dak, a conserved DegV serine residue (discussed in Sequence-based support for Dak classification) forms a hydrogen bond with the head of the lipid. The side chain of another conserved DegV residue (Thr60), which is located in the small inserted domain unique to this family, forms an additional hydrogen bond with the bound lipid. The role of this Thr residue is mimicked by a conserved histidine (His66) in Dak. Finally the side chain of a conserved histidine residue from the C-terminal domain (His270) falls in an identical structural position to the Dihydroxyacetone-bound C-terminal domain Histidine of Dak, although contributed from a different loop.
Considering the active-site makeup of DegV and its evolutionary relationship to other EDD domain-containing proteins, a tempting speculation for the function of this unknown family of proteins is phosphoryl transfer. Both the Nɛ2 position of His270 and the carboxylate of the palmitate are surface-exposed and provide potential phosphoryl acceptor sites. The presence of Dak2 domains fused to some DegV family members provides additional support for hypothesis, as Dak2 is required for the phosphotransfer reaction to Dihydroxyacetone of Dak (Gutknecht et al. 2001; Siebold et al. 2003a,b).
Materials and methods
Identifying potential distant homologs with SCOPmap
The evolutionary relationship between Dak and EIIA-man was first suggested by SCOPmap results for the Dak query structure (PDB ID 1oi2). SCOPmap determines the appropriate SCOP superfamily for a query protein by identifying homologs among a library of representative domains that have known SCOP superfamily assignments. The library used for this initial SCOPmap job was based on a previous version of SCOP (v1.63), as these results were then being used as an evaluation of SCOPmap performance.
In cases where hits from multiple superfamilies correspond to the same domain of a query protein, SCOPmap attempts to choose only one correct superfamily assignment. Consequently, distant yet correct evolutionary relationships may be disregarded in favor of closer homologs. In order to evaluate more remote evolutionary relationships with SCOPmap, the assignment strategy was modified to ignore any hits found to library domains belonging to the same SCOP superfamily as the query protein. This modified program was used to identify potential homologs of Dak (PDB IDs 1oi2 and 1un8). The library used for these SCOPmap jobs was based on SCOP v1.65.
Sequence similarity searches
To detect sequence homologs of each family, we searched the nonredundant database (nr, Jul 9, 2004; 1,918,886 sequences, filtered for low complexity regions) with PSI-BLAST (Altschul and Koonin 1998) using query sequences (gi|1827887 range 1..135 for EIIA-Man; gi|49176086 range 51..198 for Dak; and gi|42543340 range 1..161 for DegV) with defined parameters (E-value threshold 0.005, maximum 20 iterations). Found homologs were grouped using linkage clustering (score of 1 bit per site threshold, about 50% identity), and representative sequences from each group were used as new queries for subsequent rounds of PSI-BLAST. The iterations were repeated until no new sequences were detected. We used the COG database (http://www.ncbi.nlm.nih.gov/COG/; Tatusov et al. 2003) to define orthologous groups of the detected sequences, the PFAM database (http://pfam.wustl.edu/index.html; Bateman et al. 2004) to define additional domains, and the STRING database (http://string.embl.de/; von Mering et al. 2003) to evaluate genomic neighborhood.
Multiple sequence alignments and Euclidian space mapping
We constructed multiple sequence alignments of EDD domains from detected groups (corresponding to COG2376 for Dak, COG2893 for EIIA-man, COG3412 for EIIA-dak, COG3933 for transcription antiterminators, and COG1307 for DegV) using PCMA (Pei et al. 2003) with manual adjustments. The multiple alignments of each group were merged into a global alignment using structure superpositions, secondary-structure predictions (JPRED server; Cuff et al. 1998), hydrophobicity patterns, and paired BLAST hit alignments as guides. The global multiple sequence alignment was used as input for Euclidian space mapping using the previously described formula (Dij = 1/Uij − 1) for distance calculation (Grishin and Grishin 2002). Groups are colored according to the most stable configurations in the mapping procedure.
Structural similarity searches
DaliLite (Holm and Park 2000) was used to determine the closest structural neighbors of Dak, EIIA-man, and DegV representative structures in a library of PDB structures. Clustering at 90% sequence identity of all protein chains (minimum length of 20 amino acids) included in the PDB as of July 20, 2004 was obtained at ftp://ftp.rcsb.org/pub/pdb/derived_data/NR. A library of representative chains (8653 representatives) was assembled from the best representative of each cluster, which is defined as the chain with rank 1 in the cluster. Comparison of each query chain (Dak, EIIA-man, and DegV) with each library chain was performed by DaliLite (Holm and Park 2000). For each query, all pairwise comparisons were then ranked in order of descending Z-score to determine the closest structural neighbors.
Acknowledgments
This work was supported in part by the NIH grant GM67165 to N.V.G.
Abbreviations
Dak, dihydroxyacetone kinase
EIIA-man, mannose transporter IIA domain superfamily
PEP, phosphoenolpyruvate
PTS, PEP, sugar phosphotransferase system
HPR, histidine-containing phosphoryl carrier protein
ATP, adenosine triphosphate
DegV, DegV-like protein fold
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041114805.
References
- Altschul, S.F. and Koonin, E.V. 1998. Iterated profile searches with PSI-BLAST—a tool for discovery in protein databases. Trends Biochem. Sci. 23 444–447. [DOI] [PubMed] [Google Scholar]
- Anantharaman, V. and Aravind, L. 2004. The SHS2 module is a common structural theme in functionally diverse protein groups, like Rpb7p, FtsA, GyrI, and MTH1598/TM1083 superfamilies. Proteins 56 795–807. [DOI] [PubMed] [Google Scholar]
- Bateman, A., Coin, L., Durbin, R., Finn, R.D., Hollich, V., Griffiths-Jones, S., Khanna, A., Marshall, M., Moxon, S., Sonnhammer, E.L., et al. 2004. The Pfam protein families database. Nucleic Acids Res. 32 D138–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheek, S., Qi, Y., Krishna, S.S., Kinch, L.N., and Grishin, N.V. 2004. SCOP-map: Automated assignment of protein structures to evolutionary superfamilies. BMC Bioinformatics 5 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuff, J.A., Clamp, M.E., Siddiqui, A.S., Finlay, M., and Barton, G.J. 1998. JPred: A consensus secondary structure prediction server. Bioinformatics 14 892–893. [DOI] [PubMed] [Google Scholar]
- Esnouf, R.M. 1999. Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr. D Biol. Crystallogr. 55 938–940. [DOI] [PubMed] [Google Scholar]
- Grishin, V.N. and Grishin, N.V. 2002. Euclidian space and grouping of biological objects. Bioinformatics 18 1523–1534. [DOI] [PubMed] [Google Scholar]
- Gutknecht, R., Beutler, R., Garcia-Alles, L.F., Baumann, U., and Erni, B. 2001. The dihydroxyacetone kinase of Escherichia coli utilizes a phosphoprotein instead of ATP as phosphoryl donor. EMBO J. 20 2480–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm, L. and Park, J. 2000. DaliLite workbench for protein structure comparison. Bioinformatics 16 566–567. [DOI] [PubMed] [Google Scholar]
- Hu, K.Y. and Saier Jr., M.H., 2002. Phylogeny of phosphoryl transfer proteins of the phosphoenolpyruvate-dependent sugar-transporting phosphotransferase system. Res. Microbiol. 153 405–415. [DOI] [PubMed] [Google Scholar]
- Kinch, L.N. and Grishin, N.V. 2002. Evolution of protein structures and functions. Curr. Opin. Struct. Biol. 12 400–408. [DOI] [PubMed] [Google Scholar]
- Markovic-Housley, Z., Balbach, J., Stolz, B., and Genovesio-Taverne, J.C. 1994. Predicted topology of the N-terminal domain of the hydrophilic subunit of the mannose transporter of Escherichia coli. FEBS Lett. 340 202–206. [DOI] [PubMed] [Google Scholar]
- Murzin, A.G., Brenner, S.E., Hubbard, T., and Chothia, C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247 536–540. [DOI] [PubMed] [Google Scholar]
- Nunn, R.S., Markovic-Housley, Z., Genovesio-Taverne, J.C., Flukiger, K., Rizkallah, P.J., Jansonius, J.N., Schirmer, T., and Erni, B. 1996. Structure of the IIA domain of the mannose transporter from Escherichia coli at 1.7 Å resolution. J. Mol. Biol. 259 502–511. [DOI] [PubMed] [Google Scholar]
- Pei, J., Sadreyev, R., and Grishin, N.V. 2003. PCMA: Fast and accurate multiple sequence alignment based on profile consistency. Bioinformatics 19 427–428. [DOI] [PubMed] [Google Scholar]
- Robillard, G.T. and Broos, J. 1999. Structure/function studies on the bacterial carbohydrate transporters, enzymes II, of the phosphoenolpyruvate-dependent phosphotransferase system. Biochim. Biophys. Acta. 1422 73–104. [DOI] [PubMed] [Google Scholar]
- Schulze-Gahmen, U., Pelaschier, J., Yokota, H., Kim, R., and Kim, S.H. 2003. Crystal structure of a hypothetical protein, TM841 of Thermotoga maritima, reveals its function as a fatty acid-binding protein. Proteins 50 526–530. [DOI] [PubMed] [Google Scholar]
- Siebold, C., Arnold, I., Garcia-Alles, L. F., Baumann, U., and Erni, B. 2003a. Crystal structure of the Citrobacter freundii dihydroxyacetone kinase reveals an eight-stranded alpha-helical barrel ATP-binding domain. J. Biol. Chem. 278 48236–48244. [DOI] [PubMed] [Google Scholar]
- Siebold, C., Garcia-Alles, L.F., Erni, B., and Baumann, U. 2003b. A mechanism of covalent substrate binding in the x-ray structure of subunit K of the Escherichia coli dihydroxyacetone kinase. Proc. Natl. Acad. Sci. 100 8188–8192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz, B., Huber, M., Markovic-Housley, Z., and Erni, B. 1993. The mannose transporter of Escherichia coli. Structure and function of the IIABMan subunit. J. Biol. Chem. 268 27094–27099. [PubMed] [Google Scholar]
- Tatusov, R. L., Fedorova, N.D., Jackson, J.D., Jacobs, A.R., Kiryutin, B., Koonin, E.V., Krylov, D.M., Mazumder, R., Mekhedov, S.L., Nikolskaya, A.N., et al. 2003. The COG database: An updated version includes eukaryotes. BMC Bioinformatics 4 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd, A.E., Orengo, C.A., and Thornton, J.M. 1999. Evolution of protein function, from a structural perspective. Curr. Opin. Chem. Biol. 3 548–556. [DOI] [PubMed] [Google Scholar]
- von Mering, C., Huynen, M., Jaeggi, D., Schmidt, S., Bork, P., and Snel, B. 2003. STRING: A database of predicted functional associations between proteins. Nucleic Acids Res. 31 258–261. [DOI] [PMC free article] [PubMed] [Google Scholar]