

Abstract
The viral serpin, crmA, is distinguished by its small size and ability to inhibit both serine and cysteine proteases utilizing a reactive loop shorter than most other serpins. Here, we characterize the mechanism of crmA inhibition of serine proteases and probe the reactive loop length requirements for inhibition with two crmA reactive loop variants. P1 Arg crmA inhibited the trypsin-like proteases, thrombin, and factor Xa, with moderate efficiencies (~102–104 M−1sec−1), near equimolar inhibition stoichiometries, and formation of SDS-stable complexes which were resistant to dissociation (kdiss ~10−7 sec−1), consistent with a serpin-type inhibition mechanism. Trypsin was not inhibited, but efficiently cleaved the variant crmA as a substrate (kcat/KM of ~106 M−1 sec−1). N-terminal sequencing confirmed that the P1 Arg–P1′Cys bond was the site of cleavage. Altering the placement of the Arg in a double mutant P1 Gly-P1′Arg crmA resulted in minimal ability to inhibit any of the trypsin family proteases. This variant was cleaved by the proteases ~10-fold less efficiently than P1 Arg crmA. Surprisingly, pancreatic elastase was rapidly inhibited by wild-type and P1 Arg crmAs (105–106 M−1sec−1), although with elevated inhibition stoichiometries and higher rates of complex dissociation. N-terminal sequencing showed that elastase attacked the P1′Cys–P2′Ala bond, indicating that crmA can inhibit proteases using a reactive loop length similar to that used by other serpins, but with variations in this inhibition arising from different effective P2 residues. These results indicate that crmA inhibits serine proteases by the established serpin conformational trapping mechanism, but is unusual in inhibiting through either of two adjacent reactive sites.
Keywords: serpin, protease, crmA, protease inhibitor, serine protease, mutagenesis
The serpin superfamily of proteins plays key roles in the physiology of all types of organisms ranging from viruses and bacteria to plants and humans, reflecting their early evolutionary origins (Silverman et al. 2001; Cole et al. 2004). Members of the family share a common tertiary structure consisting of a core domain of ~350 amino acids and function in most cases to regulate proteolytic enzymes in complex physiologic processes such as wound healing, host defense, and apoptosis (Gettins 2002). Serpins regulate the activity of proteolytic enzymes by acting as pseudosubstrates which initially bind their target enzymes but are induced by protease cleavage to undergo conformational changes which trap the enzymes in highly stable complexes. The conformational changes provide signals for initiating or shutting down biologic responses, and are the likely reason why serpins have evolved such a sophisticated mechanism of protease inhibition that differs from the simpler lock-and-key type mechanism used by other protein protease inhibitor families (Gettins 2002).
A particularly interesting member of the family is the cowpox virus serpin, cytokine response modifier A or crmA (Ray et al. 1992). This serpin efficiently inhibits both the cytotoxic T cell serine protease, granzyme B, as well as a number of caspase-type cysteine proteases. The latter proteases have proinflammatory and proapoptotic functions in common (Komiyama et al. 1994; Quan et al. 1995; Zhou et al. 1997). The inhibition of these proteases enables the virus to subvert the defensive cellular responses to infection and survive. The ability of crmA to inhibit both serine and cysteine proteases and the fact that it is the smallest known member of the serpin family (341 amino acids) make this serpin a valuable model for understanding the minimal structural requirements for serpin function and for providing insight into the unprecedented ability of this and other serpins to inhibit proteases of two distinct mechanistic classes (Komiyama et al. 1994; Björk et al. 1998; Schick et al. 1998).
While crmA has received much attention as a tool in apoptosis research, few studies have focussed on the structural determinants of crmA function. Insights into these determinants have recently come from the elucidation of the X-ray structure of reactive loop cleaved forms of crmA by two groups (Renatus et al. 2000; Simonovic et al. 2000). The structures have shown that the protein undergoes the conformational changes typical of inhibitory members of the family wherein cleavage in the exposed reactive center loop induces the loop to insert as a new strand into the center of the major β-sheet of the serpin, sheet A. This evidence, together with the ability to inactivate crmA function by a mutation known to block the serpin conformational change (Tewari et al. 1995), have suggested that crmA functions as a protease inhibitor by the same conformational trapping mechanism used by other serpins (Stratikos and Gettins 1999; Huntington et al. 2000). However, this mechanism was elucidated for serpins that inhibit serine proteases. No comparable mechanistic studies have been done for the reaction of crmA with its only known serine protease target, granzyme B, nor has it been shown whether this mechanism extends to the inhibition of the cysteine protease targets, the caspases, which do not form the characteristic SDS-stable complexes observed with serine proteases (Komiyama et al. 1994; Zhou et al. 1997). Finally, crmA differs from most other inhibitory serpins in having a shorter reactive loop, suggesting that crmA may have different reactive loop length requirements for its inhibitory function (Simonovic et al. 2000).
To further understanding of the structural determinants of crmA function and provide a thorough biochemical characterization of the mechanism of crmA inhibition of serine proteases, we have mutated the principal P1 specificity determining residue from Asp to Arg and altered the placement of this Arg residue in the reactive center loop. These mutations were designed to alter crmA specificity to trypsin-like proteases and thereby allow determination of whether crmA functions by the same mechanism of recognition and conformational trapping demonstrated for serpins that inhibit trypsin-like serine proteases. Our results indicate that crmA recognizes and inhibits serine proteases by the established serpin inhibitory mechanism. Moreover, they suggest that the unusual reactive loop length in crmA arises from the atypically small size of this serpin and its ability to trap proteases in SDS-stable complexes through recognition of two adjacent reactive sites. These studies validate crmA’s utility in understanding the minimal requirements for serpin inhibitory function and provide a foundation for studies of the mechanism by which crmA functions as an inhibitor of cysteine proteases.
Results
Reactivity of P1 Arg crmA with proteases
Mutation of the P1 Asp residue of crmA to Arg (Table 1) profoundly affected the reactivity of the serpin with proteases. The mutant crmA was unable to inhibit the usual target proteases of the wild-type serpin, granzyme B and caspase-1 (not shown) (Komiyama et al. 1994; Quan et al. 1995), but acquired the ability to inhibit the trypsin-like proteases, thrombin and factor Xa (Fig. 1 ▶). The stoichiometries of inhibition (SI) of thrombin and factor Xa by P1 Arg crmA were 3.3 ± 0.3 and 1.4 ± 0.1 mole inhibitor/mole protease, respectively. The greater than equimolar inhibition stoichiometries for reactions of the variant crmA with the two proteases suggested that a significant fraction of the variant escaped the conformational change-dependent trapping of the acyl-intermediate by completing a normal substrate reaction (Lawrence et al. 2000). SDS-PAGE analyses confirmed that the P1 Arg crmA reactions with thrombin and factor Xa resulted in the formation of SDS-stable high molecular weight complexes as well as cleaved crmA as products of the reaction (Fig. 2 ▶). The relative amounts of the two products were consistent with the SIs measured by activity assay. The kinetics of thrombin and factor Xa inhibition by P1 Arg crmA were analyzed under pseudo first-order conditions by discontinuous assay of residual protease activity after varying times of reaction. Protease activity decayed as a single exponential process to a zero-activity endpoint under these conditions (Fig. 3 ▶) and yielded observed pseudo first-order rate constants (kobs) that were proportional to the crmA concentration, consistent with simple second-order kinetic behavior. Apparent second-order rate constants of 300 ± 10 M−1sec−1 and 9300 ± 200 M−1sec−1 for thrombin and factor Xa inhibition, respectively, were obtained from these data. Correction for the different extents of substrate reaction of the P1 Arg crmA with the two proteases by multiplying the apparent rate constant by the SI (Hood et al. 1994) showed that the variant crmA inhibited factor Xa ~10-fold faster than thrombin. The stability of P1 Arg crmA–protease complexes was examined by forming complexes with a molar ratio of serpin to protease greater than the SI and then extensively diluting them into a reporter protease chromogenic substrate at several times KM to induce complex dissociation and block reassociation of liberated protease with excess serpin (Calugaru et al. 2001). These experiments were done at a higher temperature (37°C) because of the slow rates of complex dissociation. Dilution of the complexes caused a progressive increase in the rate of chromogenic substrate hydrolysis due to generation of active protease from the complex (Fig. 4A ▶). Initial rates of complex dissociation were measured from these progress curves that were proportional to the complex concentration (Fig. 4B ▶). Complex dissociation rate constants of 1.5 ± 0.1 × 10−7 sec−1 and 3.2 ± 0.2 × 10−7 sec−1 were determined for thrombin and factor Xa complexes with P1 Arg crmA, respectively, from the slopes of these linear plots.
Table 1.
Comparison of reactive loop sequences of wild-type and variant crmAs with antithrombin
| P4 | P3 | P2 | P1 | P1′ | P2′ | P3′ | P4′ | |
| Wild-type crmA | L | V | A | D | C ↓ | A | S | T |
| P1R crmA | L | V | A | R ↓ | C ↓ | A | S | T |
| P1G P1′R crmA | L | V | A | G | R ↓ | A | S | T |
| Antithrombin | I | A | G | R ↓ | S | L | N | P |
Reactive loop amino acids are designated by the single-letter code and numbered P1, P2, P3, etc., from the N-terminal side and P1′, P2′, P3′, etc., from the C-terminal side of the P1–P1′ reactive bond based on standard nomenclature. Principal cleavage sites of thrombin, factor Xa, and trypsin are designated by bold arrows and of elastase by unbolded arrows.
Figure 1.

Stoichiometry of P1 Arg crmA inhibition of thrombin and factor Xa. Reaction mixtures containing 2 μM thrombin (▴) or 0.2 μM factor Xa (•) and P1 Arg crmA at the indicated molar ratios were incubated for 5 h or 3 h, respectively, at 25°C and residual enzyme activity measured from the initial rate of cleavage of specific chromogenic substrates as described in Materials and Methods. Activities were normalized to those measured in the absence of inhibitor and plotted vs. the molar ratio of inhibitor to enzyme. Solid lines represent linear regression fits of the data.
Figure 2.

SDS-PAGE analysis of P1 Arg crmA reactions with proteases. (A,B) Thrombin (IIa) or factor Xa (1 μM) were incubated with varying molar ratios of P1 Arg crmA for 2 h or 30 min, respectively, followed by addition of SDS sample buffer and denaturing at 100°C prior to electrophoresis. CrmA alone (lanes 1,10), protease alone (lane 2), crmA/protease ratios of 0.5, 1, 1.5, 2, 2.5, 3, and 5 for thrombin reactions and 0.25, 0.5, 0.75, 1, 1.5, 2, and 5 for factor Xa reactions (lanes 3–9). (C) P1 Arg crmA (5 μM) was incubated with 100 nM trypsin for 1, 2, 4, 6, 8, 10, 15, 20, and 30 min (lanes 2–10) and then reactions quenched with 10 μM FFR-chloromethylketone prior to denaturation and electrophoresis. Untreated crmA is shown in lane 1. The rate of disappearance of the native crmA band (cmAN) and appearance of the cleaved crmA (crmAC) was analyzed by integration of band intensities.
Figure 3.

Kinetics of P1 Arg crmA inhibition of thrombin and factor Xa. Shown are the reactions of 0.5 (•) or 1 μM (○) P1 Arg crmA with 10 nM factor Xa or of 2.5 (▴) or 5 μM (▵) P1 Arg crmA with 10 nM thrombin. Residual enzyme activity was measured after the indicated reaction times by dilution into chromogenic substrates as in Figure 1 ▶. Activities are normalized to those measured in the absence of inhibitor. Solid lines are fits to a single exponential function with zero endpoint.
Figure 4.
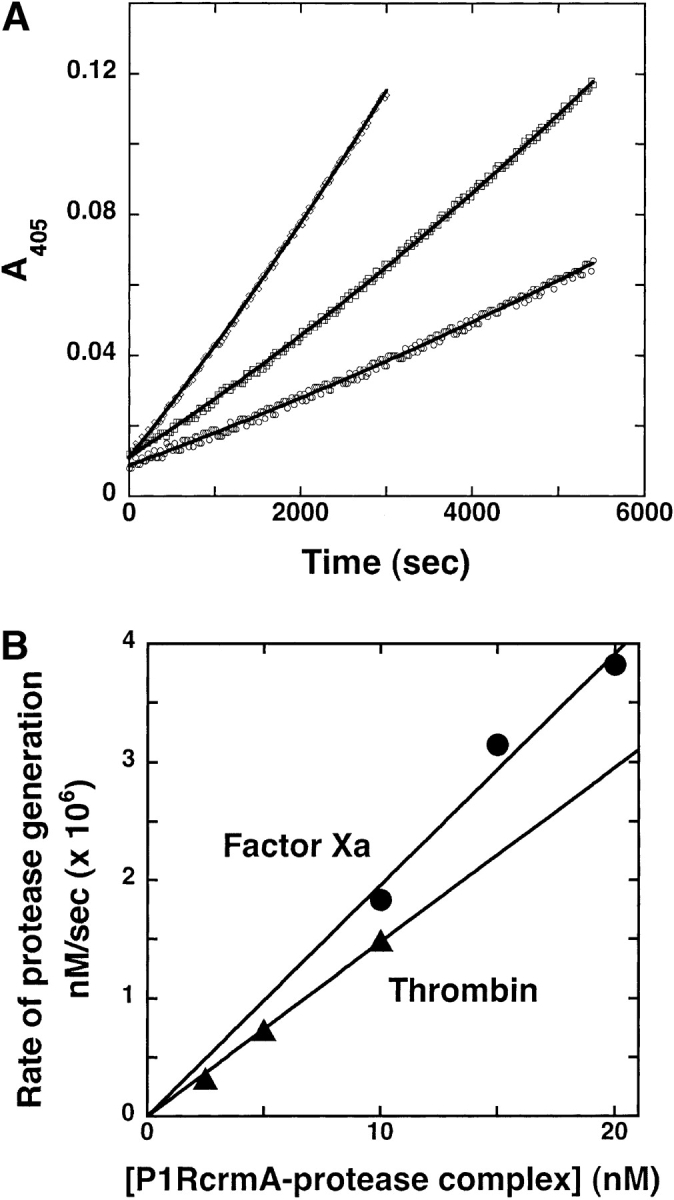
Kinetics of dissociation of P1 Arg crmA–thrombin complex. (A) P1 Arg crmA (12 μM) was mixed with 2 μM thrombin and incubated for 1 h at 25°C to form a complex. The complex was then diluted into 200 μM S-2238 substrate preincubated at 37°C to yield concentrations of 2.5 (○), 5 (□), and 10 nM (▵). Regeneration of protease from the complex was continuously monitored from the parabolic increase in absorbance at 405 nm. Solid lines are fits to the parabolic equation given in Materials and Methods. (B) Secondary plots of the fitted Δv/Δt parameter of the parabolic equation (converted to the rate of protease generation using the measured turnover number for substrate hydrolysis) are shown as a function of the complex concentration for both thrombin and factor Xa complexes, from which the first-order complex dissociation rate constant was determined.
Contrasting the reaction of P1 Arg crmA with thrombin and factor Xa, reaction of the variant crmA with β-trypsin resulted in no inhibition of proteolytic activity. However, SDS-PAGE analysis showed that the crmA variant reacted efficiently as a trypsin substrate, as evidenced by its rapid cleavage by substoichiometric levels of the enzyme (Fig. 2C ▶). Analysis of the kinetics of cleavage of 5 μM variant by 100 nM trypsin indicated an initial rate of ~0.3 μM crmA cleaved per minute. To determine the kinetic constants for the cleavage reaction, we analyzed the competitive effect of P1 Arg crmA on trypsin hydrolysis of the chromogenic substrate, S-2222 (Blake et al. 1989). P1 Arg crmA progressively reduced the rate of trypsin hydrolysis of the chromogenic substrate as the serpin concentration was increased, thereby increasing the time required for complete hydrolysis of the substrate (Fig. 5 ▶). The initial velocity of hydrolysis decreased in accordance with simple hyperbolic competitive inhibition and yielded a KM value of 0.078 ± 0.04 μM for the competitor crmA variant reaction with trypsin. The kcat for trypsin cleavage of the variant crmA was analyzed from the increase in the area under full progress curves for trypsin hydrolysis of the chromogenic substrate produced by the competitor crmA. A kcat value of 0.15 ± 0.06 sec−1 was determined from these data from which a kcat/KM of 1.9 ± 0.8 × 106 M−1 sec−1 could be calculated. N-terminal sequencing of the trypsin-treated crmA variant after removal of trypsin showed, in addition to the normal N-terminal sequence of MDIFR, a second sequence of CASTV that corresponded to cleavage at the P1 Arg–P1′ Cys bond in the reactive center loop (Table 1).
Figure 5.
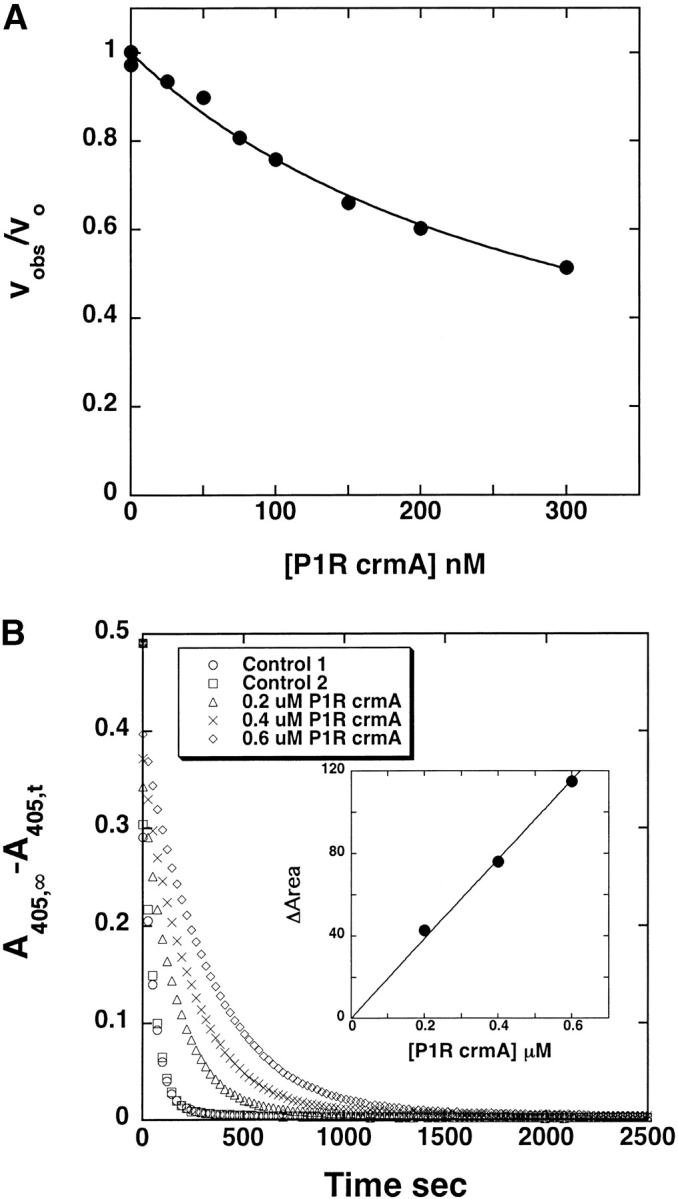
Determination of KM and kcat values for trypsin cleavage of P1 Arg crmA. (A) Dependence of the initial rate of cleavage of 100 μM S-2222 by 0.5 nM trypsin on the concentration of added P1 Arg crmA competitor substrate. The solid line represents the computer fit to the hyperbolic equation for inhibition of S-2222 hydrolysis by a purely competitive substrate interaction as given in Materials and Methods. (B) Full progress curves for the hydrolysis of 50 μM S-2222 by 5 nM trypsin in the absence and presence of the indicated concentrations of P1 Arg crmA with the ordinate axis depicting the disappearance of substrate. The inset shows the proportional dependence of the change in area under the curves measured in the presence and absence of competitor as a function of the competitor P1 Arg crmA concentration, from which the kcat for trypsin cleavage of P1 Arg crmA was determined as described in Materials and Methods.
Reactivity of P1 Gly-P1′ Arg crmA variant with proteases
The effect of moving the P1 arginine of the crmA variant one residue further from the P15 reactive loop hinge to produce an N-terminal reactive loop length similar to that found in most inhibitory serpins, i.e., 16 rather than 15 residues, was evaluated by preparing a double mutant crmA in which the P1 Asp was changed to Gly and the P1′ Cys was changed to Arg (Table 1). The reactivity of the P1 Gly-P1′ Arg crmA with trypsin family proteases differed from that of P1 Arg crmA. Reaction of the double mutant with thrombin, factor Xa, and β-trypsin resulted principally in cleavage of the variant by all proteases as a substrate, based on SDS-PAGE analyses (Fig. 6 ▶). However, inhibition of thrombin and factor Xa by the variant was detectable at high molar ratios of crmA variant (1–3 μM) to protease (ratios of 50–150 tested) and reaction times of 6 h, from which SI values >100 for the inhibition were estimated for both enzymes. Small amounts of complex bands were detectable on SDS-PAGE when high concentrations of enzyme and inhibitor were employed (Fig. 6 ▶). Unlike the P1 Arg variant, the P1 Gly-P1′ Arg crmA produced no detectable inhibition of the rate of trypsin cleavage of the chromogenic substrate, S-2222, at levels up to 0.5 μM, indicating a KM for cleavage much greater than 1 μM. Kinetic analysis of the initial rates of cleavage of the double mutant crmA by the three proteases showed that trypsin cleaved the variant crmA reasonably efficiently (kcat/KM = 2 ± 1 × 105 M−1 sec−1), factor Xa cleaved it significantly more slowly (kcat/KM = 1.1 ± 0.2 × 103 M−1 sec−1) and thrombin cleaved the variant even more slowly (kcat/KM = 6 ± 1 × 101 M−1 sec−1). Interestingly, the cleavage rates were in each case about an order of magnitude less than corrected kass or kcat/KM values determined for the P1 Arg crmA reactions with the same proteases, indicating a similar reduced recognition and/or rate of cleavage of the Arg in the P1′ position by the three proteases. N-terminal sequencing confirmed that trypsin cleaved the variant crmA at the P1′Arg–P2′Ala bond (Table 1).
Figure 6.

SDS-PAGE analysis of the reaction of P1 Gly-P1′Arg crmA with proteases. The variant crmA (5 μM) was incubated alone (lane 1), with 0.1 μM trypsin (lanes 3,4) 1 μM thrombin (lanes 6,7) or 1 μM factor Xa (lanes 9,10) for 5 or 30 min (left and right lanes of each pair) and then quenched with SDS sample buffer prior to heat denaturation and electrophoresis. Trypsin (1 μM), thrombin, or factor Xa control samples were run (lanes 2,5,8). The initial rate of crmA cleavage by these proteases was analyzed from the time-dependent appearance of the cleaved crmA band in separate gels in which 5 μM crmA was reacted with 1 nM trypsin, 5 μM thrombin or 0.1 μM factor Xa.
Reactivity of wild-type and variant crmAs with elastase
The presence of potentially elastase-cleavable Ala bonds adjacent to the P1–P1′ scissile bond in the reactive loop prompted us to investigate the reaction of wild-type and variant crmAs with porcine pancreatic elastase. Wild-type and P1 Arg crmAs but not the double mutant crmA were surprisingly found to inhibit porcine elastase activity (Fig. 7 ▶). The SI values for the inhibition were 4.7 ± 0.2 and 69 ± 4 moles inhibitor/mole protease for wild-type and P1 Arg variant crmAs, respectively, indicating that the inhibition was not efficient. SDS-PAGE analysis of the products of the reactions showed that SDS-stable high molecular weight complexes were formed in the reaction of wild-type and P1 Arg crmAs with elastase in addition to cleaved crmA as the predominant product (Fig. 8 ▶). The double mutant was cleaved by elastase as a substrate. The kinetics of elastase inhibition were studied using crmA concentrations at least five times the product of the SI and elastase concentration to achieve pseudo first-order conditions. Second-order rate constants, corrected for the fraction of crmA reacting through the substrate pathway, indicated reasonably efficient inhibition rate constants of 6.4 ± 0.4 × 105 M−1sec−1 and ~6 × 106 M−1 sec−1 for wild-type and P1 Arg crmA reactions with elastase. The former reaction appeared to obey second order kinetics, whereas the latter showed saturation behavior and yielded a poorly determined KM of 10 ± 10 nM, but well-determined limiting inhibition rate constant of 0.001 ± 0.0001 sec−1. Inducing dissociation of the crmA–elastase complexes by dilution into an elastase fluorogenic substrate at 37°C enabled measurement of the initial rates of dissociation of these complexes. Dissociation rate constants of 2.2 ± 0.1 × 10−6 sec−1 and 6.2 ± 0.7 × 10−5 sec−1 were determined for wild-type and P1 Arg crmA complexes indicating that the wild-type complex was considerably more stable than the mutant complex and that both complexes were less stable than P1Arg crmA complexes with thrombin and factor Xa. In contrast to our expectations, N-terminal sequencing of elastase-complexed or cleaved wild-type crmA showed that the primary cleavage was at the P1′Cys–P2′Ala reactive loop bond for both crmA species (Table 1). In keeping with this cleavage site being responsible for elastase inhibition, a crmA variant with the P1′ Cys changed to Ser poorly inhibited elastase activity and was mainly cleaved as a substrate like the double P1Gly–P1′Arg variant. These findings suggest that cleavage at the P1′Cys–P2′Ala bond leads to elastase inhibition whereas cleavage at other sites do not.
Figure 7.

Stoichiometry of wild-type and variant crmA inhibition of elastase. Porcine pancreatic elastase (50 nM) was incubated with the indicated molar ratios of wild-type (•) or P1 Arg (▴) crmAs for 1 h or 15 min, respectively. Residual elastase activity was then measured by 10-fold dilution into 50 μM succinyl-Ala-Ala-Ala-7-amido-4-methylcoumarin and monitoring the initial rate of substrate cleavage at 380 nm excitation and 440 nm emission wavelengths. Solid lines are linear regression fits of the data.
Figure 8.

SDS-PAGE analysis of crmA-elastase reactions. Elastase (5 μM) was incubated with wild-type (lanes 3,4), P1 Arg (lanes 6,7), and P1 Gly-P1′Arg (lanes 9,10) crmAs (5 μM) for 5 min and 30 min and reactions quenched with SDS sample buffer before heat denaturation and electrophoresis. Elastase alone (lane 1) and wild-type (lane 2), P1 Arg (lane 5), and P1 Gly-P1′ Arg (lane 8) crmAs alone.
Discussion
CrmA is the smallest known member of the serpin superfamily and one of few serpins that can inhibit both serine and cysteine proteases. Its compact size results mainly from a shorter A helix and the complete absence of the D helix (Simonovic et al. 2000). In the present study we altered crmA specificity to enable a thorough biochemical characterization of the mechanism of crmA inhibition of trypsin-type serine proteases and of the unusual reactive loop length requirements for this inhibition. The latter is of particular interest since crmA differs from most other inhibitory serpins in having a reactive loop that is one residue shorter between the scissile bond and the N-terminal hinge (Simonovic et al. 2000). In keeping with the well-established role of the P1 residue of serpins in determining target protease specificity (Huber and Carrell 1989; Gettins 2002), changing the crmA P1 residue from the wild-type Asp to Arg abolished the reactivity of crmA with its normal target proteases, granzyme B and caspase-1. These proteases are known to have a strict specificity for a negatively charged P1 Asp (Thornberry et al. 1997; Harris et al. 1998) resulting from a positively charged residue at the base of the S1 pocket (Harris et al. 1998), although P2–P4 residues also make significant contributions to recognition specificity (Thornberry et al. 1997; Zhou et al. 1997; Ekert et al. 1999). Replacing the P1 Asp with an Arg residue produced the anticipated change in the serpin’s specificity from P1 Asp to P1 Arg-specific proteases. The variant crmA efficiently inhibited factor Xa, less efficiently inhibited thrombin and was unable to inhibit trypsin. The different inhibition efficiencies reflect varying degrees of reaction of the variant serpin as a substrate of these proteases. The association rate constants for either inhibitor or substrate reactions of P1 Arg crmA with the three proteases are compatible with the extended P4–P3′ sequence of the variant serpin reactive center loop (Table 1) and the known extended sequence specificities of these proteases (Harris et al. 2000; Bianchini et al. 2002). The reactivity with factor Xa and trypsin approaches that of the best synthetic substrates, in keeping with crmA P1 and P2 residues being compatible with the minimal specificity requirements of these proteases, whereas the lower reactivity with thrombin is understandable given the deviations of P2 and P1′ residues of crmA from optimal residues for this protease (Harris et al. 2000). α1PI and antichymotrypsin variants in which P1 Met or Leu were changed to Arg exhibited similar transformations of their specificity from neutrophil elastase and cathepsin G to thrombin, factor Xa and trypsin, but with the difference that all the latter proteases were inhibited by the variant serpins (Travis et al. 1986; Djie et al. 1996).
Our studies provide compelling evidence that crmA employs a normal serpin mechanism for inhibition of serine proteases. In this mechanism the serpin traps the protease at the acyl-intermediate stage of cleaving the inhibitor as a regular substrate by undergoing a major conformational change (Lawrence et al. 1995; Stratikos and Gettins 1999; Huntington et al. 2000; Peterson and Gettins 2001). P1 Arg crmA forms high molecular weight SDS-stable complexes with thrombin and factor Xa like those formed with wild-type crmA and the target serine protease, granzyme B (Quan et al. 1995). This is a diagnostic feature of the trapped serpin–protease acyl-intermediate complex (Lawrence et al. 1995). Moreover, the observed half-lives for dissociation of the variant crmA–protease complexes of ~50 days at 37°C suggest that the acyl-intermediates have been stabilized to an extent comparable to that of other serpin complexes with serine proteases (Calugaru et al. 2001). Attempts to analyze the stability of the wild-type–granzyme B complex under similar conditions showed no detectable complex dissociation, indicating an even longer half-life for this complex. The marked stability of the acyl-intermediate arises from the remarkable conformational trapping mechanism utilized by serpins to inhibit proteases. In this mechanism the cleavage and acylation of the reactive loop by the protease induces the N-terminal fragment of the loop to insert into the center of β-sheet A of the serpin and drag the covalently tethered protease to the opposite serpin pole where steric constraints force the protease to be deformed and inactivated (Stratikos and Gettins 1999; Huntington et al. 2000; Peterson and Gettins 2001). The ability of crmA to undergo this reactive loop conformational change which is necessary to stabilize the acyl-intermediate is clear from two reported X-ray structures of cleaved crmA which show a striking resemblance to structures of other cleaved inhibitory serpins (Renatus et al. 2000; Simonovic et al. 2000). The inability of P1Arg crmA to trap trypsin in a stable acyl-intermediate complex and to instead be cleaved by the protease is in keeping with the alternative inhibition and substrate pathways available for reaction of a protease with a serpin in the serpin inhibitory mechanism (Lawrence et al. 2000). Thus, a substrate reaction will dominate when the rate of deacylation of the acyl-intermediate exceeds the rate of the serpin conformational change which traps the intermediate. A similar preferred reaction of other P1 Arg variant serpins as substrates of trypsin (Djie et al. 1996) suggests that trypin’s deacylation rate approaches the rate of the serpin conformational change with other serpins as well.
The reactive loop length requirements for crmA to function as an inhibitor are compatible with a typical serpin inhibitory mechanism and are also indicative of an unusual ability of this serpin to inhibit proteases through two adjacent reactive sites (Potempa et al. 1988). This was evident from the effects of increasing the reactive loop length between the N-terminal hinge and the P1 residue to the 16 residues found in most other inhibitory serpins by placing an Arg one residue more C-terminal from the normal P1 position (Huber and Carrell 1989). None of the trypsin-type proteases were significantly inhibited by the loop-lengthened crmA variant, indicating that the longer loop did not allow the acyl-intermediate to be sufficiently stabilized to prevent deacylation. That a longer reactive loop is still compatible with protease inhibition, however, was shown by the finding that wild-type and P1 Arg variant crmAs, but not the P1 Gly P1′-Arg crmA variant, was able to inhibit pancreatic elastase by cleavage at the P1′–P2′ Cys–Ala bond. The inhibition by wild-type crmA was reasonably fast, although a substrate reaction was preferred. An SDS-stable complex was formed and the stability of the complex under native conditions was consistent with a marked stabilization of the acyl-intermediate complex. A substrate reaction was preferred even more by the P1 Arg crmA variant, but the net rate of inhibition corrected for the substrate reaction was still rapid and the complexes formed were considerably stabilized, although not to the extent seen with complexes formed by protease attack at the P1–P1′ bond. These findings are in keeping with elastase reaction at the crmA P1′– P2′ bond causing protease inhibition by a normal serpin mechanism. Our observation that the trapping of elastase by wild-type and P1 Arg crmAs is more efficient with an Asp versus an Arg residue in what is effectively the P2 position for the reactive Cys–Ala bond suggests that the nature of the P2 residue may account for the poor trapping of trypsin-like proteases by the P1 Gly–P1′Arg crmA. Gly is in the effective P2 position in the latter variant, and consequently, may lack the necessary side chain to stabilize the complex sufficiently when the distal loop inserts into sheet A (Chaillan-Huntington et al. 1997; Huntington et al. 2000).
Intriguingly, inhibition of elastase by the P1 Arg crmA was characterized by an unusually low KM as was cleavage of this variant crmA by trypsin, suggesting that an optimal substrate-like canonical interaction of the crmA reactive loop with these proteases may be formed (Dementiev et al. 2003). In this regard, the substrate behavior and high KM value of the double variant may derive from the increased flexibility of the loop when the P1 residue is changed to Gly. Consistent with this idea, the specificity constants for cleavage of the loop-lengthened variant by trypsin, factor Xa, and thrombin were similarly reduced by about 10-fold from rate constants for inhibition or cleavage of the variant with a wild-type loop length by these proteases, suggesting that recognition and/or acylation of the reactive loop Arg was similar but equally less efficient at the P1′ position as compared to the normal P1 position.
The finding that either the normal 15 residue or an extended 16 residue reactive loop length are compatible with crmA functioning as an inhibitor of serine proteases fits well with the proposed mechanism of serpin inhibition of proteases through conformational deformation (Huntington et al. 2000; Peterson and Gettins 2001). According to this mechanism the reactive loop must be just long enough to insert into sheet A and translocate the covalently linked protease to the distal edge of the serpin so that the residual loop tether pulls and crushes the protease against the serpin body. A loop longer than the optimum would allow the protease additional space and flexibility to prevent this deformation and promote deacylation of the acyl-intermediate. In support of this mechanism, serpins engineered to have longer loops or loops shortened by more than two residues fail to trap proteases in stable acyl-intermediate complexes (Zhou et al. 2001; Plotnick et al. 2002). However, serpins with reactive loops shortened by one or two residues retain the ability to conformationally trap proteases, although with reduced efficiency, indicating that a narrow range of reactive loop lengths are compatible with the protease deformation mechanism. Modeling of fully loop inserted crmA and α1PI based on X-ray structures of cleaved forms of these serpins has shown that the 15-residue loop of crmA extends to the distal end of the more compact A sheet of this serpin just short of the position occupied by the more common 16-residue loop fragment of α1PI (Simonovic et al. 2000); i.e., the reactive loop length required for full protease translocation in crmA is shortened to between 15 and 16 residues. We conclude that crmA has evolved a shorter reactive loop to allow optimal stressing of the translocated protease at the distal end of sheet A, and hence, maximal stabilization of the acyl-intermediate. However, because the distance for full protease translocation is reduced by less than a single residue length, a one-residue longer loop can still stress the protease sufficiently to cause some deformation, although this produces a less stable complex (Calugaru et al. 2001).
CrmA thus appears to inhibit serine proteases by the established mechanism for inhibitory serpins, and has undergone structural modifications to accommodate this mechanism within the context of its more compact structure. The P1 residue provides the primary basis for recognition of target proteases and the serpin is able to trap proteases at the acyl-intermediate stage of cleavage of the P1–P1′ bond as a normal substrate by undergoing a major conformational change in which the cleaved reactive loop inserts into β-sheet A. The conformational change transforms the serpin from a metastable to a stable folded state and provides the energy to translocate and deform the protease at the distal end of the serpin. A 15-residue reactive loop length appears optimal for this conformational trapping mechanism due to the compactness of crmA relative to other serpins, although a 16-residue loop is also compatible with the mechanism. The findings that the reactive loop conformational change is also required for crmA inhibition of cysteine proteases (Tewari et al. 1995) and that a serpin can be transformed into an inhibitor of cysteine proteases through changes in the reactive loop sequence (Irving et al. 2002) support a similar mechanism for cysteine protease inhibition.
Materials and methods
Proteins
Wild-type and variant forms of crmA were expressed in Escherichia coli and purified after refolding from inclusion bodies as described previously (Simonovic et al. 2000) except for the inclusion of an additional monoQ chromatography step. After initial purification by DE52 anion exchange chromatography the refolded protein was dialyzed against, and applied to, the monoQ column, in 20 mM sodium phosphate buffer plus 1 mM dithiothreitol (pH 7.4), washed for 15 min in this buffer, and the protein then eluted with a NaCl gradient from 0–1 M. An initial sharp peak containing active protein was well separated from a subsequent broad peak containing aggregated inactive protein. Mutagenesis of wild-type crmA was done by annealing oligonucelotides encoding the mutation to the wild-type crmA pQE-60 plasmid (Qiagen) and using PCR to synthesize plasmid carrying the mutated gene. Concentrations of crmA were calculated from absorbance measurements at 280 nm using an extinction coefficient of 31,860 calculated from the amino acid composition (Gill and von Hippel 1989). β-Trypsin was isolated from commercial bovine trypsin (Sigma type XIII) by affinity chromatography on soybean trypsin inhibitor-agarose (Yung and Trowbridge 1975). Granzyme B purified from cyto-toxic T cells was a gift of Dr. Chris Froelich, Northwestern University, and recombinant caspase-1 was a gift of Nancy Thornberry of Merck. Thrombin was obtained by activation of prothrombin with Taipan snake venom (Owen and Jackson 1973) after purification of the proenzyme from human plasma (Miletich et al. 1980). The activated enzyme was then purified as described (Owen and Jackson 1973). Human factor Xa was purchased from Enzyme Research. Porcine pancreatic elastase (PPE) was obtained from Worthington Biochemical. Active protease concentrations were determined by standard enzymatic assays which had been previously calibrated with active-site titrated enzyme except for PPE whose active concentration was determined by titration with re-combinant α1-protease inhibitor (α1PI) prepared as described (Kwon et al. 1995).
SDS-PAGE
The purity of all proteins and analysis of the products of the reactions of crmA with proteases were assessed by SDS denaturing gel electrophoresis using the Laemmli discontinuous buffer system (Laemmli 1970). Kinetic experiments of protease cleavage of crmAs were done by incubating 2.5 μM or 5 μM crmA with 1–100 nM trypsin, 100 nM factor Xa, or 5 μM thrombin for varying times and then quenching reactions with SDS sample buffer prior to heat denaturation and electrophoresis. Reactions with trypsin were quenched with 10 μM FFR-chloromethylketone before denaturation in SDS buffer. Bands on gels were quantitated after scanning using Scion Image 4.0 software (Scion Corp.).
Experimental conditions
Experiments were done in either 0.02 M sodium phosphate or 0.1 M HEPES buffers containing 0.1 M NaCl, 0.1 mM EDTA, 0.1% PEG 8000, and 0.1 mM dithtiothreitol (pH 7.4) at 25°C or 37°C. An exception was experiments involving caspase-1, which were done in 0.1 M HEPES, 0.1 mM EDTA, 0.1% CHAPS, 1 mM dithiothreitol (pH 7.5), 25°C. Dithiothreitol present in the former buffers caused minimal enzyme inactivation over the time frame of experiments.
Stoichiometry of crmA–protease interactions
CrmA was added to fixed levels of protease (0.2–2 μM) at varying molar ratios and incubated for a time sufficient to yield essentially complete inhibition based on measured association rate constants. Residual enzyme activity was then assayed by dilution of an aliquot of the reaction mixture into chromogenic or fluorogenic substrates specific for the protease and measuring the initial linear rate of substrate cleavage at 405 nm for the colorimetric substrates or at 440 nm emission and 380 nm excitation wavelengths for fluorescent substrates. The substrates employed were acetyl-YVAD-7-amido-4-methylcoumarin (25 μM) for caspase-1, IETD-p-nitroanilide (100 μM) for granzyme B, S-2238 (100 μM) for thrombin, Spectrozyme FXa (100 μM) for factor Xa, S-2222 (100 μM) for trypsin, and succinyl-AAA-7-amido-4-methylcoumarin (50 μM) for PPE. The inhibition stoichiometry was determined from the X-axis intercept of linear plots of residual enzyme activity versus the molar ratio of inhibitor/protease.
Kinetics of crmA inhibition of proteases
The kinetics of inhibition of proteases by crmA was analyzed under pseudo first-order conditions in which the inhibitor was present at concentrations at least fivefold greater than the product of the enzyme concentration and the stoichiometry of inhibition. Reactions were initiated by adding protease to a solution of crmA. The time-dependent loss in protease activity was measured by assaying aliquots of the reaction mixture at varying times by dilution into appropriate substrates and then measuring the initial rate of substrate cleavage as outlined above in measurements of inhibition stoichiometry. The decay of protease activity was computer fit by a single exponential function with an endpoint of zero activity. The second-order association rate constant was calculated by dividing the fitted pseudo first-order rate constant by the crmA concentration.
For PPE reactions with P1 Arg crmA, a continuous inhibition assay was employed in which 0.1–1 nM PPE was mixed with 50–1000 nM crmA variant in the presence of 50 μM PPE substrate and the exponential decrease in the rate of fluorescence change due to PPE inhibition was continuously monitored for about 10 half-lives. Progress curves were fit to an exponential function with a linear endpoint to obtain the observed pseudo first-order rate constant (Björk et al. 1998). The dependence of kobs on inhibitor concentration was fit by the hyperbolic function, kobs = klim × [I]o/(KI × (1 + [S]o/KS) + [I]o), where [I]o and [S]o are the nominal inhibitor and substrate concentrations, KI and KS are Michaelis constants for inhibitor and substrate binding to the enzyme, respectively, and klim is the limiting inhibition rate constant when the enzyme is saturated with inhibitor. The value for KS was independently measured to be 280 μM and values for KI and klim were the fitted parameters. The ratio, klim/KI, in this case represents the second-order rate constant for crmA inhibition of PPE.
Kinetics of dissociation of crmA–protease complexes
CrmA–protease complexes were prepared by mixing inhibitor with enzyme using an inhibitor concentration sufficient to fully complex the enzyme and yield a molar excess, based on measurements of inhibition stoichiometry, to prevent protease degradation in the complex. Complexes were then diluted >100-fold into specific protease substrates at varying concentrations and the initial rate of complex dissociation was continuously monitored from the parabolic increase in absorbance or fluorescence resulting from the linear rate of protease generation. Data were fit by the parabolic function (Calugaru et al. 2001),
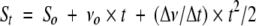 |
where St and So represent the spectroscopic signal (absorbance or fluorescence) at time, t, or time, zero, vo is the initial velocity of substrate hydrolysis at time zero, and Δv/Δt is the initial rate of change of the substrate hydrolysis rate due to generation of active protease. The first-order rate constant for complex dissociation was determined from the slope of a linear plot of fitted values of Δv/Δt versus the concentration of complex using the expression (Calugaru et al. 2001),
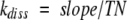 |
where TN is the turnover number for substrate hydrolysis under the conditions of the dissociation experiment. The latter was experimentally determined by measuring and plotting the initial rate of change of the spectroscopic signal as a function of known concentrations of enzyme and calculating the slope of the plot by linear regression. The proportional dependence of Δv/Δt on complex concentration provided validation that a first-order dissociation rate constant for complex dissociation was being measured without any contribution from reassociation of protease with residual inhibitor (Calugaru et al. 2001).
Kinetics of protease cleavage of crmA as a substrate
To solutions of 100 μM S-2222 prepared in the absence and presence of varying concentrations of crmA variants were added 0.5 nM trypsin, and the initial rate of chromogenic substrate cleavage was measured at 405 nm. The decrease in cleavage rate produced by the competitor crmA was analyzed by fitting to the hyperbolic equation for competitive substrate inhibition,
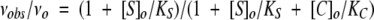 |
where vo and vobs are the initial velocities of chromogenic substrate cleavage in the absence and presence of crmA, [C]o and [S]o are the total crmA and chromogenic substrate concentrations, and KC and KS are the Michaelis constants for crmA and S-2222 cleavage by trypsin. The kcat for trypsin cleavage of crmA was measured by the method of Blake et al. (1989). Full progress curves of S-2222 hydrolysis by trypsin were measured in the absence and presence of varying concentrations of crmA. The area under progress curves were analyzed by integration using Kaleidagraph 3.6 (Synergy Software). The difference in areas determined in the presence and absence of competitor crmA was plotted as a function of the crmA concentration and the slope of the proportional dependence determined by linear regression. This slope was used to calculate kcat based on known values of KM and kcat for trypsin cleavage of the chromogenic substrate and the dissociation constant, KP, for the enzyme interaction with the product of this cleavage. The latter kinetic parameters were determined from full progress curves of trypsin cleavage of S-2222 as a function of substrate concentration using the integrated Michaelis-Menten equation.
Sequencing of cleavage sites in crmA
Solutions of single and double variant crmAs were treated with trypsin for a time sufficient to fully cleave the protein and then trypsin was removed by adsorption to soybean inhibitor-agarose. Wild-type crmA was treated with PPE using a molar excess of serpin over that required to fully inhibit the protease. The reaction mixture was then chromatographed first on a monoQ column following the protocol used to purify crmA and second on a Superdex 200 column to isolate pure cleaved serpin. The wild-type crmA-PPE complex was isolated from a similar reaction mixture by separation on a nondenaturing 7% Laemmli gel followed by transfer to a PVDF membrane and cutting out the complex band after detection by Coomassie staining. N-terminal sequencing was done by the UIC sequencing facility using an Applied Biosystems sequenator.
Acknowledgments
This work was supported by NIH grant P01-HL-64013 to P.G.W.G. and S.T.O. We thank Jill Bayliss and Scott Suda for mutagenesis of crmA and Inder Chand for expression and purification of wild-type and variant crmAs.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041104905.
References
- Bianchini, E.P., Louvain, V.B., Marque, P.-E., Juliano, M.A., Juliano, L., and Le Bonniec, B.F. 2002. Mapping of the catalytic groove preferences of factor Xa reveals an inadequate selectivity for its macromolecule substrates. J. Biol. Chem. 277 20527–20534. [DOI] [PubMed] [Google Scholar]
- Björk, I., Nordling, K., Raub-Segall, E., Hellman, U., and Olson, S.T. 1998. Inactivation of papain by antithrombin due to autolytic digestion: A model of serpin inactivation of cysteine proteinases. Biochem. J. 335 701–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake II, R.C., Vassall, R.F., and Blake, D.A. 1989. The Michaelis constants of a nonchromatogenic substrate may be determined using a chromogenic substrate. Arch. Biochem. Biophys. 272 52–68. [DOI] [PubMed] [Google Scholar]
- Calugaru, S.V., Swanson, R., and Olson, S.T. 2001. The pH dependence of serpin–proteinase complex dissociation reveals a mechanism of complex stabilization involving inactive and active conformational states of the proteinase which are perturbable by calcium. J. Biol. Chem. 276 32446–32455. [DOI] [PubMed] [Google Scholar]
- Chaillan-Huntington, C.E., Gettins, P.G.W., Huntington, J.A., and Patston, P.A. 1997. The P6–P2 region of serpins is critical for proteinase inhibition and complex stability. Biochemistry 36 9562–9570. [DOI] [PubMed] [Google Scholar]
- Cole, E.B., Miller, D., Rometo, D., Greenberg, R.M., Brömme, D., Cataltepe, S., Pak, S.C., Mills, D.R., Silverman, G.A., and Luke, C.J. 2004. Identification and activity of a lower eularyotic serine proteinase inhibitor (serpin) from Cyanea capillata: Analysis of a jellyfish serpin, jellypin. Biochemistry 43 11750–11759. [DOI] [PubMed] [Google Scholar]
- Dementiev, A., Simonovic, M., Volz, K., and Gettins, P.G.W. 2003. Canonical inhibitor-like interactions explain reactivity of a1-proteinase inhibitor Pittsburgh and antithrombin with proteinases. J. Biol. Chem. 278 37881–37887. [DOI] [PubMed] [Google Scholar]
- Djie, M.Z., Le Bonniec, B.F., Hopkins, P.C.R., Hipler, K., and Stone, S.R. 1996. Role of the P2 residue in determining the specificity of serpins. Biochemistry 35 11461–11469. [DOI] [PubMed] [Google Scholar]
- Ekert, P.G., Silke, J., and Vaux, D.L. 1999. Inhibition of apoptosis and clonogenic survival of cells expressing crmA variants: Optimal caspase substrates are not necessarily optimal inhibitors. EMBO J. 18 330–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gettins, P.G.W. 2002. Serpin structure, mechanism, and function. Chem. Rev. 102 4751–4803. [DOI] [PubMed] [Google Scholar]
- Gill, S.C. and von Hippel, P.H. 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182 319–326. [DOI] [PubMed] [Google Scholar]
- Harris, J.L., Peterson, E.P., Hudig, D., Thornberry, N.A., and Craik, C.S. 1998. Definition and redesign of the extended substrate specificity of granzyme B. J. Biol. Chem. 273 27364–27373. [DOI] [PubMed] [Google Scholar]
- Harris, J.L., Backes, B.J., Leonetti, F., Mahrus, S., Ellman, J.A., and Craik, C.S. 2000. Rapid and general profiling of protease specificity by using combinatorial fluorogenic substrate libraries. Proc. Natl. Acad. Sci. 97 7754–7759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood, D.B., Huntington, J.A., and Gettins, P.G.W. 1994. α1-Proteinase inhibitor variant T345R. Influence of P14 residue on substrate and inhibitory pathways. Biochemistry 33 8538–8547. [DOI] [PubMed] [Google Scholar]
- Huber, R. and Carrell, R.W. 1989. Implications of the three-dimensional structure of α1-antitrypsin for structure and function of serpins. Biochemistry 28 8951–8966. [DOI] [PubMed] [Google Scholar]
- Huntington, J.A., Read, R.J., and Carrell, R.W. 2000. Structure of a serpin–protease complex shows inhibition by deformation. Nature 407 923–926. [DOI] [PubMed] [Google Scholar]
- Irving, J.A., Pike, R.N., Dai, W., Brömme, D., Worrall, D.M., Silverman, G.A., Coetzer, T.H.T., Dennison, C., Bottomley, S.P., and Whisstock, J.C. 2002. Evidence that serpin architecture intrinsically supports papain-like cysteine protease inhibition: Engineering a1-antitrypsin to inhibit cathepsin proteases. Biochemistry 41 4998–5004. [DOI] [PubMed] [Google Scholar]
- Komiyama, T., Ray, C.A., Pickup, D.J., Thronberry, N.A., Peterson, E.P., and Salvesen, G. 1994. Inhibition of interleukin-1β coverting enzyme by the cowpox virus serpin crmA. J. Biol. Chem. 269 19331–19337. [PubMed] [Google Scholar]
- Kwon, K.-S., Lee, S., and Yu, M.-H. 1995. Refolding of α1-antitrypsin expressed as inclusion bodies in Escherichia coli: Characterization of aggregation. Biochim. Biophys. Acta 1247 179–184. [DOI] [PubMed] [Google Scholar]
- Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227 680–685. [DOI] [PubMed] [Google Scholar]
- Lawrence, D.A., Ginsburg, D., Day, D.E., Berkenpas, M.B., Verhamme, I.M., Kvassman, J., and Shore, J.D. 1995. Serpin–protease complexes are trapped as stable acyl-enzyme intermediates. J. Biol. Chem. 270 25309–25312. [DOI] [PubMed] [Google Scholar]
- Lawrence, D.A., Olson, S.T., Muhammad, S., Day, D.E., Kvassman, J.-O., Ginsburg, D., and Shore, J.D. 2000. Partitioning of serpin–proteinase reactions between stable inhibition and substrate cleavage is regulated by the rate of serpin reactive center loop insertion into β-sheet A. J. Biol. Chem. 275 5839–5844. [DOI] [PubMed] [Google Scholar]
- Miletich, J.P., Broze Jr., G.J., and Majerus, P.W. 1980. The synthesis of sulfated dextran beads for isolation of human plasma coagulation factors II, IX and X. Anal. Biochem. 105 304–310. [DOI] [PubMed] [Google Scholar]
- Owen, W.G. and Jackson, C.M. 1973. Activation of prothrombin with Oxyuranos scutellatus scutellatus (Taipan snake) venom. Thromb. Res. 3 705–714. [Google Scholar]
- Peterson, F.C. and Gettins, P.G.W. 2001. Insight into the mechanism of serpin–proteinase inhibition from 2D (1H-15N) NMR studies of the 69 kDa α1-proteinase inhibitor Pittsburgh–trypsin covalent complex. Biochemistry 40 6284–6292. [DOI] [PubMed] [Google Scholar]
- Plotnick, M.I., Rubin, H., and Schechter, N.M. 2002. The effects of reactive site location on the inhibitory properties of the serpin α1-antichymotrypsin. J. Biol. Chem. 277 29927–29935. [DOI] [PubMed] [Google Scholar]
- Potempa, J., Shieh, B.-H., and Travis, J. 1988. α-2-antiplasmin: A serpin with two separate but overlapping reactive sites. Science 241 699–700. [DOI] [PubMed] [Google Scholar]
- Quan, L.T., Caputo, A., Bleackley, R.C., Pickup, D.J., and Salvesen, G.S. 1995. Granzyme B is inhibited by the cowpox virus serpin cytokine response modifier A. J. Biol. Chem. 270 10377–10379. [DOI] [PubMed] [Google Scholar]
- Ray, C.A., Black, R.A., Kronheim, S.R., Greenstreet, T.A., Sleath, P.R., Salvesen, G.S., and Pickup, D.J. 1992. Viral inhibition of inflammation: Cowpox virus encodes an inhibitor of the interleukin-1-β converting enzyme. Cell 69 597–604. [DOI] [PubMed] [Google Scholar]
- Renatus, M., Zhou, Q., Stennicke, H.R., Snipas, S.J., Turk, D., Bankston, L.A., Liddington, R.C., and Salvesen, G.S. 2000. Crystal structure of the apoptotic suppressor crmA in its cleaved form. Structure 8 789–797. [DOI] [PubMed] [Google Scholar]
- Schick, C.S., Pemberton, P.A., Shi, G.-P., Kamachi, V., Cataltepe, S., Bartuski, A.J., Gornstyein, E.R., Bromme, D., Chapman, H.A., and Silverman, G.A. 1998. Cross-class inhibition of the cysteine proteinases cathepsins K, L, and S by the serpin squamous cell carcinoma antigen 1: A kinetic analysis. Biochemistry 37 5258–5266. [DOI] [PubMed] [Google Scholar]
- Silverman, G.A., Bird, P.I., Carrell, R.W., Church, F.C., Coughlin, P.B., Gettins, P.G.W., Irving, J.A., Lomas, D.A., Luke, C.J., Moyer, R.W., et al. 2001. The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. J. Biol. Chem. 276 33293–33296. [DOI] [PubMed] [Google Scholar]
- Simonovic, M., Gettins, P.G.W., and Volz, K. 2000. Crystal structure of viral serpin crmA provides insights into its mechanism of cysteine proteinase inhibition. Protein Sci. 9 1423–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratikos, E. and Gettins, P.G.W. 1999. Formation of the covalent serpin–proteinase complex involves translocation of the proteinase by more than 70 A and full insertion of the reactive center loop into β-sheet A. Proc. Natl. Acad. Sci. 96 4808–4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari, M., Quan, L.T., O’Rourke, K., Desnoyers, S., Zeng, Z., Beidler, D.R., Poirier, G.G., Salvesen, G.S., and Dixit, V.M. 1995. YAMA/CPP32β, a mammalian homolog of CED-3, is a crmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell 81 801–809. [DOI] [PubMed] [Google Scholar]
- Thornberry, N.A., Rano, T.A., Peterson, E.P., Rasper, D.M., Timkey, T., Garcia-Calvo, M., Houtzager, V.M., Nordstrom, P.A., Roy, S., Vaillancourt, J.P., et al. 1997. A combinatorial approach defines specificities of members of the caspase family and granzyme B. J. Biol. Chem. 272 17907–17911. [DOI] [PubMed] [Google Scholar]
- Travis, J., Matheson, N.R., George, P.M., and Carrell, R.W. 1986. Kinetic studies on the interaction of α1-proteinase inhibitor (Pittsburgh) with trypsin-like serine proteinases. Biol. Chem. Hoppe-Seyler 367 853–859. [DOI] [PubMed] [Google Scholar]
- Yung, B.Y. and Trowbridge, C.G. 1975. Resolution of α and β anhydrotrypsin by affinity chromatography. Biochem. Biophys. Res. Commun. 65 927–930. [DOI] [PubMed] [Google Scholar]
- Zhou, Q., Snipas, S., Orth, K., Muzio, M., Dixit, V.M., and Salvesen, G.S. 1997. Target protease specificity of the viral serpin crmA. Analysis of five caspases. J. Biol. Chem. 272 7797–7800. [DOI] [PubMed] [Google Scholar]
- Zhou, A., Carrell, R.W., and Huntington, J.A. 2001. The serpin inhibitory mechanism is critically dependent on the length of the reactive center loop. J. Biol. Chem. 276 27541–27547. [DOI] [PubMed] [Google Scholar]