

Abstract
Growth-limiting stresses in bacteria induce the general stress response to protect the cells against future stresses. Energy stress caused by starvation conditions in Bacillus subtilis is transmitted to the σB transcription factor by stress-response regulators. RsbP, a positive regulator, is a phosphatase containing a PAS (Per-ARNT-Sim) domain and requires catalytic function of a putative α/β hydrolase, RsbQ, to be activated. These two proteins have been found to interact with each other. We determined the crystal structures of RsbQ in native and inhibitor-bound forms to investigate why RsbP requires RsbQ. These structures confirm that RsbQ belongs to the α/β hydrolase superfamily. Since the catalytic triad is buried inside the molecule due to the closed conformation, the active site is constructed as a hydrophobic cavity that is nearly isolated from the solvent. This suggests that RsbQ has specificity for a hydrophobic small compound rather than a macromolecule such as RsbP. Moreover, structural comparison with other α/β hydrolases demonstrates that a unique loop region of RsbQ is a likely candidate for the interaction site with RsbP, and the interaction might be responsible for product release by operating the hydrophobic gate equipped between the cavity and the solvent. Our results support the possibility that RsbQ provides a cofactor molecule for the mature functionality of RsbP.
Keywords: α/β hydrolase superfamily, energy stress, stress response, σ factor, PAS domain, cavity, phenylmethanesulfonyl fluoride (PMSF), catalytic triad
In Bacillus subtilis, a wide variety of growth-limiting stresses induce the transcription of the general stress response regulon to adapt itself to the presence of the stresses or to repair the damage caused by them (for review, see Hecker and Volker 1998, Price 2001). This regulon comprises more than 100 genes under the control of the σB transcription factor. Growth limitation by carbon, phosphate, or oxygen starvation causes energy stress (Price 2001) (or nutritional stress [Zhang and Haldenwang 2003]), resulting in release of σB from RsbW by the partner-switching mechanism as shown in Figure 1 ▶. Energy stress is accompanied by a decrease of cellular ATP concentration, and subsequently a decline of RsbW’s kinase activity (Delumeau et al. 2002). As a result, population of the unphosphorylated RsbV increases and the partner switching is induced. In contrast, RsbP induces the partner switching by dephosphorylating RsbV (Vijay et al. 2000). RsbP contains a PAS (Per-ARNT-Sim) domain at its N-terminal region (Vijay et al. 2000) that is expected to be responsible for energy stress sensing (Price 2001).
Figure 1.

The energy stress response regulation pathway. RsbW binds the σB transcription factor in the unstressed state. Growth-limiting stress triggers the activation of phosphatases RsbP (energy stress) or RsbU (environmental stress [not shown]) to dephosphorylate RsbV. RsbW changes its binding partner to RsbV and sets σB free. This partner switching is controlled by the tension between the kinase RsbW and the phosphatases RsbP and RsbU (Price 2001). RelA, known as a (p)ppGpp synthetase, is also reported to be a component of this pathway (Zhang and Haldenwang 2003).
The rsbQ gene is cotranscribed with rsbP from the rsbQP operon (Brody et al. 2001). The sequence of RsbQ suggests that it belongs to the α/β hydrolase superfamily with a putative catalytic triad composed of Ser96, His247, and Asp219. Yeast two-hybrid analysis revealed that RsbQ interacts with RsbP. Although the catalytic function of RsbQ as a putative hydrolase has yet to be elucidated, mutating either Ser96 or His247 abolishes the energy stress response in vivo. Brody et al. (2001) proposed the following model: RsbP is expressed as an inactive form and converted to the active form by RsbQ that modifies either RsbP or its potential cofactor. Here, we determined the native and inhibitor-bound crystal structures of RsbQ, and we discuss the molecular mechanism of RsbQ functioning as a potential modifier for RsbP.
Results
Crystallography
Bacillus subtilis RsbQ was overexpressed in Escherichia coli in a tag-fused form, and the tag was removed during the purification step. The purified protein consists of the full-length RsbQ protein except for the first four residues (Met1–Ala4), and six plasmid-derived residues at the N terminus. The estimated molecular mass derived from a gel filtration experiment is 32 kDa (data not shown). Although several α/β hydrolases homologous with RsbQ have been reported to form homo-oligomers such as a trimer (Hecht et al. 1994) or octamer (Nandhagopal et al. 2001), the estimated mass is consistent with a monomer’s calculated mass of 30.2 kDa.
We solved two crystal structures of RsbQ: the native structure solved up to 2.5 Å resolution, and the one reacted with phenylmethanesulfonyl fluoride (PMSF), a widely used serine protease inhibitor, solved up to 2.6 Å resolution. As a result of the reaction, a PMS group covalently attaches itself to the Oγ atom of a serine side chain (Fig. 2A ▶). The structures were solved by a molecular replacement method employing an ensemble model (see Materials and Methods). An asymmetric unit contains two RsbQ molecules, referred to as chain A and chain B. Crystallographic statistics are shown in Table 1. The catalytic nucleophile, Ser96, is positioned in a generously allowed region in the Ramachandran plot in both the native and the PMS-bound structures. This is common among the α/β hydrolases and is due to the formation of a sharp turn, called the “nucleophile elbow,” that pushes the nucleophile outward away from the hydrolase domain (Nardini and Dijkstra 1999; see below). The temperature factors are quite high, especially for the chain B atoms. The average temperature factors calculated for the main chain atoms of the native structure are 40 Å2 for chain A and 60 Å2 for chain B. This is probably due to a relatively high solvent content of the crystal (65%) and poor crystal packing. The buried surface area contributing to intermolecular contacts in the crystal is 2392 Å2 for chain A, whereas it is 1433 Å2 for chain B, which occupies only 13% of the total molecular surface of the RsbQ molecule.
Figure 2.
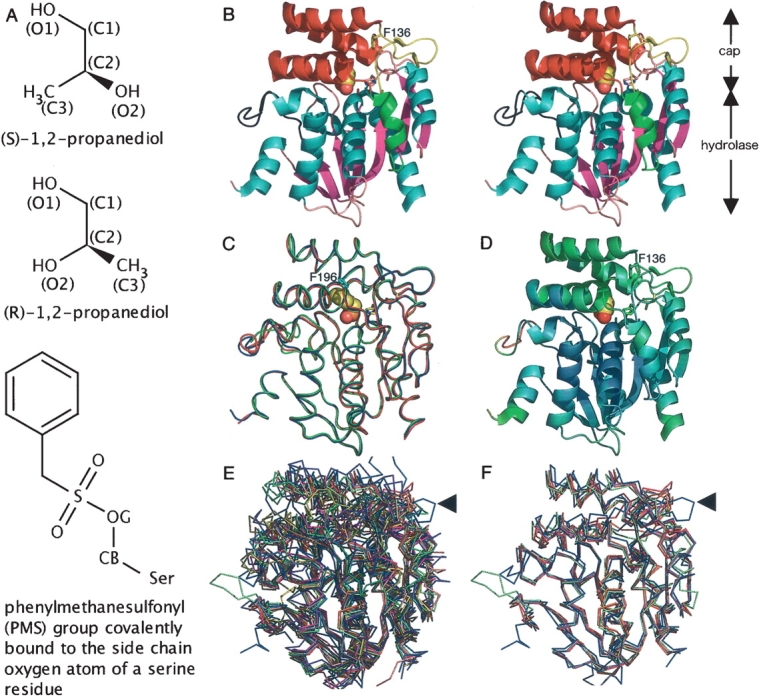
Overall structure and structural comparison with other α/β hydrolases. (A) Chemical structures of propylene glycol isomers and a phenylmethanesulfonyl (PMS) group covalently bound to the serine Oγ atom (denoted as OG). (B) Stereo side view of the native RsbQ (chain A). The propylene glycol molecule at the active site is shown in sphere representation. (C) Comparison of a native RsbQ molecule (chain A, blue) with the other molecule in the asymmetric unit (chain B, red) as well as with a PMS-bound molecule (chain A, green). The PMS group is shown in sphere representation. Phe196 is shown in cyan. (D) Ribbon representation of the native chain A molecule colored according to the temperature factors from blue (low) to red (high). (E) Twelve superposed structures. RsbQ is shown in blue. The unique loop region of RsbQ is indicated with an arrow. (F) RsbQ superposed with four cofactor-free haloperoxidases. Color usage is same as that in E.
Table 1.
Crystallographic statistics
| Crystal (PDB ID) | Native (1WOM) | PMS-bound (1WPR) |
| Data collection statistics | ||
| Space group | P212121 | P212121 |
| Unit cell (Å) | ||
| a = | 77.5 | 77.2 |
| b = | 82.4 | 81.0 |
| c = | 137.4 | 136.9 |
| Resolution range (Å) | 31.23–2.50 (2.59–2.50)a | 35.86–2.60 (2.69–2.60) |
| Number of unique reflections | 31,002 | 26,989 |
| Average redundancy | 7.05 (7.21) | 6.83 (6.93) |
| Completeness | 0.995 (100.0) | 0.995 (0.998) |
| Rmerge | 0.064 (0.333) | 0.061 (0.343) |
| I/σ (I) | 6.3 (1.9) | 7.0 (1.9) |
| Refinement statistics | ||
| Rcryst | 0.217 | 0.231 |
| Rfreeb | 0.262 | 0.292 |
| Number of protein residues | 271 | 271 |
| Number of molecules | ||
| Water | 148 | 170 |
| Malonic acid | 1 | 0 |
| Cryo-protectant | 12 | 4 |
| PMS group | 0 | 2 |
| Root-mean-square deviations from ideal values | ||
| Bond length (Å) | 0.006 | 0.011 |
| Bond angle (°) | 1.20 | 1.50 |
| Dihedral angle (°) | 22.60 | 22.90 |
| Improper angle (°) | 0.80 | 0.98 |
| Average temperature factor (Å2) | ||
| Main-chain protein atoms | 50.0 | 55.0 |
| Side-chain protein atoms | 52.0 | 56.2 |
| Water molecules | 48.5 | 52.0 |
| PMS group | 70.9 | |
| Other heterocompounds | 66.7 | 76.7 |
| Ramachandran plot (%) | ||
| In most-favored regions | 89.8 | 85.2 |
| In additionally allowed regions | 9.6 | 14.2 |
| In generously allowed regions | 0.6 | 0.7 |
| In disallowed regions | 0 | 0 |
a Data for high-resolution bins are in parentheses.
b Each Rfree factor was calculated for a randomly chosen 5% of the total reflections that was not included in the refinement.
Overall structure of RsbQ
A ribbon representation of the RsbQ structure is shown in Figure 2B ▶. It consists of an α/β hydrolase domain and a cap domain. The secondary structure elements contributing to the conserved “canonical” α/β hydrolase fold (Heikinheimo et al. 1999; Nardini and Dijkstra 1999) are colored in cyan and magenta for α helices and β strands, respectively. The cap domain is drawn using two colors: yellow for a loop region containing the short antiparallel β sheet and red for the region consisting of four α helices. The catalytic triad residues, Ser96, Asp219, and His247, are positioned in the hydrolase domain and are covered by the cap domain, limiting the access to the active site from the solvent. Electron density corresponding to a loop region (Fig. 2B ▶, shown in black) was weak or not visible, especially for chain B. No significantly large structural difference was found between the two RsbQ molecules in the asymmetric unit, as well as between the native and the PMS-bound RsbQ molecules, except for the poorly defined loop region (Fig. 2C ▶). Temperature factor distribution shows relatively high perturbation for the cap domain (Fig. 2D ▶). To compare the structure of RsbQ with those of other proteins belonging to the α/β hydrolase superfamily, we chose 11 proteins that have high sequence identities with RsbQ from a BLAST (Altschul et al. 1997) search result against the sequence data set deposited in the Protein Data Bank (Berman et al. 2000) (see Materials and Methods for the 11 PDB IDs). Sequence identities between RsbQ and each of the 11 proteins are between 13% and 24%. Multiple structural alignment trials were performed on an alignment server, MASS (Dror et al. 2003; Shatsky et al. 2004). These 11 structures and the structure of RsbQ are well superposed on one another for the “canonical” hydrolase domain, whereas the structures corresponding to the cap domain and an α helix (shown in green in Fig. 2B ▶) of RsbQ are very diverse (Fig. 2E ▶). Root-mean-square deviation of the 12 structures is 1.4 Å for 126 Cα atoms from each structure. Among them, four cofactor-free haloperoxidases (1BRO: Hecht et al. 1994; 1A88, 1A8S, and 1A8Q: Hofmann et al. 1998) are superposable especially well with RsbQ. Root-mean-square deviation of these five structures is 1.2 Å for 219 Cα atoms, including the α helices in the cap domain and the noncanonical α helix (Fig. 2F ▶). This structural similarity with the haloperoxidases may imply the possibility of a catalytic function for RsbQ. The loop region (arrowed in Fig. 2E,F ▶), however, is unique to RsbQ. This loop protrudes from the other superposed α/βhydrolase structures.
The catalytic triad
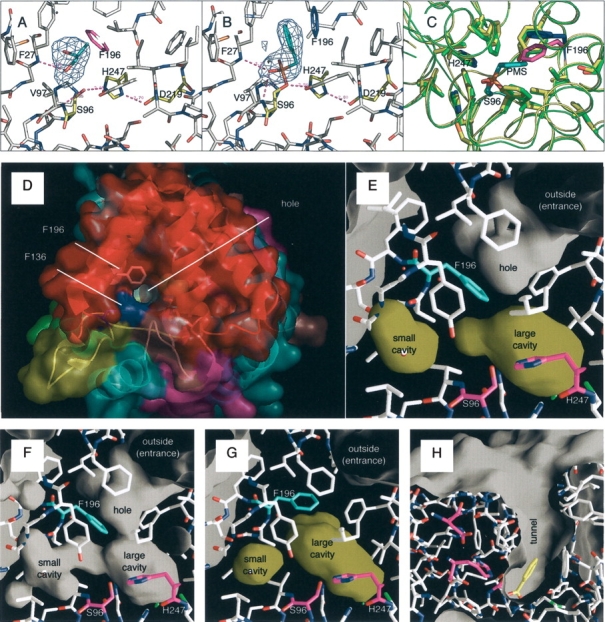
Figure 3A ▶ shows the active site of the native RsbQ molecule. The catalytic triad consists of Ser96, His247, and Asp219. Brody et al. (2001) did not make the conclusion that Asp219 is one of the triad residues from their sequence alignment results, because the nearby residue is also an aspartic acid residue, Asp218. Our structure now clearly shows that the acidic residue is Asp219. The nucleophile, Ser96, forms two hydrogen bonds, one connecting the Oγ atom of itself with the side chain nitrogen atom Nɛ of His247, and the other with the backbone amino nitrogen atom of Val97 (Fig. 3A ▶). This result is consistent with other α/β hydrolase structures: the backbone nitrogen atom of the residue following the nucleophile contributes to the formation of an oxyanion hole that stabilizes the negatively charged nucleophile during the transition state (Nardini and Dijkstra 1999). The native RsbQ captures a propylene glycol molecule supplemented as cryoprotectant (see Materials and Methods). The O1 atom of the molecule is connected with the backbone nitrogen atom of Phe27 by a hydrogen bond. Figure 3B ▶ shows the active site structure of the PMS-bound RsbQ. The PMS group is covalently bound to the nucleophile, thus completely inhibiting its activity. In this structure, two hydrogen bonds connect the backbone nitrogen atoms of either Val97 or Phe27 with an oxygen atom of the PMS group. These results suggest that the backbone nitrogen atom of Phe27 may function as a site for substrate binding. A major structural difference caused by the binding of the PMS group is seen in a large movement of Phe196 (Fig. 3C ▶). Steric repulsion with the PMS group pushes the residue outward, although the backbone φ, ψ angles do not differ largely.
Figure 3.
The active site cavity. (A) A propylene glycol captured in the active site of the native RsbQ. Fo–Fc omit map of the molecule countered at 3 σ is also shown here. The catalytic triad is shown in yellow. Phe196 is shown in magenta. (B) A PMS group covalently bound to the Ser96 side chain. Fo–Fc omit map of the group countered at 3 σ is also shown. Phe196 is shown in blue. (C) Comparison of the native and PMS-bound structures. The native and PMS-bound RsbQ structures are shown in green and yellow, respectively. The corresponding colors for Phe196 are magenta and blue, respectively. (D) The top view of the native RsbQ surface. Molecular coloring is in accordance with that in Figure 2B ▶ except Phe136 and Phe196, which are colored here in blue and white, respectively. (E) The inside of the native RsbQ calculated with a probe radius of 1.4 Å. Cavities are colored yellow. Ser96 and His247 are colored magenta; Phe196 is cyan. In (E–G), each molecule is illuminated from the inside. (F) The inside of the native RsbQ calculated with a probe radius of 1.2 Å. (G) The inside of the PMS-bound RsbQ calculated with a probe radius of 1.4 Å. (H) The molecular surface of Streptomyces aureofaciens haloperoxidase (PDB ID 1A8U). A benzoate molecule is caught at the bottom of the tunnel. The catalytic triad residues are colored magenta.
The hole and the active site cavity
A hole can be seen on the cap domain surface (Fig. 3D ▶). It does not, however, reach the catalytic triad buried beneath the cap domain. Figure 3E ▶ shows the inside of the native RsbQ molecule drawn with a probe radius of 1.4 Å. The bottom of the hole is lined with hydrophobic residues including Phe196, providing a hydrophobic gate. Two cavities, a large cavity and a small cavity, are found near the active site. The propylene glycol molecule is trapped in the large cavity. By drawing with a probe radius of 1.2 Å, corridors connecting these cavities with the hole can be seen (Fig. 3F ▶). This suggests that only a molecule as small as a water molecule can squeeze into the cavities. Obviously, the corridors are too narrow for a molecule such as a propylene glycol or a PMSF to go through. Figure 3G ▶ shows the cavities in the PMS-bound RsbQ by removing the PMS group from calculation, with a probe radius of 1.4 Å. The PMS group is in the large cavity. The large cavity is bulkier than that of the native molecule, and Phe196 is pushed outward as shown in Figure 3C ▶. It is noted, however, that the movement of Phe196 does not result in opening the gate. The gate is still closed but the entrance hole has become much shallower by a rise of the gate position. This may suggest that the hydrophobic gate closes after trapping a substrate molecule, and the hole becomes shallower in order to reject any approaches of other molecules during the catalytic reaction. On the contrary, each of the haloperoxidase structures used for structural comparison in Figure 2F ▶ has an open tunnel connecting the catalytic triad with the solvent outside to welcome its substrate molecules to come in (Fig. 3H ▶). Structural comparison between RsbQ and haloperoxidase molecules clearly shows that, in addition to the gate, Phe136 of RsbQ is also responsible for the narrow entrance (Fig. 3D ▶, shown in blue). In spite of the overall structural similarity of the cap domain, the substrate entrance of RsbQ is designed very differently from those of haloperoxidases.
Discussion
Many proteins with a diverse variety of functions are assigned to the α/β hydrolase superfamily. For example, a screening experiment by Sanishvili et al. (2003) identified that an E. coli α/β hydrolase, BioH (PDB ID 1M33), has carboxyesterase and thioesterase activities, as well as low lipase and aminopeptidase activities in vitro. Another example is a Pseudomonas fluorescens esterase. Pelletier and Altenbuchner (1995) found that this esterase is highly homologous with the haloperoxidases shown in Figure 2F ▶ and is actually bifunctional in vitro. Although our preliminary experiment shows that RsbQ also has esterase activity (T. Kaneko and T. Kumasaka, unpubl.), it is uncertain that this biochemical activity reflects its biological function in vivo.
The crystal structures of the native and PMS-bound RsbQ molecules strongly suggest that its potential substrate(s) is small and hydrophobic. Therefore, it is unlikely that RsbQ hydrolyzes the RsbP protein itself for its conversion to the active form. Rather, RsbQ may possibly provide a small molecule as the catalyzed product that is required for the function or folding of the active RsbP. As discussed by Brody et al. (2001), the RsbP PAS domain is a candidate that receives a product, or a cofactor, from RsbQ. Among PAS domains identified thus far, a subset of them bind a cofactor such as heme, FMN, or FAD (for review, see Gilles-Gonzalez and Gonzalez 2004). Although no cofactor has ever been identified for the RsbP PAS domain (Brody et al. 2001), our structural results greatly support the possibility that RsbP binds a cofactor for its activity.
To function as a hydrolase, RsbQ must take in at least two molecules: a substrate molecule and a water molecule used for hydrolysis. Our crystal structures, however, show that the active site of RsbQ is nearly isolated from the solvent, and the hydrophobic gate denies solvent molecules any easy access to the active site. Nevertheless, RsbQ caught a propylene glycol or a PMSF molecule during the experimental process. Thus, the gate is not always closed. Considering that the uptake of the propylene glycol must have happened during soaking of the crystal in the cryoprotectant solution at room temperature, the RsbQ molecules fluctuate between the open and closed forms even being packed in a crystal at room temperature. A high solvent content of the crystal may help preserve this mobility. All the same, the apparent structural differences from the haloperoxidases lead to the suggestion that RsbQ exhibits much higher specificity in substrate recognition than the haloperoxidases. Another speculation is that the gate may be equipped to hold the catalyzed product inside and release it only when RsbQ reaches its destination. Identification of a substrate of RsbQ in vivo will be instrumental to a proper understanding of the differences.
The yeast two-hybrid analysis revealed that RsbQ interacts with RsbP (Brody et al. 2001). Biological significance of the interaction is, however, unclear. Although there is no obvious interaction site on the structure of RsbQ, we propose the loop region of the cap domain (colored in yellow in Figs. 2B ▶, 3D ▶) as a possible interaction site. This region is unique among the α/β hydrolases used for comparison (Fig. 2E ▶). Once RsbP binds it, Phe136 that forms the narrow intake is expected to move (Fig. 3D ▶, in blue). The nearby α helices of the cap domain that possess the gate-forming residues such as Phe196 will also be perturbed. Here, an interpretation of the function of the closed gate may be made. Once RsbQ captures a substrate molecule, the gate closes from its flexible state to hold the molecule. Then RsbP attaches itself to the interaction site on RsbQ to open the gate. This enables RsbP to receive the catalyzed product from RsbQ. In this way, RsbP prompts RsbQ to provide a catalyzed product for RsbP itself. Although this interpretation is attractive, it is currently a speculative explanation. There is also the not yet studied, interesting question of whether RsbQ and RsbP can make a stable complex, or whether their interaction is transient as a part of the stress-response regulation. Our results provide fundamental information for further investigation into the interaction of RsbQ with RsbP as well as the regulation mechanism of energy stress response in Bacillus subtilis.
Materials and methods
Protein preparation
The rsbQ gene, corresponding to the region from Ile5 to Val269 of the RsbQ protein, was cloned from a single colony of Bacillus subtilis 168 strain by the colony-PCR method. The PCR product was inserted into the E. coli expression vector pTYB12 supplied in the IMPACT-CN system (New England BioLabs) with the SpeI and EcoRI sites. The tag-fused recombinant protein was overexpressed in E. coli ER2566 at 18°C overnight by induction with 0.5 mM β-D-1-thiogalactopyranoside. The cells were then collected and lysed by sonication in buffer A (20 mM HEPES-NaOH [pH 8.2] and 0.5 M NaCl) supplemented with 0.1% Triton-X 100. After loading on a chitin bead column (New England BioLabs), the column was washed with buffer A, and the tag fused to the protein was removed by on-column cleavage reaction with the buffer A supplemented with 50 mM dithiothreitol at 25°C for 2 d. The product cleaved from the tag was then eluted from the column with buffer A, dialyzed against 20 mM Tris-HCl (pH 8.0), and further purified with the anion exchange column MonoQ (Amersham Biosciences) in 20 mM Tris-HCl (pH 8.0) and an NaCl gradient. Preparation for RsbQ sample inhibited by PMSF (Fig. 2A ▶) was performed as follows. In the dialysis step described above, PMSF with a final calculated concentration of 10 mM, a concentration well beyond its solubility, was added into the dialysis buffer. The sample was dialyzed for 2 d at 4°C and further purified with the MonoQ column.
Crystallization and data collection
Both native and PMSF-inhibited RsbQ crystals were grown by the hanging-drop vapor diffusion method at 25°C within 2 d; 1.2 M sodium malonate was used as the precipitant for both samples. For collection of X-ray diffraction data at 100K, the native crystal was soaked in the precipitant solution containing 25% propylene glycol for several minutes at room temperature. The PMSF-inhibited crystal was soaked in the precipitant solution containing 20% glycerol. The data sets for the native crystal and the PMSF-inhibited crystal were collected at the beam line BL38B1 at SPring-8 and then processed with CrystalClear (Rigaku/MSC). The statistics for each data set are listed in Table 1.
Structure determination
Solving the crystal structure of RsbQ by the molecular replacement method was somewhat complicated, since none of the structures solved thus far shares a sufficiently high sequence identity with RsbQ. Finally, two RsbQ molecules were found in an asymmetric unit by means of the following procedure. We chose five structures as the molecular replacement search model candidates (PDB IDs 1M33, 1A88, 1BRO, 1C4X, and 1IUN). The sequence identity between RsbQ and each of the candidate proteins is between 19% and 24%. The diffraction data set from the PMSF-inhibited crystal was used. First, a structure prediction trial was performed on the I-SITES server (Bystroff and Shao 2002). It generated a predicted structure of RsbQ based on the coordinates of 1BRO (Hecht et al. 1994). However, molecular replacement trials using this predicted model failed. Therefore, an ensemble model was prepared in the following way. Coordinates of the five candidate proteins were superposed on one another to construct a “pseudo-NMR” model (Chen et al. 2000). This model was then applied to the molecular replacement trials with MolRep (Vagin and Teplyakov 1997). The result gave a solution corresponding to one molecule that could be assumed correct. The second molecule was uncertain at this stage. Therefore, model building with Xtal-View (McRee 1999) and refinement with CNS (Brunger et al. 1998) were performed for the first molecule with the help of the predicted structure described above. The refined structure was used as a search model for the molecular replacement trials to find the second molecule, which was done with EPMR (Kissinger et al. 1999). The calculated Matthews coefficient and solvent content are 3.5 and 65%, respectively. The structure of the native crystal was solved based on the PMSF-inhibited structure. Although we found 12 propylene glycol molecules in the asymmetric unit, it is impossible to distinguish between the two propylene glycol isomers since they have quite similar chemical structures (Fig. 2A ▶), and the resolution is also far from high enough to differentiate an oxygen atom from a carbon atom in the electron density map. Therefore, all the propylene glycol molecules are assigned as (s)-1,2-propanediol. Both structures were refined with XtalView and CNS. The refinement statistics for both structures are shown in Table 1. Molecular graphics were performed with Pymol (http://pymol.sourceforge.net), except for Figure 3 ▶, E–H, drawn with GRASP (Nicholls et al. 1991). The 11 PDB IDs whose structures were used for structural comparison in Figure 2E ▶ are as follows: 1BRO (bromoperoxidase; Hecht et al. 1994), 1A88, 1A8S, 1A8Q (chloroperoxidases; Hofmann et al. 1998), 1M33 (E. coli BioH; Sanishvili et al. 2003), 1C4X (Nandhagopal et al. 2001), 1EHY, 1Q0R, 1BN7, 1IUP, and 1J1I.
Data deposition
The atomic coordinates and structure factors have been deposited in the Protein Data Bank with PDB ID codes 1WOM and 1WPR for the native and PMS-bound structures, respectively.
Acknowledgments
We thank Masaki Yamamoto and Go Ueno for data collection at SPring-8 BL26B1, Ihsanawati for technical assistance, and Teh Aik Hong for useful discussion. This study was supported in part by a grant from the National Project on Protein Structural and Functional Analyses, the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Abbreviations
PAS domain, Per-ARNT-Sim domain
PMSF, phenyl-methanesulfonyl fluoride
FMN, flavin mononucleotide
FAD, flavin adenine dinucleotide
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041170005.
References
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. 2000. The Protein Data Bank. Nucleic Acids Res. 28 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody, M.S., Vijay, K., and Price, C.W. 2001. Catalytic function of an α/β hydrolase is required for energy stress activation of the σB transcription factor in Bacillus subtilis. J. Bacteriol. 183 6422–6428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR systems: A new software suite for macro-molecular structure determination. Acta Crystallogr. D. Biol. Crystallogr. 54 (Pt. 5) 905–921. [DOI] [PubMed] [Google Scholar]
- Bystroff, C. and Shao, Y. 2002. Fully automated ab initio protein structure prediction using I-SITES, HMMSTR and ROSETTA. Bioinformatics 18 (Suppl. 1) S54–S61. [DOI] [PubMed] [Google Scholar]
- Chen, Y.W., Dodson, E.J., and Kleywegt, G.J. 2000. Does NMR mean “not for molecular replacement”? Using NMR-based search models to solve protein crystal structures. Structure Fold. Des. 8 R213–R220. [DOI] [PubMed] [Google Scholar]
- Delumeau, O., Lewis, R.J., and Yudkin, M.D. 2002. Protein-protein interactions that regulate the energy stress activation of σB in Bacillus subtilis. J. Bacteriol. 184 5583–5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror, O., Benyamini, H., Nussinov, R., and Wolfson, H.J. 2003. Multiple structural alignment by secondary structures: Algorithm and applications. Protein Sci. 12 2492–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilles-Gonzalez, M.A. and Gonzalez, G. 2004. Signal transduction by heme-containing PAS-domain proteins. J. Appl. Physiol. 96 774–783. [DOI] [PubMed] [Google Scholar]
- Hecht, H.J., Sobek, H., Haag, T., Pfeifer, O., and van Pee, K.H. 1994. The metal-ion-free oxidoreductase from Streptomyces aureofaciens has an α/β hydrolase fold. Nat. Struct. Biol. 1 532–537. [DOI] [PubMed] [Google Scholar]
- Hecker, M. and Volker, U. 1998. Non-specific, general and multiple stress resistance of growth-restricted Bacillus subtilis cells by the expression of the σB regulon. Mol. Microbiol. 29 1129–1136. [DOI] [PubMed] [Google Scholar]
- Heikinheimo, P., Goldman, A., Jeffries, C., and Ollis, D.L. 1999. Of barn owls and bankers: A lush variety of α/β hydrolases. Structure Fold. Des. 7 R141–R146. [DOI] [PubMed] [Google Scholar]
- Hofmann, B., Tolzer, S., Pelletier, I., Altenbuchner, J., van Pee, K.H., and Hecht, H.J. 1998. Structural investigation of the cofactor-free chloroperoxidases. J. Mol. Biol. 279 889–900. [DOI] [PubMed] [Google Scholar]
- Kissinger, C.R., Gehlhaar, D.K., and Fogel, D.B. 1999. Rapid automated molecular replacement by evolutionary search. Acta Crystallogr. D. Biol. Crystallogr. 55 (Pt. 2) 484–491. [DOI] [PubMed] [Google Scholar]
- McRee, D.E. 1999. XtalView/Xfit—A versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 125 156–165. [DOI] [PubMed] [Google Scholar]
- Nandhagopal, N., Yamada, A., Hatta, T., Masai, E., Fukuda, M., Mitsui, Y., and Senda, T. 2001. Crystal structure of 2-hydroxyl-6-oxo-6-phenylhexa-2,4-dienoic acid (HPDA) hydrolase (BphD enzyme) from the Rhodococcus sp. strain RHA1 of the PCB degradation pathway. J. Mol. Biol. 309 1139–1151. [DOI] [PubMed] [Google Scholar]
- Nardini, M. and Dijkstra, B.W. 1999. α/β hydrolase fold enzymes: The family keeps growing. Curr. Opin. Struct. Biol. 9 732–737. [DOI] [PubMed] [Google Scholar]
- Nicholls, A., Sharp, K.A., and Honig, B. 1991. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11 281–296. [DOI] [PubMed] [Google Scholar]
- Pelletier, I. and Altenbuchner, J. 1995. A bacterial esterase is homologous with nonhaem haloperoxidases and displays brominating activity. Microbiology 141 (Pt. 2) 459–468. [DOI] [PubMed] [Google Scholar]
- Price, C.W. 2001. General stress response. In Bacillus subtilis and its closest relatives (eds. A.L. Sonenshein et al.), pp. 369–384. ASM Press, Washington, D.C.
- Sanishvili, R., Yakunin, A.F., Laskowski, R.A., Skarina, T., Evdokimova, E., Doherty-Kirby, A., Lajoie, G.A., Thornton, J.M., Arrowsmith, C.H., Savchenko, A., et al. 2003. Integrating structure, bioinformatics, and enzymology to discover function: BioH, a new carboxylesterase from Escherichia coli. J. Biol. Chem. 278 26039–26045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatsky, M., Dror, O., Schneidman-Duhovny, D., Nussinov, R., and Wolfson, H.J. 2004. BioInfo3D: A suite of tools for structural bioinformatics. Nucleic Acids Res. 32 W503–W507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin, A. and Teplyakov, A. 1997. MOLREP: An automated program for molecular replacement. J. Appl. Cryst. 30 1022–1025 [Google Scholar]
- Vijay, K., Brody, M.S., Fredlund, E., and Price, C.W. 2000. A PP2C phosphatase containing a PAS domain is required to convey signals of energy stress to the σB transcription factor of Bacillus subtilis. Mol. Microbiol. 35 180–188. [DOI] [PubMed] [Google Scholar]
- Zhang, S. and Haldenwang, W.G. 2003. RelA is a component of the nutritional stress activation pathway of the Bacillus subtilis transcription factor σB. J. Bacteriol. 185 5714–5721. [DOI] [PMC free article] [PubMed] [Google Scholar]