

Abstract
An Escherichia coli plasmid vector for the high-level expression of hydrophobic membrane proteins is described. The plasmid, pBCL, directs the expression of a target polypeptide fused to the C terminus of a mutant form of the anti-apoptotic Bcl-2 family protein, Bcl-XL, where the hydrophobic C terminus has been deleted, and Met residues have been mutated to Leu to facilitate CNBr cleavage after a single Met inserted at the beginning of the target sequence. Fusion protein expression is in inclusion bodies, simplifying the protein purification steps. Here we report the high-level production of PLM, a membrane protein that is a member of the FXYD family of tissue-specific and physiological-state–specific auxiliary subunits of the Na,K-ATPase, expressed abundantly in heart and skeletal muscle. We demonstrate that milligram quantities of pure, isotopically labeled protein can be obtained easily and in little time with this system.
Keywords: membrane protein expression, FXYD, PLM, Mat-8, CHIF, Bcl-XL, NMR, lipid
The most versatile and widely used method for obtaining recombinant proteins is by expression in Escherichia coli. The ability to express proteins in bacteria is particularly useful for NMR structure determination, because it allows milligram quantities of isotopically labeled proteins to be obtained relatively economically, and a variety of isotopic labeling schemes to be incorporated in the NMR experimental strategy.
Some polypeptides, however, are toxic to the bacterial hosts that express them. For example, some membrane proteins and peptides, including some of bacterial origin, congest the cell membranes when they are overexpressed, and act as toxic, antibacterial agents, regardless of their actual biological functions. For these difficult polypeptides, solid-phase synthesis is not a practical alternative, because it is typically limited to sequences shorter than 50 amino acids, and while this size limit can be extended through the use of chemical ligation methods (Dawson et al. 1994; Kochendoerfer 2001) that can also be applied to membrane proteins (Kochendoerfer et al. 1999, 2004), NMR studies still require bacterial expression of the polypeptide precursors for the practical introduction of various isotopic labels.
Several E. coli cell strains and expression strategies have been developed to address this problem (Miroux and Walker 1996; Rogl et al. 1998; Jones et al. 2000; Majerle et al. 2000; Opella et al. 2001; Sharon et al. 2002; Bannwarth and Schulz 2003; Booth 2003; Kiefer 2003; Lindhout et al. 2003; Smith and Walker 2003; Wiener 2004), and more recently, cell-free expression has been used to obtain milligram quantities of isotopically labeled membrane proteins (Klammt et al. 2004). Many strategies rely on the use of fusion protein tags to improve expression and facilitate purification, and many involve protein expression in inclusion bodies, to keep the hydrophobic polypeptide away from the bacterial membranes, and thus increase the level of expression. The formation of inclusion bodies also limits proteolytic degradation and simplifies protein purification, which can be further assisted by the incorporation of an engineered His tag for metal affinity chromatography.
The TLE (a portion of the Trp ΔLE 1413 polypeptide) (Miozzari and Yanofsky 1978; Kleid et al. 1981; Staley and Kim 1994), and KSI (Kuliopulos et al. 1994) fusion partners promote the accumulation of expressed proteins as inclusion bodies, and have been used to express several membrane peptides and proteins ranging in size from 20 to 200 amino acids (Opella et al. 2001; Opella and Marassi 2004). We have found TLE to be useful for the production of Mat-8 (mammary tumor protein 8 kDa, or FXYD3) and CHIF (corticosteroid-hormone–induced factor, or FXYD4), two 67-residue membrane proteins that belong to the FXYD family of tissue-specific and physiological-state–specific auxiliary subunits of the Na,K-ATPase, with yields of purified protein that range between 3 and 4 mg/L cell culture in M9 minimal medium (Crowell et al. 2003).
The FXYD proteins are widely expressed in mammalian tissues that specialize in fluid and solute transport or that are electrically excitable (Sweadner and Rael 2000; Crambert and Geering 2003), and we are using NMR spectroscopy to determine their structures in lipid micelles and bilayers environments. They are characterized by the short amino acid sequence Phe-X-Tyr-Asp (or FXYD, hence the name) before their single trans-membrane helix, which is conserved in all examples, and where X is usually Tyr, but can also be Thr, Glu, or His (Sweadner and Rael 2000). The ability to produce milligram quantities of pure FXYD proteins also facilitates functional studies that, together with structure determination, can provide important structure–activity correlations. Since Mat-8 and CHIF could be purified in sufficiently high quantities, we were able to obtain resonance assignments for their solution NMR spectra in micelles, as well as two-dimensional solid-state NMR spectra in lipid bilayers (Franzin et al. 2005). However, phospholemman (PLM, or FXYD1), a 72-residue FXYD family member that is the major substrate of hormone-stimulated phosphorylation by cAMP-dependent protein kinase A and C in heart and skeletal muscle, resists high-level expression with either the TLE or KSI fusion protein system, yielding amounts of purified material (0.5–1 mg/L cell culture) that are insufficient for NMR structural studies.
In the course of our separate studies with the apoptosis regulatory proteins of the Bcl-2 family (named after B-cell leukemia/lymphoma where they were first discovered), we have found that the anti-apoptotic protein Bcl-XL (B-cell leukemia/lymphoma extra long), which contains a hydrophobic C-terminal sequence of ~20 amino acids, can be overexpressed in E. coli and purified in high-yields. In nature, Bcl-XL localizes primarily to the outer mitochondrial and ER membranes, presumably through its hydrophobic C terminus, which is sufficiently long to span the lipid bilayer membrane (Adams and Cory 1998; Reed 1998), and while a deletion mutant of Bcl-XL, lacking the C-terminal sequence, can be expressed in bacteria as a soluble protein (Muchmore et al. 1996), the full-length protein accumulates as insoluble inclusion bodies. These observations prompted us to explore the usefulness of Bcl-XL as a fusion partner for the expression of hydrophobic polypeptides that, similar to PLM, are difficult to express and purify.
To test this possibility, we constructed a fusion protein expression plasmid (pBCL) where the hydrophobic C terminus of Bcl-XL was deleted to be replaced with a hydrophobic polypeptide gene of interest by insertion at an engineered cloning site. The plasmid utilizes a T7 expression system (Studier et al. 1990), and a single Met residue, inserted at the beginning of the target polypeptide sequence, enables its release from Bcl-XL by CNBr cleavage. Here we report the high-level production of PLM with this plasmid, and we demonstrate that milligram quantities of pure, isotopically labeled protein can be obtained easily and in little time.
Results and Discussion
The pBCL plasmid vector that we constructed directs the expression of a target polypeptide fused to the C terminus of BCL, a mutant form of Bcl-XL lacking the hydrophobic C-terminal domain of the wild-type protein. The configuration of pBCL is shown in Figure 1A ▶, and the amino acid sequences of wild-type Bcl-XL, mature PLM and the two other FXYD proteins, Mat-8 and CHIF, are shown in Figure 1B ▶.
Figure 1.

(A) Construction of the pBCL173 and pBCL99 fusion protein expression plasmids. The target sequence with N-terminal Met is inserted between the AflII and XhoI cloning sites. BCL173 has a cleavable Met after the His tag, while BCL99 does not. (B) Amino acid sequences of wild-type Bcl-XL and of the FXYD proteins, PLM (human), Mat-8 (human), and CHIF (rat), that were inserted in the pBCL plasmid vectors. Met residues that were changed to Leu are marked with asterisks. Both BCL173 and BCL99 lack the Bcl-XL hydrophobic C terminus (highlighted in the gray box), and BCL173 (solid-underline) also lacks the flexible loop of Bcl-XL while BCL99 (dashed underline) lacks the first 116 residues. The trans-membrane domains of PLM, Mat-8, and CHIF are highlighted in the gray boxes.
We prepared and tested two mutant forms of Bcl-XL, BCL173 and BCL99, as fusion tags for the high-level expression of PLM and the other FXYD proteins. To obtain BCL173, the 173-residue 21.5-kDa fusion tag (Fig. 1B ▶, solid underline), we deleted the C-terminal domain (residues 213–233) and the flexible loop (residues 44–84) of Bcl-XL, we changed the two remaining Met residues (Met159 and Met170) to Leu, and we introduced an AflII/XhoI cloning site (Leu-Lys) after Arg212 at the C-terminal end of the gene to facilitate the insertion of target polypeptide sequences. To obtain BCL99, the 99-residue 12.9-kDa tag (Fig. 1B ▶, dashed underline), we further deleted residues 1–116, spanning the loop as well as helices 1–4 in the structure of the soluble form of Bcl-XL (Muchmore et al. 1996). Both fusion tags have an N-terminal (His)6 sequence to allow protein purification by Ni-affinity chromatography.
Reaction with CNBr cleaves the expressed fusion protein after the single Met residue introduced at the start of the inserted target sequence, releasing the protein of interest intact. In cases where the target protein contains Met residues that cannot be mutated, separation from BCL can be obtained by introducing amino acid sequences specific for cleavage by other chemical means, such as hydroxylamine (Asn-Gly), or for cleavage by one of the commonly used proteases: thrombin, factor Xa, enterokinase, and tobacco etch virus protease. Chemical cleavage is an attractive option because it eliminates the difficulties—poor specificity and enzyme inactivation—often encountered with protease treatment of hydrophobic proteins in detergents.
The expression of PLM using pBCL173 and pBCL99, as well as the two other commonly used plasmids, pKSI and pTLE, is compared in Figure 2A ▶ by SDS-PAGE. For this study, each of the four plasmids containing the PLM gene insert were used to transform E. coli C41(DE3) cells, a mutant strain selected for the expression of insoluble and toxic proteins (Miroux and Walker 1996), and the positive clones were grown to a cell density of OD600 = 0.7 before induction with IPTG for 4 h. The total lysate from cells transformed with pBCL173-PLM, before induction with IPTG, is shown in the first lane of the gel, while IPTG induction of each of the four clones is shown in the following lanes. In all four cases, fusion protein overexpression is marked by the appearance of a distinct band at the calculated molecular weight of the corresponding fusion protein, albeit with different intensities (Fig. 2A ▶, solid arrows). To obtain a quantitative estimate of protein expression, we compared the intensity of each fusion protein band to the intensity of the band from a 39-kDa protein that is expressed in the cells without IPTG induction (Fig. 2A ▶, dashed arrow). In Figure 2A ▶, the intensity of each band is reported as %Irel, the percentage relative to the intensity of the band at 39 kDa.
Figure 2.
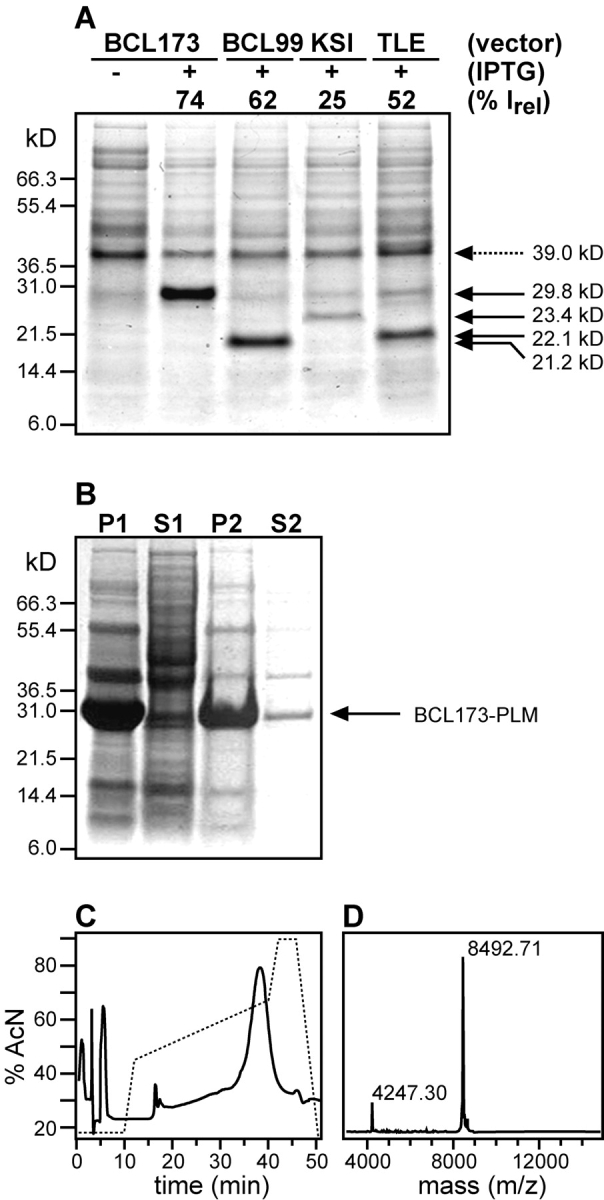
(A) Expression of PLM with four different fusion protein plasmid vectors: pBCL173, pBCL99, pKSI, and pTLE. The gel shows total lysates from cells, transformed with each plasmid, and harvested before (−) or after (+) induction with IPTG. Fusion protein overexpression is marked by the appearance of a distinct band (solid arrows) at the molecular weight of the corresponding fusion protein: BCL173-PLM (29.8 kDa), BCL99-PLM (21.2 kDa), KSI-PLM (23.4 kDa), and TLE-PLM (22.1 kDa). The intensity of each band is reported as (%Irel) the percentage relative to the intensity of the band at 39 kDa (dashed arrow). (B) Isolation of BCL173-PLM inclusion bodies (29.8 kDa) after cell lysis. The gel shows the pellet (P1) and supernatant (S1) obtained after centrifugation of the total cell lysate, and the pellet (P2) and supernatatnt (S2) obtained after resuspending the inclusion bodies pellet (P1) in buffer A and centrifuging again to separate it from the soluble fraction (S2). (C) Reverse-phase HPLC trace for the purification of PLM after CNBr cleavage and ion-exchange chromatography. PLM elutes at 65% acetonitrile. (D) MALDI-TOF mass spectrum of purified 15N-labeled PLM at 8.5 kDa. The peak at 4.3 kDa is from the doubly charged protein.
Induction of BCL173-PLM and BCL99-PLM both yield very strong bands at 29.8 kDa and 21.2 kDa, as expected from their calculated molecular weights. Both these bands have significantly greater intensity than those observed for the expression of KSI-PLM at 23.4 kDa or of TLE-PLM at 22.1 kDa, demonstrating the superior performance of pBCL for the expression of PLM. Similar expression levels were obtained for the other FXYD family proteins Mat-8 and CHIF in pBCL.
Upon overexpression, both full-length Bcl-XL and BCL-PLM accumulate as insoluble proteins and cannot be driven to the soluble cellular fraction by lowering the temperature of the cell culture to 15°C. This is demonstrated in Figure 2B ▶, where the insoluble fraction of the cell lysate (lane P1) is highly enriched in BCL173-PLM (arrow), while the soluble fraction (lane S1) contains very little. In the initial steps of fusion protein isolation, the N-terminal His tag can be used for purification by Ni-affinity chromatography; however, in practice, we have found that the isolation of inclusion bodies already yields very pure protein (Fig. 2B ▶, lane P2) and that the purity is not improved by Ni-affinity. Therefore, after isolating the inclusion bodies by centrifugation, we dissolved them in acidic GdnHCl and proceeded directly to CNBr cleavage. The cleavage reaction was monitored by SDS-PAGE and found to be virtually 100% complete, releasing two fragments corresponding to BCL and PLM, which could be separated chromatographically.
After CNBr cleavage, PLM was separated from the BCL fusion partner by using ion exchange chromatography, to exploit the large difference in isoelectric point (pI) between the basic PLM (pI = 9.3) and its acidic fusion partner BCL173 (pI = 4.9) or BCL99 (pI = 5.7). This procedure is also useful for the other FXYD family members. PLM was further purified by reverse-phase HPLC (Fig. 2C ▶), and its mass and purity were confirmed by mass spectrometry (Fig. 2D ▶). The HPLC trace demonstrates that PLM elutes as a single well-resolved peak, in ~65% acetonitrile, as observed previously for purification from the TLE fusion partner (Crowell et al. 2003). The major peak in the MALDI-TOF mass spectrum matches the calculated mass of uniformly 15N-labeled PLM (8.490 kDa), while the small peak at half mass arises from the doubly charged species. The spectrum shows no evidence of degradation or chemical modifications, and the absence of other intensity demonstrates that this procedure yields very pure protein. The typical yields of PLM obtained with the pBCL173 plasmid are in the range of 10 mg of purified PLM per liter of culture in M9 minimal medium.
We have also purified PLM by reverse-phase HPLC directly after CNBr cleavage, bypassing the ion-exchange step, and this method also yields pure protein but is more taxing on the HPLC column. Alternatively, Ni-affinity chromatography could be used to separate PLM from the BCL99 fusion partner, which retains its N-terminal His tag after cleavage, but not to separate it from BCL173, which has a cleavable Met residue, not mutated to Leu, after the His tag.
The two-dimensional 1H/15N HSQC spectrum of purified uniformly 15N-labeled PLM, in SDS micelles, is shown in Figure 3 ▶. The spectrum is identical to that previously obtained from PLM prepared using the TLE expression system (Crowell et al. 2003), and the presence of one well-defined resonance for each amide site in the protein is indicative of a high-quality micelle sample. The resonances are well dispersed and clearly resolved, demonstrating that PLM is folded and adopts a unique conformation in micelles. The limited chemical shift dispersion reflects the helical structure of this protein, also observed by circular dichroism (Crowell et al. 2003). Helical secondary structures have also been determined by NMR for Mat-8 and CHIF, for which we have completed the resonance assignments and measured several structural parameters (Franzin et al. 2005).
Figure 3.

Solution NMR 1H/15N HSQC spectrum of uniformly 15N-labeled PLM in SDS micelles.
Previously, we had assigned several resonances in the HSQC spectrum of PLM, including those from the six Gly residues labeled in Figure 3 ▶, using uniformly and selectively 15 N-labeled protein produced with the TLE system, and HSQC–NOESY data. However, the low yields attainable with the TLE system precluded us from preparing sufficient quantities of isotopically labeled protein for complete resonance assignment and structure determination, and prompted us to explore alternative expression strategies. The ability to produce large quantities of isotopically labeled PLM with BCL, makes it feasible to perform multidimensional, triple-resonance experiments for resonance assignment and structure determination of this protein in lipid micelles. The high-level production of PLM also enables structure determination in lipid bilayers, using solid-state NMR spectroscopy, which will be important for understanding the mechanism with which this integral membrane protein binds and regulates the Na,K-ATPase.
The solid-state NMR spectra of membrane proteins, associated with planar lipid bilayers oriented perpendicular to the magnetic field, trace out characteristic patterns that reflect the protein structure and orientation within the membrane, and thus provide very useful structural restraints. Helical structures give 15N chemical shift/1H-15N dipolar coupling correlation PISEMA spectra, where the resonances from amide sites in the protein track helical wheel projections (Schiffer and Edmunson 1967) that contain information on helix tilt and helix rotation within the membrane (Marassi and Opella 2000; Wang et al. 2000; Marassi 2001). Typically, trans-membrane helices have spectra with 15N chemical shifts between 150 and 200 ppm, and 1H-15N dipolar couplings between 2 and 10 kHz, while helices that bind parallel to the membrane surface have spectra with shifts between 70 and 100 ppm and couplings between 0 and 5 kHz.
The solid-state NMR spectra of PLM in lipid bilayers, shown in Figure 4 ▶, demonstrate that it can be reconstituted in membranes, in its trans-membrane orientation. The 15N chemical shift spectrum of PLM in unoriented lipid bilayer vesicles is a powder pattern (Fig. 4B ▶, solid line) that spans the full range of the amide 15N chemical shift interaction (Fig. 4B ▶, dotted line) and provides no resolution of individual amide sites. The additional intensity at the isotropic resonance frequencies (100–130 ppm) is from amino acid sites that are mobile on the time scale of the 15N chemical shift interaction. The peak at 35 ppm is from the amino groups at the N terminus and side chains of the protein.
Figure 4.

Solid-state NMR 15N chemical shift spectra of uniformly 15N-labeled PLM in DOPC/DOPG lipid bilayers. (A) Spectrum in oriented planar bilayers. (B) Spectrum in unoriented bilayer vesicles (solid line), and powder pattern calculated for a rigid 15N amide site (dotted line).
The spectrum of PLM in oriented planar lipid bilayers is very different (Fig. 4A ▶), with some of the amide resonances centered at a frequency (80 ppm) associated with NH bonds in helices parallel to the membrane surface, and a distinct set of resonances observed in a relatively narrow band of frequencies (200 ppm) typically associated with NH bonds in trans-membrane helices. This is consistent with a conformation where the trans-membrane helix of PLM crosses the bilayer with a small tilt angle, similar to CHIF, whose trans-membrane helix crosses the membrane with a tilt of 15° (±2.5°) (Franzin et al. 2005). Thus, purified PLM can be refolded in micelles and in lipid bilayers, where it adopts a unique trans-membrane conformation.
Conclusions
Producing milligram amounts of protein for structural studies often involves screening for protein expression levels in a variety of expression vectors, since no single system is equally well suited for all target proteins. The BCL fusion protein plasmid vector proved to be very useful for the high-level production of integral membrane proteins of the FXYD family, and particularly for PLM, a family member that had previously been difficult to produce in E. coli. The FXYD proteins, PLM, Mat-8, and CHIF, could not be expressed at substantial levels using pET plasmids without an auxiliary fusion protein partner, and while the TLE system has been used successfully for the production of membrane peptides and proteins ranging in length from ~20 to 200 amino acids (Opella et al. 2001) and is useful for the production of Mat-8 and CHIF (Crowell et al. 2003), the yields of purified PLM obtained with TLE are at least 10 times lower than those obtained with BCL. The KSI system, which is designed for the expression of either tandem repeats of 10–25 residues peptides, separated by Met residues for CNBr cleavage, or of individual 25–75 residues polypeptides, works very well for the production of some shorter trans-membrane peptides (Opella and Marassi 2004) but does not yield high expression of PLM, whose length of 72 residues is close to the limit for optimal expression with KSI. The GB-1 (B1 immunoglobulin binding domain of streptococcal protein G) fusion protein system was recently developed for the high-level expression of peptides with 10–25 amino acids of varying degrees of hydrophobicity (Lindhout et al. 2003) and offers another attractive expression alternative, although we did not test its usefulness for the production of PLM or other integral membrane proteins.
The ability to produce PLM and other FXYD family proteins, isotopically labeled and in large amounts, facilitates their structural study by NMR in lipid environments, and their functional characterization as regulators of the Na,K-ATPase. Given the efficiency of BCL as a fusion protein tag for the high-level production of PLM, we anticipate that it may also be useful for the production of other single-spanning membrane proteins with similar size and topology, and of shorter hydrophobic peptides, that can be substituted for the hydrophobic C terminus of Bcl-XL.
Materials and methods
Preparation of the plasmid vectors and inserts
We prepared two fusion protein plasmids, pBCL173 and pBCL99, which differ in the amino acid sequence of the Bcl-XL fusion partner (Fig. 1 ▶). For both pBCL173 and pBCL99, we deleted amino acids 213–233, spanning the hydrophobic C terminus of human Bcl-XL (accession: Z23115); we mutated two Met residues, M159 and M170, to Leu; and we introduced an AflII/XhoI cloning site at the C terminus of the construct. In addition, for pBCL173, we deleted residues 44–84, spanning the long flexible loop of human Bcl-XL, while for pBCL99 we deleted residues 1–116 spanning the loop as well as helices 1–4 of the protein (Muchmore et al. 1996). These mutant genes, BCL173 and BCL99, were each inserted in the EcoRI and XhoI cloning sites of a pET-21d(+) (Novagen) plasmid vector that had been previously modified to encode an N-terminal (His)6 tag. The wild-type Bcl-XL gene in the modified pET-21d(+) plasmid was a gift from John Reed (The Burnham Institute).
The FXYD genes had been previously constructed by PCR, with synthetic, overlapping, single-stranded oliogonucleotides, optimized for E. coli codons (Crowell et al. 2003). There are no Met residues in the amino acid sequence of PLM (accession: NP_005022), but those in the Mat-8 (accession: NP_068710; Met31 and Met36) and CHIF (accession: NP_071783; Met22 and Met35) sequences were mutated to Leu.
For cloning into the pBCL vectors, we introduced AflII and XhoI restriction sites at the 5′ (AflII) and 3′ (XhoI) ends of each FXYD gene, as well as a Met codon at the start of the sequences to allow CNBr cleavage. The plasmids and inserts were each digested with AflII and XhoI, purified by agarose gel electrophoresis, and ligated together. The cloning and production of Mat-8, CHIF, and PLM, using the TLE vector were described previously (Crowell et al. 2003). For cloning in the KSI vector, we introduced AlwNI and XhoI restriction sites at the ends of the FXYD genes, digested them with the enzymes, and ligated them with the similarly digested pET-31b(+) plasmid (Novagen). For each of the four systems, the resulting plasmid was transformed in DH5α, and positive clones were selected by PCR screening and DNA sequencing. For protein expression, the plasmid obtained from positive DH5α colonies was transformed in E. coli C41(DE3) (www.overexpress.com; Miroux and Walker 1996).
Protein expression
For protein expression, 5–10 μL of transformed C41(DE3) cells, from a frozen glycerol stock, were used to inoculate 10 mL of LB media and grown for 5 h at 37°C with vigorous shaking; then 1 mL of this starter culture was added to 100 mL of minimal M9 media and grown overnight. All media contained 100 μg/mL of ampicillin. In the morning, 1 L of fresh M9 media was inoculated with the overnight culture, and the cells were grown to a cell density of OD600 = 0.7. Protein expression was induced by the addition of 1 mM IPTG for 4 h at 37°C. The cells were subsequently harvested by centrifugation and stored overnight at −20°C. For uniformly 15N-labeled proteins, (15NH4)2SO4 (Cambridge Isotope Laboratories) was supplied to the M9 salts as the sole nitrogen source. SDS-PAGE was performed with the Tris-Tricine system (Schagger and von Jagow 1987), and gels were stained with Coomassie Blue G250. The band intensities were quantified by using SigmaScan Pro5.0 (SPSS).
Protein purification
Frozen cells from 1 L of culture were lysed by French press in 30 mL of buffer A (50 mM Tris HCl at pH 8.0, 15% glycerol). The soluble fraction (Fig. 2B ▶, lane S1) was removed by centrifugation (48,000g, 4°C, 30 min), and the pellet (Fig. 2B ▶, lane P1) was washed twice by resuspension in 30 mL of buffer A, followed by centrifugation (48,000g, 4°C, 30 min) to remove the soluble fraction (Fig. 2B ▶, lane S2). The resulting pellet (Fig. 2B ▶, lane P2) was dissolved in 30 mL of 6 M GdnHCl and again centrifuged (48,000g, 4°C, 2 h) to remove any insoluble materials. The 6 M GdnHCl protein solution was adjusted to 0.1 N HCl (pH 0.2), a 100-fold molar excess of solid CNBr was added, and the mixture was allowed to react overnight in the dark at room temperature. In the morning, the reaction mixture was dialyzed against water until the pH reached ~5.0 (6 h with several changes of 4 L of water, in a dialysis membrane with molecular weight cutoff of 1 kDa), lyophilized to powder, and dissolved in buffer B (20 mM Tris HCl at pH 7.0, 8 M urea). PLM was purified by ion exchange chromatography with a NaCl gradient (FF-S column, Amersham Biosciences), followed by exchange in buffer C (20 mM Tris HCl at pH 7.0, 4 mM SDS), and preparative reverse-phase HPLC (Delta-Pak C4 column, Waters), with a gradient of acetonitrile in water and 0.1% trifluoroacetic acid. Purified protein was stored as lyophilized powder at −20°C. Alternatively, the lyophilized cleavage mixture was dissolved directly in buffer C, and PLM was purified with reverse-phase HPLC.
Mass spectrometry
The molecular weight of PLM was verified by MALDI-TOF mass spectrometry. Approximately 0.2 mg of lyophilized protein were dissolved in 10 μL of solution 1 (75% acetonitrile, 25% water, 0.1% TFA), and 1 μL of this protein solution was mixed with 1 L of solution II (15 mg/mL sinnapinic acid, 300 μL acetonitrile, 200 μL methanol, 500 μL Milli-Q ultra purified water). The resulting solution was spotted onto a seed layer spot on the MALDI target. The seed layer was prepared from matrix solution (6 mg sinnapinic acid, 600 μL methanol, 390 μL acetone, 10 μL of 0.1% aqueous TFA). The MALDI-TOF mass spectrum was collected in linear mode on a Voyager DE-PRO mass spectrometer (Applied Biosystems). Typically, 250–500 laser pulses were averaged for each spectrum.
Solution NMR spectroscopy
Samples were prepared by dissolving 4 mg of 15N-labeled protein in 300 μL of NMR buffer (500 mM SDS, 10 mM DTT, 13% D2O, 20 mM sodium citrate at pH 5.0). Solution NMR experiments were performed on a Bruker AVANCE 600 spectrometer with a 600/54 Magnex magnet, equipped with a triple-resonance 5-mm probe with three-axis field gradients. The two-dimensional 1H/15N HSQC (Mori et al. 1995) spectrum was obtained at 40°C. The 15N and 1H chemical shifts were referenced to 0 ppm for liquid ammonia and tetramethylsilane, respectively. The NMR data were processed using NMR Pipe (Delaglio et al. 1995) and rendered in SPARKY (Goddard and Kneller 2004) on a Dell Precision 330 MT Linux workstation.
Solid-state NMR spectroscopy
The lipids, 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-dioleoyl-sn-glycero-3-(phospho-rac-[1-glycerol]) (DOPG) were from Avanti. Samples of PLM in lipid bilayers were prepared by adding 100 mg of the lipids (DOPC/DOPG in 8:2 molar ratio) dissolved in 1 mL of chloroform to 6 mg of 15N-labeled protein. After 5 min of bath sonication and the addition of two to five drops of trifluoroethanol, to obtain an optically clear solution, the lipid and protein mixture was distributed over the surface of 15 glass slides (11 × 11, #00, Marienfeld). After allowing the organic solvents to evaporate under vacuum overnight, the slides were stacked and equilibrated for 24 h at 40°C and 93% relative humidity, to form oriented planar lipid bilayers. The samples were wrapped in parafilm and then sealed in thin polyethylene film prior to insertion in the NMR probe. Lipid bilayer vesicles with membrane-inserted PLM were prepared by crushing the glass slides supporting the protein-reconstituted lipid bilayers in additional water, and resealing this sample in a plastic bag.
Solid-state NMR experiments were performed on a Bruker AVANCE 500 spectrometer with a 500/89 AS Magnex magnet. The home-built 1H/15N double-resonance probe had a square radiofrequency coil (11 × 11 × 3 mm) wrapped directly around the samples. The one-dimensional 15N chemical shift spectra were obtained with single contact CPMOIST (Pines et al. 1973; Levitt et al. 1986), with a cross polarization contact time of 1 msec, a 1H 90° pulse width of 5 μsec, and continuous 1H decoupling of 63 kHz. The 15N chemical shifts were referenced to 0 ppm for liquid ammonia. The data were processed using NMR Pipe (Delaglio et al. 1995) and rendered in Sparky (Goddard and Kneller 2004) on a Dell Precision 330 MT Linux workstation.
Acknowledgments
We thank John Reed for his gift of the wild-type Bcl-XL gene. We thank Xiao-Min Gong for helpful discussions, Jinghua Yu for her assistance with NMR experiments, Andrey Bobkov for his assistance with the SDS-PAGE image analysis, and Dario Miranda for performing MALDI-TOF mass spectrometry. This research was supported by grants from the National Institutes of Health (R01 CA082864), the Department of the Army Breast Cancer Research Program (DAMD17-02-1-0313), and the California Breast Cancer Research Program (8WB0110). The NMR studies utilized the Burnham Institute NMR Facility, supported by a grant from the National Institutes of Health (P30 CA030199).
Abbreviations
BCL, Bcl-XL mutant lacking the hydrophobic C terminus adapted as fusion partner
Bcl-2, B-cell leukemia/lymphoma 2
Bcl-XL, B-cell leukemia/lymphoma extra long
CHIF, corticosteroid-hormone-induced factor, FXYD4
CNBr, cyanogen bromide
CPMOIST, cross polarization with mismatch-optimized IS polarization transfer
DOPC, 1,2-dioleoyl-sn-glycero-3-phosphocholine
DOPG, 1,2-dioleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)]
GdnHCl, guanidinium HCl
HPLC, high-performance liquid chromatography
HSQC, heteronuclear single quantum coherence
HSQC-NOESY, HSQC–nuclear overhauser effect spectroscopy
IPTG, isopropyl β-D-thiogalactopyranoside
KSI, ketosteroid isomerase
ER, endoplasmic reticulum
MALDI-TOF, matrix-assisted laser desorption ionization–time of flight
Mat-8, mammary tumor protein 8 kDa, FXYD3
NMR, nuclear magnetic resonance
PISEMA, polarization inversion with spin exchange at the magic angle
PLM, phospholemman, FXYD1
PAGE, polyacrylamide gel electrophoresis
pI, isoelectric point
SDS, sodium dodecyl sulfate
TLE, a portion of the Trp ΔLE 1413 polypeptide
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041244305.
References
- Adams, J.M. and Cory, S. 1998. The Bcl-2 protein family: Arbiters of cell survival. Science 281 1322–1326. [DOI] [PubMed] [Google Scholar]
- Bannwarth, M. and Schulz, G.E. 2003. The expression of outer membrane proteins for crystallization. Biochim. Biophys. Acta 1610 37–45. [DOI] [PubMed] [Google Scholar]
- Booth, P.J. 2003. The trials and tribulations of membrane protein folding in vitro. Biochim. Biophys. Acta 1610 51–56. [DOI] [PubMed] [Google Scholar]
- Crambert, G. and Geering, K. 2003. FXYD proteins: New tissue-specific regulators of the ubiquitous Na,K-ATPase. Sci. STKE 2003: RE1. [DOI] [PubMed]
- Crowell, K.J., Franzin, C.M., Koltay, A., Lee, S., Lucchese, A.M., Snyder, B.C., and Marassi, F.M. 2003. Expression and characterization of the FXYD ion transport regulators for NMR structural studies in lipid micelles and lipid bilayers. Biochim. Biophys. Acta 1645 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson, P.E., Muir, T.W., Clark-Lewis, I., and Kent, S.B. 1994. Synthesis of proteins by native chemical ligation. Science 266 776–779. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6 277–293. [DOI] [PubMed] [Google Scholar]
- Franzin, C.M., Yu, J., and Marassi, F.M. 2005. Solid-state NMR of the FXYD family membrane proteins in lipid bilayers. In NMR Spectroscopy of biological solids (ed. A. Ramamoorthy). Marcel Dekker, New York, NY (in press).
- Goddard, T.D. and Kneller, D.G. 2004. SPARKY 3. University of California, San Francisco.
- Jones, D.H., Ball, E.H., Sharpe, S., Barber, K.R., and Grant, C.W. 2000. Expression and membrane assembly of a transmembrane region from Neu. Biochemistry 39 1870–1878. [DOI] [PubMed] [Google Scholar]
- Kiefer, H. 2003. In vitro folding of α-helical membrane proteins. Biochim. Biophys. Acta 1610 57–62. [DOI] [PubMed] [Google Scholar]
- Klammt, C., Lohr, F., Schafer, B., Haase, W., Dotsch, V., Ruterjans, H., Glaubitz, C., and Bernhard, F. 2004. High level cell-free expression and specific labeling of integral membrane proteins. Eur. J. Biochem. 271 568–580. [DOI] [PubMed] [Google Scholar]
- Kleid, D.G., Yansura, D., Small, B., Dowbenko, D., Moore, D.M., Grubman, M.J., McKercher, P.D., Morgan, D.O., Robertson, B.H., and Bachrach, H.L. 1981. Cloned viral protein vaccine for foot-and-mouth disease: responses in cattle and swine. Science 214 1125–1129. [DOI] [PubMed] [Google Scholar]
- Kochendoerfer, G.G. 2001. Chemical protein synthesis methods in drug discovery. Curr. Opin. Drug Discov. Devel. 4 205–214. [PubMed] [Google Scholar]
- Kochendoerfer, G.G., Salom, D., Lear, J.D., Wilk-Orescan, R., Kent, S.B., and DeGrado, W.F. 1999. Total chemical synthesis of the integral membrane protein influenza A virus M2: Role of its C-terminal domain in tetramer assembly. Biochemistry 38 11905–11913. [DOI] [PubMed] [Google Scholar]
- Kochendoerfer, G.G., Jones, D.H., Lee, S., Oblatt-Montal, M., Opella, S.J., and Montal, M. 2004. Functional characterization and NMR spectroscopy on full-length Vpu from HIV-1 prepared by total chemical synthesis. J. Am. Chem. Soc. 126 2439–2446. [DOI] [PubMed] [Google Scholar]
- Kuliopulos, A., Nelson, N.P., Yamada, M., Walsh, C.T., Furie, B., Furie, B.C., and Roth, D.A. 1994. Localization of the affinity peptide-substrate inactivator site on recombinant vitamin K-dependent carboxylase. J. Biol. Chem. 269 21364–21370. [PubMed] [Google Scholar]
- Levitt, M.H., Suter, D., and Ernst, R.R. 1986. Spin dynamics and thermodynamics in solid-state NMR cross-polarization. J. Chem. Phys. 84 4243–4255. [Google Scholar]
- Lindhout, D.A., Thiessen, A., Schieve, D., and Sykes, B.D. 2003. High-yield expression of isotopically labeled peptides for use in NMR studies. Protein Sci. 12 1786–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majerle, A., Kidric, J., and Jerala, R. 2000. Production of stable isotope enriched antimicrobial peptides in Escherichia coli: An application to the production of a 15N-enriched fragment of lactoferrin. J. Biomol. NMR 18 145–151. [DOI] [PubMed] [Google Scholar]
- Marassi, F.M. 2001. A simple approach to membrane protein secondary structure and topology based on NMR spectroscopy. Biophys. J. 80 994–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marassi, F.M. and Opella, S.J. 2000. A solid-state NMR index of helical membrane protein structure and topology. J. Magn. Reson. 144 150–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miozzari, G.F. and Yanofsky, C. 1978. Translation of the leader region of the Escherichia coli tryptophan operon. J. Bacteriol. 133 1457–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miroux, B. and Walker, J.E. 1996. Over-production of proteins in Escherichia coli: Mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260 289–298. [DOI] [PubMed] [Google Scholar]
- Mori, S., Abeygunawardana, C., Johnson, M.O., and Vanzijl, P.C.M. 1995. Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation. J. Magn. Reson. B 108 94–98. [DOI] [PubMed] [Google Scholar]
- Muchmore, S.W., Sattler, M., Liang, H., Meadows, R.P., Harlan, J.E., Yoon, H.S., Nettesheim, D., Chang, B.S., Thompson, C.B., Wong, S.L., et al. 1996. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature 381 335–341. [DOI] [PubMed] [Google Scholar]
- Opella, S.J., and Marassi, F.M. 2004. Structure determination of membrane proteins by NMR spectroscopy. Chem. Rev. 104 3587–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opella, S.J., Ma, C., and Marassi, F.M. 2001. Nuclear magnetic resonance of membrane-associated peptides and proteins. Methods Enzymol. 339 285–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines, A., Gibby, M.G., and Waugh, J.S. 1973. Proton-enhanced NMR of dilute spins in solids. J. Chem. Phys. 59 569–590. [Google Scholar]
- Reed, J.C. 1998. Bcl-2 family proteins. Oncogene 17 3225–3236. [DOI] [PubMed] [Google Scholar]
- Rogl, H., Kosemund, K., Kuhlbrandt, W., and Collinson, I. 1998. Refolding of Escherichia coli produced membrane protein inclusion bodies immobilised by nickel chelating chromatography. FEBS Lett. 432 21–26. [DOI] [PubMed] [Google Scholar]
- Schagger, H. and von Jagow, G. 1987. Tricine-sodium dodecyl sulfate–poly-acrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166 368–379. [DOI] [PubMed] [Google Scholar]
- Schiffer, M. and Edmunson, A.B. 1967. Use of helical wheels to represent the structures of proteins and to identify segments with helical potential. Biophys. J. 7 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, V.R. and Walker, J.E. 2003. Purification and folding of recombinant bovine oxoglutarate/malate carrier by immobilized metal-ion affinity chromatography. Protein Exp. Purif. 29 209–216. [DOI] [PubMed] [Google Scholar]
- Staley, J.P. and Kim, P.S. 1994. Formation of a native-like subdomain in a partially folded intermediate of bovine pancreatic trypsin inhibitor. Protein Sci. 3 1822–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier, F.W., Rosenberg, A.H., Dunn, J.J., and Dubendorff, J.W. 1990. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 185 60–89. [DOI] [PubMed] [Google Scholar]
- Sweadner, K.J. and Rael, E. 2000. The FXYD gene family of small ion transport regulators or channels: cDNA sequence, protein signature sequence, and expression. Genomics 68 41–56. [DOI] [PubMed] [Google Scholar]
- Wang, J., Denny, J., Tian, C., Kim, S., Mo, Y., Kovacs, F., Song, Z., Nishimura, K., Gan, Z., Fu, R., et al. 2000. Imaging membrane protein helical wheels. J. Magn. Reson. 144 162–167. [DOI] [PubMed] [Google Scholar]
- Wiener, M.C. 2004. A pedestrian guide to membrane protein crystallization. Methods 34 364–372. [DOI] [PubMed] [Google Scholar]