

Abstract
The Critical Assessment of PRedicted Interactions (CAPRI) experiment was designed in 2000 to test protein docking algorithms in blind predictions of the structure of protein–protein complexes. In four years, 17 complexes offered by crystallographers as targets prior to publication, have been subjected to structure prediction by docking their two components. Models of these complexes were submitted by predictor groups and assessed by comparing their geometry to the X-ray structure and by evaluating the quality of the prediction of the regions of interaction and of the pair wise residue contacts. Prediction was successful on 12 of the 17 targets, most of the failures being due to large conformation changes that the algorithms could not cope with. Progress in the prediction quality observed in four years indicates that the experiment is a powerful incentive to develop new procedures that allow for flexibility during docking and incorporate nonstructural information. We therefore call upon structural biologists who study protein–protein complexes to provide targets for further rounds of CAPRI predictions.
Keywords: protein–protein interaction, protein docking, structure prediction, conformation change
The Critical Assessment of PRedicted Interactions (CAPRI) is a community-wide experiment designed on the model of the Critical Assessment of Techniques for Protein Structure Prediction (CASP). Both are blind prediction experiments that rely on the willingness of structural biologists to provide unpublished experimental structures as targets. CASP evaluates fold prediction algorithms (Venclovas et al. 2003), CAPRI tests docking algorithms that predict the structure of protein–protein complexes. CASP targets are single proteins, and predictors are given amino acid sequences. CAPRI targets are protein–protein complexes, and the prediction starts from the three-dimensional structure of the component proteins (Janin et al. 2003; Wodak and Mendez 2004).
Macromolecular interaction is a central theme of functional genomics, subject to large-scale genetic and biochemical studies in model organisms. Most gene products, whether protein or RNA, perform their functions in cells by interacting with other gene products. In yeast, the set of macromolecular interactions—the interactome—is at least an order of magnitude larger than the set of gene products. In humans, there may be hundreds of thousands of physical interactions, either pair wise contacts or multicomponent assemblies of polypeptide chains, that have physiological and possibly medical relevance. These assemblies are poorly represented in the Protein Data Bank (PDB), and they are likely to remain so in coming years. Structural genomics programs are designed to determine X-ray/NMR structures on a genome scale for individual gene products, not for multicomponent assemblies. Specific research programs combining crystallography, NMR, and cryoelectron microscopy, are being set up to do that, but the sampling of the interactome is likely to remain sparse in coming years while the space of individual protein structures are being progressively filled. Thus, there is a good case for testing in silico methods that generate structural models of the assemblies by docking their components. In many cases, we cannot access their atomic structures by experiment. In all cases, reliable models will greatly help in designing experiments, but we need objective estimates of the model quality and of the performance of docking methods.
The targets of CAPRI
Protein–protein complexes are a subset of the interactome in which the components exist both in free and bound form. A complex taken from the PDB may be dissociated in the computer and reconstituted by docking the component molecules, still in their “bound” conformation. This may be useful as a test of the performance of a docking algorithm, but not of its predictive value: the answer is known in advance, conformational changes are ignored, “bound” docking is biased toward to correct answer by the exact complementarity of the two surfaces in contact. A realistic prediction must start with structural models of the free or “unbound” components. Cases where the PDB contains entries for both a complex and its free components are relatively few, and a selection of these is available to benchmark docking methods (Chen et al. 2003). The prediction of permanent assemblies such as oligomeric proteins, a closely related problem, cannot be assessed in this way because the subunit structures is not independently known, but some CAPRI targets were proteins which exist in two different oligomeric forms, one present in the PDB, the other to be predicted.
The ideal CAPRI target is the unpublished X-ray structure of a complex between two proteins of known structure, submitted for “unbound” docking prediction. Due to the paucity of those, “bound/unbound” complexes between a protein of known structure and a novel protein are also acceptable. In less than 4 years, 17 complexes have been submitted as targets, two to four at a time, in five rounds of predictions (Table 1). Five targets were “unbound,” the others, “bound/unbound.” On day zero of each round, component atomic coordinates are communicated to registered predictor groups who have a few weeks to submit models of the complexes to the http://capri.ebi.ac.uk Web site managed by K. Henrick at the European Bioinformatics Institute (Hinxton, UK). After that deadline, the CAPRI assessors (S. Wodak and R. Mendez, Free University of Brussels, Belgium) may start evaluating the models against the target X-ray structures.
Table 1.
CAPRI prediction rounds 2001–2004
| Rounda | Targets | Predictorsb | Submissionsc | |
| 1 | July–Sept. 2001 | 3 | 19 | 276 |
| 2 | Jan.–March 2002 | 4 | 16 | 281 |
| 3 | Jan. 6–Feb. 2, 2003 | 2 | 22 | 344 |
| 4 | Sept. 1–Oct. 12, 2003 | 4 | 25 | 820 |
| 5 | Jan. 19–March 21, 2004 | 4 | 29 | 850 |
a The results of Rounds 1 and 2 and the algorithms used by predictor groups were presented at the First CAPRI Evaluation Meeting held in La Londe des Maures (France) in September 2002, and published in a special issue of Proteins, Vol. 52, dated July 1, 2003.
b Registered predictor groups in Rounds 3–5 included the ClusPro (http://nrc.bu.edu/cluster) Web server.
c In Rounds 1–2, each group was allowed to submit five models of each target; in Rounds 3–4, 10 models were allowed.
Assessing docking predictions
A specific procedure was developed in early rounds of CAPRI to assess geometric and biological properties of the models (Mendez et al. 2003). It was approved at the First CAPRI Evaluation Meeting in September 2002, and used in subsequent rounds. For convenience, we shall call the larger component of a complex the receptor R, the other component, the ligand L. The R and L epitopes are the regions of the R and L surface that make the interaction, comprising all residues that have atoms less than 5 Å apart in the target X-ray structure. After least-square superposition of R in the model and target, the geometric quality of the model is judged from the RMS distance Lrms between Cα atoms of L in the model and target, and by the rotation angle θL and translation dL needed to further superimpose L. However, Lrms, θL and dL concern the whole ligand, often a large protein in CAPRI targets, and they may not represent the quality of the fit where it matters, that is, in the contact regions. Thus, another useful parameter in assessing the geometry of the model is the interface RMS distance Irms, calculated on Cα’s of the epitopes only.
The biological quality of the models was judged on the prediction of, first, the R and L epitopes, then, of the pairwise contacts between R and L residues. If the R epitope comprises NR residues in the target, nR of which make ligand contacts in the model, the ratio fR = nR/NR measures how well the model predicts the R epitope; its equivalent fL does the same for the L epitope. A good model should also say which residues of R are in contact with which residues of L. This is measured by the fraction of native contacts fnc = nc/Nc, where Nc is the number of residue pairs in contact in the target, and nc the number of those native contacts that are present in the model. Because nc can be artificially increased by pushing the ligand into the receptor, we also reject all models that have too many nonnative contacts: more than the average number in other models plus two standard deviations.
The parameters Irms, Lrms, and fnc were combined to rank models. In models of the “high-quality” and “good” categories, a majority of epitope residues and at least 30% of the contact pairs are correctly predicted (fnc > 0.3), and the L epitope is very close to its position in the X-ray structure (Irms < 2 Å). Such models reproduce the overall geometry and many biologically significant features of the interaction, but not necessarily its atomic details. Models with 10%–30% of the native contact pairs and Irms between 2 Å and 4 Å, are placed in the “acceptable” category. Although their geometry is poor, they should still be useful for site-directed mutagenesis and other experiments, because a large part of the epitopes must be correctly identified to yield fnc > 0.1.
Success and failure on CAPRI targets
Figure 1 ▶ represents a successful prediction of target T08, a complex between a fragment of laminin and domain G3 of nidogen (Takagi et al. 2003). The model shown here is at the limit of the “high-quality” and “good” categories. Three-quarters of the two epitopes and nearly half of the residue pairs in contact are correctly predicted, and the epitopes are at the right position in spite of a noticeable tilt relative to the X-ray structure. No attempt was made by the predictors to reproduce the conformation change at the N-terminus of the laminin fragment, but this change affects the contact only marginally. Nine predictor groups who use quite different docking algorithms submitted high- or good-quality models of T08. In contrast, the best models of target T01 were only “acceptable.” T01 is a complex of a bacterial protein kinase with its substrate protein HPr (Fieulaine et al. 2002). The kinase is a hexamer that binds six HPr molecules in a symmetrical way (only one is drawn in Fig. 2 ▶). In the best models, HPr is shifted by over 5 Å relative to the X-ray structure, and only 20% of the residue pairs are reproduced, a disappointing result given that the interaction could safely be assumed to involve the active site of the kinase and the phosphorylated Ser46 of HPr. The reason for this failure is apparent on Figure 2B ▶: in the X-ray structure, HPr binding implicates not one, but two kinase subunits, one through its active site as expected, the other through its C-terminal helix. The helix rotates in the complex to make with HPr a set of contacts that none of the groups predicted. A much better docking model was obtained when the C-terminal helix was left free to move, but that simulation could only be done “a posteriori” (Schneidman-Duhovny et al. 2003).
Figure 1.

Successful prediction of the laminin–nidogen complex (target T08). In T08, a fragment of laminin (entry 1KLO) was docked on the “bound” nidogen domain G3. Nidogen is drawn as a blue surface, laminin as a ribbon: green in the X-ray structure (Takagi et al. 2003), red in this “good-quality” docking model. Predicted epitope positions are within 1.1 Å of their correct position, and 47% of the residue–residue contacts are reproduced, even though the elongated laminin fragment is tilted relative to the X-ray structure and no attempt was made to reproduce the conformation change seen at the bottom end of the fragment. (Courtesy of Dr. Z. Weng, Boston University, Boston, MA.)
Figure 2.

A difficult target: the HPr kinase–HPr complex (target T01). In T01, HPr (entry 1SPH) was docked on HPr kinase (entry 1JB1). The kinase is a symmetrical hexamer that binds six HPr molecules. (A) The three subunits forming the top trimer are drawn as a surface in different shades of gray; a bound HPr is drawn as a ribbon, blue in the X-ray structure (Fieulaine et al. 2002), red in an “acceptable” docking model that reproduces 20% of the residue–residue contact. In that model, HPr is tilted 34° and 5 Å away from the X-ray structure. Conserved residues colored cyan on the kinase surface, are part of the HPr binding site. (Adapted from Ben-Zeev et al. 2003, with permission of the authors.) (B) The same trimer seen from the left; HPr at the active site of the blue kinase subunit also interacts with the C-terminal helix of the purple subunit drawn as a ribbon. The helix is green in the free kinase, purple in the complex. A simulation of its rotation yields the position drawn in gold; HPr then moves to the docking position in dark blue, closer to the crystal structure than in the original prediction. (Adapted from Schneidman-Duhovny et al. 2003, with permission from the authors.)
These two examples illustrate a major conclusion that was drawn from early rounds of CAPRI. Docking algorithms developed in the years 1990–2000 have often been reviewed (Smith and Sternberg 2002; Halperin et al. 2002; Valencia and Pazos 2002; Wodak and Janin 2002). All treat protein molecules as rigid bodies, allowing only for small changes like the surface side-chain rotations that occur in all complexes or the local movement seen in laminin. None of these algorithms can handle the larger movements seen in HPr kinase, or in targets T09 and T11 where the predictions also failed. T11, a complex between two components of the bacterial cellulosome (Carvalho et al. 2004), was “unbound.” The same complex was submitted as “bound/unbound” in T12, substituting the dockerin taken from the complex to the free molecule NMR structure used in T11. The two dockerins differed by more than 4 Å RMS, and the consequences on the prediction were obvious: a majority of the predictor groups produced high- or good-quality models of T12, but the best models of T11 were at the limit of “good” and “acceptable.”
Information from the literature could be used to guide docking in most cases. It was decisive in the case of T07, a complex between a bacterial superantigen and the T-cell receptor (Sundberg et al. 2002): a simple sequence homology search could locate a similar structure already in the PDB. In contrast, the fair amount of biochemical data that was available on HPr and the kinase in T01, or on dockerin and cohesin in T11, proved to be insufficient in the presence of conformation changes. Biochemical information frequently concerns the binding regions or residues, seldomly their pair-wise contacts or the geometry of the binding. Most docking procedures incorporate such information, either to limit the rigid-body search or to filter candidate solutions issued from the search. Experience shows that it is very useful, but not foolproof. Seven CAPRI targets were complexes of “unbound” protein antigens with “bound” antibody fragments (Table 2). Antibodies bind antigen through hypervariable loops of their VL and VH domains, which puts constraints on docking solutions. Five targets of this type have had good predictions: T02 and T03, two Fab complexes with large viral proteins, the T06 complex of α-amylase with the VHH domain of a camel single-chain antibody domain, T13, and T19. Predictors submitted models of these targets in which the epitope recognized by the antibody was correctly identified, with no help from the literature in several cases, and the geometry of binding was essentially right. In contrast, they failed entirely on two other complexes with α-amylase (T04–T05) in which the VHH domain makes a lateral contact that implicates frame-work residues, and only one hypervariable loop (Fig. 3 ▶). Conformation changes are not to be blamed here, for the antibody moiety was “bound” and the antigen essentially unchanged. “A posteriori” analysis indicates that the mode of binding seen in the X-ray structure either was outside the limits set to the search, or it had been rejected from the docking solutions as incompatible with established rules of antigen–antibody recognition.
Table 2.
Overview of the prediction quality for 17 CAPRI targets
| Model qualitya | |||||
| Target | Reference | Submitting groups | High/good | Acceptable | |
| T01 | HPr kinase/HPr | Fieulaine et al. 2002 | 16 | 0 | 8 |
| T02b | Rotavirus VP6/Fab | Thouvenin et al. 2001 | 15 | 1 | 6 |
| T03 | Flu hemagglutinin/Fab | Barbey-Martin et al. 2002 | 13 | 2 | 0 |
| T04 | Amylase/camel VHH | Desmyter et al. 2002 | 13 | 0 | 0 |
| T05 | Amylase/camel VHH | Desmyter et al. 2002 | 13 | 0 | 0 |
| T06 | Amylase/camel VHH | Desmyter et al. 2002 | 13 | 8 | 0 |
| T07c | Superantigen/TCRβ | Sundberg et al. 2002 | 14 | 12 | 8 |
| T08 | Nidogen/laminin | Takagi et al. 2003 | 18 | 11 | 18 |
| T09 | LicT dimmer | H. van Tilbeurgh and M. Graille, in prep. | 17 | 0 | 0 |
| T10 | TBE virus E trimer | Bressanelli et al. 2004 | 20 | 1 | 3 |
| T11 | Cohesin/dockerin (unbound) | Carvalho et al. 2004 | 19 | 0 | 13 |
| T12 | Cohesin/dockerin (bound) | Carvalho et al. 2004 | 22 | 20 | 10 |
| T13 | SAG1/Fab | M. Graille and F. Ducancel, in prep. | 21 | 5 | 8 |
| T14 | Phosphatase 1δ/MYPT1 | Terrak et al. 2004 | 25 | 38 | 32 |
| T15d | Colicin D/Imm D | Graille et al. 2004 | 10 | 9 | 6 |
| T18e | Xylanase/TAXI | Sansen et al. 2004 | 26 | 5 | 4 |
| T19 | Ovine prion/Fab | Eghiaian et al. 2004 | 24 | 11 | 9 |
a High-quality models must correctly predict over 50% of the residue pairs in contact (fnc > 0.5), and place either the whole ligand or its epitope to within 1 Å of its position in the X-ray structure (Lrms or Irms < 1 Å). Good models have fnc > 0.3 and Lrms < 5 Å or Irms < 2 Å; acceptable models have fnc > 0.1, and Lrms < 10 Å or Irms < 4 Å.
b An electron micrograph view of the complex had been published (Thouvenin et al. 2001).
c A complex involving homologous proteins was available in the PDB.
d Only submissions made before the Web publication of the X-ray structure were assessed.
e Targets T16 and T17 were cancelled.
Figure 3.
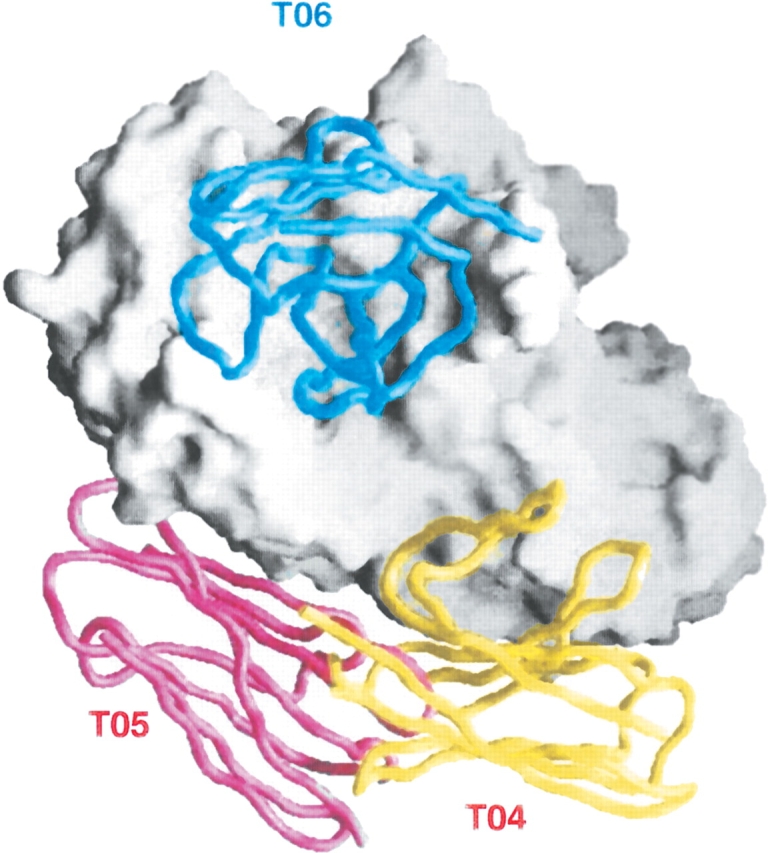
Success and failure on antigen–antibody complexes (targets T04–6). All three targets are complexes of porcine α-amylase (entry 1PIF) with the VHH domains of three different monoclonal camel antibodies (Desmyter et al. 2002). In T06, the VHH domain binds end on to the α-amylase active site. High/good-quality docking models of this target were submitted by several CAPRI predictor groups. Instead, prediction failed entirely on T04 and T05, where the VHH domain makes with the antigen a lateral contact that involves framework residues as well as the H3 hypervariable loop.
Progress in predictions
Table 2 is a summary of the results obtained on 17 CAPRI targets. It shows that high/good-quality predictions were made of all targets except the five mentioned above. Each individual predictor group missed many more than five targets, yet the prediction experiment has been a collective success. More important perhaps, there were three (collective) failures on the seven targets submitted in 2001–2002, but only two on the 10 targets of 2003–2004. Although each protein–protein complex has features of its own that make the docking prediction easy or difficult, significant progress has been achieved in less than three years. Part of it may be attributed to the participation in recent rounds of new predictor groups using novel algorithms, part to improved scoring functions, and part to the more efficient use of outside information (especially from sequences) when sorting out the many false positives that all docking algorithms generate. Thus, we are at the stage where structural predictions in silico can reliably be made in cases where the component structures are available and known to undergo no major changes upon association. We expect the docking of close homologues to behave in the same way, which will greatly expand the scope of the predictions, and intend to test that point extensively in further rounds of CAPRI.
Structural biologists, please help with targets!
The very raison d’être of CAPRI is to foster progress in prediction methods and performance. The improvement seen in the last two rounds is a strong incentive to go on with the experiment in coming years. To do that, we call upon the structural biologists who study protein–protein complexes by X-ray or NMR to provide new targets. When both components are of known structure, all we need to start a round of docking prediction is the two PDB entry codes. Alternatively, one component may be given as an entry code and the other as “bound” atomic coordinates communicated to us by the authors. In either case, a complete set of coordinates should later be forwarded to the CAPRI assessors; they will remain confidential until released by the authors or the PDB. Whether successful or not, CAPRI predictions will publicize the work of the structural biologists who submit targets, and help integrating the structural approach in the general field of protein–protein interaction.
Acknowledgments
The CAPRI Organizing Committee comprises Dr. Kim Henrick (Hinxton, UK), Joël Janin (LEBS-CNRS, Gif-sur-Yvette), John Moult (Rockville, MD), Lynn Ten Eyck (San Diego, CA), Michael Sternberg (London, UK), Sandor Vajda (Boston, MA), Ilya Vakser (Stony Brook, NY), Shoshana Wodak (Brussels, Belgium). We are grateful to all crystallographers who have provided targets: Dr. Sonia Fieulaine, Sylvie Nessler, Marcel Knossow, Carole Barbey, Frédéric Eghiaian, Herman van Tilbeurgh, Marc Graille (LEBS-CNRS, Gif-sur-Yvette, France), Felix Rey, Marie-Christine Vaney, Stéphane Bressanelli (LVMS-CNRS, Gif-sur-Yvette, France), Frederic Ducancel (CEA-Saclay, France), Christian Cambillau (AFMB, Marseille, France), Eric Sundberg, Roy Mariuzza (CARB, Rockville, MD), Timothy Springer (Harvard Medical School, Boston, MA), Maria Romao, Anna Luisa Carvalho (University of Lisbon, Portugal), Roberto Dominguez (Boston Research Institute, Boston, MA), Stefaan Sansen, Anja Rabijns (Leuwen University, Belgium). The figures in this paper are courtesies of Dr. Zhiping Weng (Boston University, Boston, MA), Miri Eisenstein (Weizmann Institute, Rehovot, Israel), and Haim Wolfson (Tel Aviv University, Israel).
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041081905.
References
- Barbey-Martin, C., Gigant, B., Bizebard, T., Calder, L.J., Wharton, S.A., Skehel, J.J., and Knossow, M. 2002. An antibody that prevents the hemagglutinin low pH fusogenic transition. Virology 294 70–74. [DOI] [PubMed] [Google Scholar]
- Ben-Zeev, E., Berchanski, A., Heifetz, A., Shapira, B., and Eisenstein, M. 2003. Prediction of the unknown: Inspiring experience with the CAPRI experiment. Proteins 52 41–6. [DOI] [PubMed] [Google Scholar]
- Bressanelli, S., Stiasny, K., Allison, S.L., Stura, E.A., Duquerroy, S., Lescar, J., Heinz, F.X., and Rey, F.A. 2004. Structure of a flavivirus envelope glycoprotein in its low pH-induced membrane fusion conformation. EMBO J. 23 728–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho, A.L., Dias, F.M.V., Prates, J.A.M., Nagy, T., Gilbert, H.J., Davies, G.J., Ferreira, L.M.A., Romao, M.J., and Fontes, C.M.G.A. 2004. Cellulosome assembly revealed by the crystal structure of the cohesin–dockerin complex. Proc. Natl. Acad. Sci. 100 13809–13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, R., Mintseris, J., Janin, J., and Weng, Z. 2003. A protein–protein docking benchmark. Proteins 52 88–91. [DOI] [PubMed] [Google Scholar]
- Desmyter, A., Spinelli, S., Payan, F., Lauwereys, M., Wyns, L., Muyldermans, S., and Cambillau, C. 2002. Three camelid VHH domains in complex with porcine pancreatic α-amylase. J. Biol. Chem. 277 23645–23650. [DOI] [PubMed] [Google Scholar]
- Eghiaian, F., Grosclaude, J., Lesceu, S., Debey, P., Doublet, B., Treguer, E., Rezaei, H., and Knossow, M. 2004. Insight into the PrPC → PrPSc conversion from the structures of antibody-bound ovine prion scrapie-susceptibility variants. Proc. Natl. Acad. Sci. 101 10254–10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieulaine, S., Morera, S., Poncet, S., Mijakovic, I., Galinier, A., Janin, J., Deutscher, J., and Nessler, S. 2002. X-ray structure of a bifunctional protein kinase in complex with its protein substrate HPr. Proc. Natl. Acad. Sci. 99 13437–13441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graille, M., Mora, L., Buckingham, R.H., van Tilbeurgh, H., and de Zamaroczy, M. 2004. Structural inhibition of the colicin D tRNase by the tRNA-mimicking immunity protein. EMBO J. 23 1474–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halperin, I., Ma, B., Wolfson, H., and Nussinov, R. 2002. Principles of docking: An overview of search algorithms and a guide to scoring functions. Proteins 47 409–443. [DOI] [PubMed] [Google Scholar]
- Janin, J., Henrick, K., Moult, J., Ten Eyck, L., Sternberg, M., Vajda, S., Wakser, I., and Wodak, S.J. 2003. CAPRI: A critical assessment of predicted interactions. Proteins 52 2–9. [DOI] [PubMed] [Google Scholar]
- Mendez, R., Leplae, R., De Maria, L., and Wodak, S.J. 2003. Assessment of blind predictions of protein–protein interactions: Current status of docking methods. Proteins 52 51–67. [DOI] [PubMed] [Google Scholar]
- Sansen, S., De Ranter, C.J., Gebruers, K., Brijs, K., Courtin, C.M., Delcour, J.A., and Rabijns, A. 2004. Structural basis for inhibition of Aspergillus niger xylanase by triticum aestivum xylanase inhibitor-I. J. Biol. Chem. 279 36022–36028. [DOI] [PubMed] [Google Scholar]
- Schneidman-Duhovny, D., Inbar, Y., Polak, V., Shatsky, M., Halperin, I., Benyamini, H., Barzilai, A., Dror, O., Haspel, N., Nussinov, R., et al. 2003. Taking geometry to its edge: Fast unbound rigid (and hinge-bent) docking. Proteins 52 107–112 [DOI] [PubMed] [Google Scholar]
- Smith, G.R. and Sternberg, M.J. 2002. Prediction of protein–protein interactions by docking methods. Curr. Opin. Struct. Biol. 12 28–35. [DOI] [PubMed] [Google Scholar]
- Sunberg, E.J., Li, H., Llera, A.S., McCormick, J.K., Tormo, J., Schlievert, P.M., Karjalainen, K., and Mariuzza, R.A. 2002. Structures of two streptococcal superantigens bound to TCR β chains reveals diversity in the architecture of T cell signalling complexes. Structure 10 687–699. [DOI] [PubMed] [Google Scholar]
- Takagi, J., Yang, Y., Liu, J.H., Wang, J.H., and Springer, T.A. 2003. Complex between nidogen and laminin fragments reveals a paradigmatic β-propeller interface. Nature 424 969–973. [DOI] [PubMed] [Google Scholar]
- Terrak, M., Kerff, F., Langsetmo, K., Tao, T., and Dominguez, R. 2004. Structural basis of protein phosphatase 1 regulation. Nature 429 780–784. [DOI] [PubMed] [Google Scholar]
- Thouvenin, E., Schoehn, G., Rey, F., Petitpas, I., Mathieu, M., Vaney, M.C., Cohen, J., Kohli, E., Pothier, P., and Hewat, E. 2001. Antibody inhibition of the transcriptase activity of rotavirus DLP: A structural view. J. Mol. Biol. 307 161–172. [DOI] [PubMed] [Google Scholar]
- Valencia, A. and Pazos, F. 2002. Computational methods for the prediction of protein interactions. Curr. Opin. Struct. Biol. 12 368–373. [DOI] [PubMed] [Google Scholar]
- Venclovas, C., Zemla, A., Fidelis, K., and Moult, J. 2003. Assessment of progress over the CASP experiments. Proteins 53 585–589. [DOI] [PubMed] [Google Scholar]
- Wodak, S. and Janin, J. 2002. The structural basis of macromolecular recognition. Adv. Protein Chem. 61 9–68. [DOI] [PubMed] [Google Scholar]
- Wodak, S.J. and Mendez, R. 2004. Predictions of protein–protein interactions: The CAPRI experiment, its evaluation and implications. Curr. Opin. Struct. Biol. 14 242–249. [DOI] [PubMed] [Google Scholar]