

Abstract
Calbindin D28k (calbindin) is a cytoplasmic protein expressed in the central nervous system, which is implied in Ca2+ homeostasis and enzyme regulation. A combination of biochemical methods and mass spectrometry has been used to identify post-translational modifications of human calbindin. The protein was studied at 37°C or 50°C in the presence or absence of Ca2+. One deamidation site was identified at position 203 (Asn) under all conditions. Kinetic experiments show that deamidation of Asn 203 occurs at a rate of 0.023 h−1 at 50°C for Ca2+-free calbindin. Deamidation is slower for the Ca2+-saturated protein. The deamidation process leads to two Asp iso-forms, regular Asp and iso-Asp. The form with regular Asp 203 binds four Ca2+ ions with high affinity and positive cooperativity, i.e., in a very similar manner to non-deamidated protein. The form with β-aspartic acid (or iso-Asp 203) has reduced affinity for two or three sites leading to sequential Ca2+ binding, i.e., the Ca2+-binding properties are significantly perturbed. The status of the cysteine residues was also assessed. Under nonreducing conditions, cysteines 94 and 100 were found both in reduced and oxidized form, in the latter case in an intramolecular disulfide bond. In contrast, cysteines 187, 219, and 257 were not involved in any disulfide bonds. Both the reduced and oxidized forms of the protein bind four Ca2+ ions with high affinity in a parallel manner and with positive cooperativity.
Keywords: calbindin D28k, deamidation, disulfide bond, calcium binding, mass spectrometry, post-translational modifications, ICAT
Calbindin D28k (calbindin) is a cytoplasmic protein mainly expressed in the central nervous system, where it constitutes 0.1%–1.5% of the total amount of protein (Christakos et al. 1989). Calbindin is noted for its distinct expression pattern restricted to certain neuronal cell types, and is also expressed in the epithelial tissue and in kidney (Andressen et al. 1993). The protein plays an important role in Ca2+ homeostasis of the central nervous system (Airaksinen et al. 1997).
Calbindin (28.9 kDa, 261 residues) (Fig. 1 ▶) contains six EF hands (Kretsinger and Nockolds 1973), of which EF hands 1, 3, 4, and 5 bind Ca2+ with high affinity (Åkerfeldt et al. 1996). The protein has exposed hydrophobic surfaces, both in the absence and presence of bound Ca2+ (Berggård et al. 2000a). Calbindin undergoes conformational changes that are induced over the cellular variation in free Ca2+ concentration, suggesting that the protein may act both as a Ca2+ buffer and a Ca2+ sensor (Berggård et al. 2002a). Recent studies have identified caspase-3 (Bellido et al. 2000), myo-inositol monophosphatase (Berggård et al. 2002b), and the Ran-binding protein (Lutz et al. 2003) as target proteins. Four out of five cysteines in human calbindin can be S-nitrosylated by NO, suggesting that the protein plays the role of a NO reservoir (Tao et al. 2002). In addition, cysteine residues are involved in redox-regulation of the protein conformation (Cedervall et al. 2005). Taken together, these results show that the regulation of calbindin is complex, necessitating further investigations to identify factors influencing the structure and functions of this protein.
Figure 1.

(A) Location of EF hands in the sequence of human calbindin D28k. Black circle represents the Ca2+ ion. Mutation sites are underlined. (B) The EF-hand motifs are shown with high affinity Ca2+ sites marked by black circles. Mutant or fragment name is given to the left, with the amino acid identity at each substitution site given to the right. Fragments borders are indicted by numbers inside the bars. (C) List and numbering of the endo-Lys C fragments, as cited in the text, which contain Q and/or N residues and which have molecular weight >700 Da. (D,E) List and numbering of the Asp and chymotrypsin fragments, respectively, which allows the mapping of Q and N inaccessible with Lys-C digestion.
Post-translational chemical modifications of proteins can be classified as enzymatic or nonenzymatic. The biological activity of the protein may be tuned by enzymatic modifications such as phosphorylation (McLachlin and Chait 2001), glycosylation (Luthra and Balasubramanian 1993), acetylation (Sen and Chakrabarti 1990), hydroxylation (Drakenberg et al. 1983), and carboxylation (Stenflo et al. 1974). Nonenzymatic processes, as well as their biological functions, are sometime difficult to evaluate in vitro, because of the impossibility of mimicking exactly the cellular environment. These processes often lead to a dramatic change in the biophysical characteristics of the protein and involve disulfide bond formation (Ozaki et al. 1987) and deamidation (Berson and Yalow 1966). Deamidation involves the spontaneous conversion of Asn to Asp or Gln to Glu (for review, see Bischoff and Kolbe 1994; Reissner and Aswad 2003). To our knowledge, no enzymatic process has been identified for conversion of Asn to Asp or iso-Asp. However, deamidation of glutamine can occur in vivo also as an enzymatic process. In celiac disease this leads to the formation of pathogenic epitopes leading to autoimmune disease (Molberg et al. 1998; Arentz-Hansen et al. 2000). Some pathogenic bacteria use catalytic deamidation as a key step in the attack (Lerm et al. 2002).
In the present work, we have studied deamidation and disulfide bond formation in human calbindin, and their effect on Ca2+ binding. Both types of modifications are compared for the apo and Ca2+ forms of calbindin at 37°C, which is the physiological temperature, and at 50°C, which is the temperature used in the NMR study (Helgstrand et al. 2004). We have used biochemical methods, including enzymatic digestion and chemical derivatization in combination with mass spectrometry (MS). The Ca2+-binding constants of deamidated and reduced versus nonreduced protein were measured using competition with a chromophoric chelator (Linse 2002).
Results
Deamidation by agarose gel electrophoresis
Calbindin has previously been suspected to deamidate at neutral or basic pH (Thulin and Linse 1999). Deamidation is a nonenzymatic post-translational modification (Fig. 2 ▶; Bornstein and Balian 1970). To delineate the region of deamidation, cloned calbindin fragments (see Fig. 1 ▶ or Abbreviations for nomenclature) were analyzed by native agarose electrophoresis (Johansson 1972) after incubation at neutral pH (Fig. 3 ▶). The method allows the separation of molecular species differing by one or more charges. Each of F12, F34, and F123 migrates as one band, whereas each of F56 and F456 exhibits two bands, indicative of deamidation in the C-terminal part of the protein.
Figure 2.

Molecular mechanism of deamidation.
Figure 3.

Native agarose gel electrophoresis of the F12, F34, F56, F123, and F456 fragments of calbindin.
Deamidation site identification by mass spectrometry
Deamidation leads to a +1-Da increment per deamidation event (Fig. 2 ▶). We have therefore used MS to perform an unbiased study in which all Asn and Gln residues of calbindin were considered potential deamidation sites. The wild-type protein was incubated at 37°C and 50°C in the presence of EDTA (ethylenediaminetetraacetic acid) or Ca2+. The occurrence of deamidation was tracked by MALDI-Tof (matrix-assisted laser desorption ionization time-of-flight) analysis of peptide mixtures generated by digestion with endoproteinase Lys-C, chymotrypsin or Asp-N. The Lys-C fragments over 700 Da and complementary fragments from chymotrypsin and Asp-N digestions that contain Asn or Gln residues are shown in Figure 1C ▶. With the exception of two fragments, labeled E1 and E2 (m/z 3874.9 and m/z 3893.9 monoisotopic [M+H]+ ions, respectively), no unassigned fragments are observed after a 3-h digestion period at 37°C. The analysis of the E1 and E2 fragments is presented in the “disulfide bond” section below.
No significant mass variation was observed for the different peptide fragments arising from the three proteases, except for fragments 194–216 (Lys-C), 199–211 (chymotrypsin), and 201–216 (Asp-N). All three fragments display a mass shift of +1 Da and include Asn 203 and Asn 209. The monoisotopic ion peak of the nondeamidated 194–216 fragment appears at m/z 2645.23 [M + H]+. No fragment arising from incomplete digestion could have this mass (simulation with Cloe 2.0 software, http://www.ibs.fr/software). The isotopic profile changes over time, with an overlap between the isotopic massifs of nondeamidated and deamidated protein (Fig. 4 ▶). The intensity of the monoisotopic ion peak at m/z 2645.2, [M + H]+ is progressively decreasing, and the relative intensity of the other peaks change upon time. After 5 d, the monoisotopic ion peak of the fragment 194–216 is completely shifted to m/z 2646.2. The mass increase could correspond to one deamidation event in the m/z 2645.1 peptide.
Figure 4.

(Left) Close-up view of the MALDI-ToF spectra of the 194–216 peptide isotopic profile at different time points during the course of deamidation (0 h, 24 h, and 72 h). (Right) Deconvolution of the isotopic profiles of nondeamidated and deamidated peptide mixture.
Localization of the deamidation site
To localize the deamidation site, the fully deamidated 194–216 peptide was sequenced by MS-MS spectrometry (Fig. 5 ▶). For this experiment, the doubly charged ion of the deamidated peptide was selected for collision-induced dissociation. The MS-MS spectrum shows unambiguously that the B and Y ions are increased by one mass unit starting from the B11 and Y14 ions, respectively, which identifies Asn 203 as the site of deamidation.
Figure 5.

(A) Tandem mass spectra of the 194–216 deamidated peptide (fragment 20 of Lys-C digestion). Ions labeled by a star contain the deamidation site. (B) Calculated and recorded mass, both in [M + 2H]2+ as for the spectrum, for the fragments of interest in B and Y series. (C) Sequence resulting from the MS/MS sequencing.
Deamidation rate
Since the mass increment due to deamidation is +1, peaks of the isotopic profiles of deamidated and nondeamidated species overlap (Fig. 4 ▶). Therefore, the experimental profiles need to be deconvoluted into the two components to estimate their relative proportions. The theoretical isotopic contributions of the deamidated and nondeamidated A194-K216 peptide was calculated using the Selket software from http://medweb.unimuenster.de/institute/impb/research/hillenkamp/ and are shown separately and as weighted sums (Fig. 4 ▶). In the sums, the peak at m/z 2645.2 corresponds to the monoisotopic ion of the nondeamidated fragment only. On the basis of the relative intensity of this peak, it is possible to calculate the isotopic contribution of the deamidated peptide to each mass, and hence the fractions of non-deamidated and deamidated form. As the deamidated and nondeamidated peptides possess almost the same amino acid composition with only a slight change in pI, the ionization yields of the two forms are assumed to be the same. For the apo form at 50°C the deamidation rate for Asn 203 is estimated to be 0.023 h−1, assuming a mono-exponential decay, A(t)/A0 = exp(−kt), where A(t) and A0 represent the amount of nondeamidated peptide at times t and zero, respectively.
Disulfide bond
During the analysis of the deamidation data, two high molecular mass peptides (E1 and E2 (m/z 3874.9 and m/z 3892.9, monoisotopic [M+H]+, respectively) were observed under all conditions. None of these peptides can be assigned from the theoretical digestion pattern and they are not affected by prolonged digestion. Therefore, the effect of the reducing agent DTT (dithiothreitol) on the peptide mixture was analyzed. Mass spectra acquired for the reduced and nonreduced mixture with Ca2+ at 37°C are shown in Figure 6, A and B ▶, with a close-up view of the E1 and E2 region in Figure 6, C–E ▶. The E2 peptide disappears upon reduction with DTT (Fig. 6D ▶) and at the same time, the intensity of the two peptides with masses of 3022.70- and 872.45-Da increases (Fig. 6B ▶). These peptides correspond to I73-K98 (Lys-C peptide 6) and S99-K105 (Lys-C peptide 7), and encompass Cys 94 and Cys 100, respectively. This means that E2 is composed of peptides 6 and 7, covalently linked by a disulfide bond. On the contrary, E1 is not affected by DTT. An MS/MS spectrum of the protonated molecule of E1 proves that this peptide contains the same I73-K105 residues as E2 (data not shown). Since reduction was effective for E2 (Fig. 6C,D ▶), the resistance to disulfide reduction is not due to the solution conditions during the desalting step. Another possibility is that aggregation of the E1 peptide prevents reduction by DTT. The solution was therefore submitted to sonication prior to reduction in order to dissociate any aggregated E1. The MALDI-ToF MS spectrum after sonication and reduction shows a +2-Da shift in E1 (Fig. 6E ▶), confirming that E1 includes I73-K105 with a disulfide bond between Cys 94 and 100. On the one hand, these results demonstrated the presence of an intramolecular disulfide bridge involving Cys 94 and Cys 100 in fragment E1. The E2 fragment on the other hand could correspond to an intramolecular species that has been cleaved at Lys98, or could originate from an intermolecular disulfide bridge between Cys 94 and Cys 100 in a protein dimer.
Figure 6.

MALDI-ToF mass spectra of the endo-LysC digest of calbindin without (A) and with DTT (B). Close-up views of the spectra of the 73–105 peptide (fragment E1) and of the 73–98/99–105 disulfide-linked peptide (fragment E2), marked by dotted circles in (A) and (B), without DTT reduction (C), after reduction with DTT (D), and after sonication and reduction with DTT (E).
The kinetic study reveals that E1 and E2 fragments are present at zero time under all conditions of the incubation experiment, in addition to the peptides T73-K98 and S99-K105, which include the reduced cysteines 94 and 100, respectively. Over time, the E1 and E2 signal intensity increases by up to five times in Ca2+, but is constant in EDTA at both temperatures. Hence, the formation and/or stabilization of the disulfide bridge between Cys 94 and Cys 100 appears to be linked to Ca2+ binding.
Alkylation of cysteine residues
MALDI-ToF MS mapping after alkylation with ICAT1 reagent (Gygi et al. 1999) was used as a complementary approach to determine the oxidation state of the cysteine residues. Denatured full-length calbindin was first incubated with ICAT in the absence of DTT, followed by proteolytic cleavage. This protocol allows the purification and detection only of cysteines that are reduced in the full-length protein (fraction 1). Reduction with DTT, followed by a second alkylation with ICAT, leads to the trapping of cysteines that were disulfide-bonded in the intact protein (fraction 2). The results are summarized in Table 1. Clearly, Cys 100 was found in both fractions 1 and 2, showing that the oxidized and reduced forms coexist in the full-length protein. The peptides encompassing cysteines 187, 219, and 257 were labeled in the absence of DTT, and were not retrieved in fraction 2, confirming unambiguously that these three cysteines are not engaged in any disulfide bonds.
Table 1.
Results of the ICAT alkylation
| Peptide | Measured mass of ICAT peptides | Calculated mass of ICAT peptides | Alkylation.Identified cysteines (Fraction 1) | Reduction and alkylation.Identified cysteines (Fraction 2) |
| 72–97 | n.d. | 3465.90 | n.d. | n.d. |
| 98–104 | 1331.6 | 1331.6 | C100 | C100 |
| 180–192 | 1971.7 | 1971.9 | C187 | — |
| 216–220 | 1050.8 | 1050.2 | C219 | — |
| 246–260 | 2094.8 | 2094.5 | C257 | — |
Fraction 1: alkylation of the peptides mixture obtained from digestion of the nonreduced wt calbindin. Fraction 2: alkylation of the previous peptides mixture after reduction.
Macroscopic Ca2+-binding constants–effect of disulfide bond
The method for Ca2+-binding constant determination uses a chelator for which the absorbance at 263 nm is reduced upon calcium binding. The quin 2 chelator was chosen because it has similar Ca2+-affinity as wild-type calbindin (Berggård et al. 2002a). In a plot of absorbance versus total Ca2+ concentration, a solution with only chelator yields a linear change until a 1:1 ratio of calcium to chelator is reached, after which no further change is seen (Fig. 7A ▶, dotted line). In the presence of a Ca2+-binding protein, this point in the titration is shifted by an amount corresponding to the concentration of Ca2+ sites having an affinity comparable to that of the chelator. Titration curves with different shapes are obtained depending on whether the protein binds Ca2+ with the same, higher, or lower affinity as the chelator. Simulated curves for a protein with one site with the same (dashed line) or twofold higher (solid line) Ca2+ affinity than quin 2 are shown in Figure 7A ▶. Sequential and cooperative binding produces curves with different shapes allowing distinction between these scenarios (lines with long dashes and solid line with symbols in Fig. 7A ▶).
Figure 7.

Ca2+-titrations of quin2 in the presence of calbindin. (A) Simulated curves for 25 μM quin2 and 25 μM protein, for five cases where the protein contains: (1) no Ca2+-binding site (dotted line); (2) one site with the same affinity as quin 2 (dashed line); (3) one site with twofold (0.3 log units) higher affinity than quin 2 (solid line); (4) three sites with same affinity as quin 2 and no cooperativity plus one site that is 100-fold weaker (line with long dashes); (5) four sites with strong positive cooperativity and on average twofold higher affinity than quin 2 (solid line with symbols). (B) Experimental data for ED-CCCCC in reduced (♦) and nonreduced (▪) forms, with fitted curves as solid lines. (C) Experimental data for wild type (•) or deamidated wild type (▾) under reducing conditions, with fitted curves as solid lines. All data are obtained in 2 mM Tris/HCl, pH 7.5. Reduced samples (prepared as described in the Methods section) contain 1 mM DTT. (A–C) The absorbance is normalized such that 1.0 corresponds to apo quin 2 and 0.0 to Ca2+bound quin2. The total calcium concentration is normalized such that 1.0 corresponds to the quin 2 concentration plus four times the protein concentration. (D) Relative error square sum (χ2) obtained for different fractions of the form with regular Asp (expressed in percent) for the cases of one (line with squares) or two (line with filled circles) high affinity sites in the form with iso-Asp. (E,F) Transformed data from panels B and C, respectively, to display the average number of bound Ca2+ ions per protein as a function of free Ca2+ concentration.
To assess the role of the disulfide bond in Ca2+-binding, titration experiments were performed for nonreduced and reduced wild type as well as the ED-CCCCC mutant (Fig. 7B ▶; for mutant nomenclature see Fig. 1 ▶). The ED mutations were incorporated to avoid inhomogeneity due to deamidation. All curves are sigmoidal, indicative of cooperative Ca2+ binding. Data points for nonreduced protein lie below the reduced protein, indicating lower Ca2+ affinity for the nonreduced protein (compare dashed and solid lines in Fig. 7A ▶). The coinciding inflection points suggest the same stoichiometry of Ca2+ binding for reduced and nonreduced protein. Equation 1 was fitted to the data to obtain estimates of the macroscopic Ca2+-binding constants. The stoichiometry of high affinity Ca2+ binding sites is four in both cases, and the overall affinity for Ca2+ is slightly higher for the reduced protein. The mean logarithm of the four macroscopic binding constants is 8.8 ± 0.05 for reduced and 8.5 ± 0.05 for nonreduced ED-CCCCC, respectively, and 8.7 ± 0.05 for reduced and 8.5 ± 0.05 for nonreduced wild type, respectively. The separate macroscopic binding constants are obtained with lower precision (±0.2 for lgK1 and lgK2 and 0.1 for lgK3 and lgK4) and are as follows (given in the order lgK1, lgK2, lgK3, lgK4); 9.0, 9.2, 8.8, 8.2 for reduced ED-CCCCC, 8.7, 8.9, 8.5, 7.9 for nonreduced ED-CCCCC, 8.9, 9.1, 8.7, 8.1 for reduced wild type and 8.7, 8.9, 8.5, 7.9 for nonreduced wild type. Hence, both proteins display a small affinity increase upon reduction. In Figure 7E ▶ the data are transformed to the number of bound Ca2+ ions versus free Ca2+ concentration. The reduced protein has higher affinity for Ca2+ (curve shifted to lower Ca2+ concentration) with maintained cooperativity (steepness of curve) and stoichiometry of four (end point).
Macroscopic Ca2+-binding constants–effect of deamidation
To assess the effect of deamidation on Ca2+-binding, titration experiments were performed for a sample of reduced wild-type calbindin for which deamidation had been forced by 10-d incubation (Fig. 7C ▶). The integrity of the protein was checked by SDS-PAGE gel electrophoresis. The deamidated protein yielded one major band with the same mobility as the nondeamidated protein, showing that the protein chain was intact in the deamidated sample. There is a dramatic effect of deamidation on the Ca2+-titration curve, which has become straighter with the inflection point at lower Ca2+ concentration (Fig. 7C ▶). This shows that the number of strong binding sites is reduced and that the Ca2+ affinity is lower. Equation 1 was fitted to the data in Figure 7C ▶ to obtain estimates of the macroscopic Ca2+-binding constants. As stated above, the nondeamidated but reduced wild-type has four high-affinity sites with an average macroscopic lgK of 8.7 ± 0.05, and positive cooperativity. The data for the reduced deamidated wild-type was fitted assuming that the sample contains two species in fractions X (Asp) and 1-X (iso-Asp). The form with normal Asp can be represented by the reduced ED-CCCCC mutant, because no difference in Ca2+-binding was detected due to the Q182E substitution (Cedervall et al. 2005). For this form, the stoichiometry was fixed at four high-affinity sites, and the four macroscopic binding constants were locked at the values obtained for reduced ED-CCCCC. The iso-Asp species was allowed to contain four, three, two, or one high-affinity site. As shown in Figure 7D ▶, the data could only be fitted if the iso-Asp species contains two or one high-affinity sites. Two solutions yield equally good fits. In the first solution the iso-Asp form represents 67% of the deamidated protein and contains two high-affinity sites (average lgK = 8.0) and two lower affinity sites (average lgK = 6.3). This number (67% iso-Asp) is close to what is generally obtained (Geiger and Clarke 1987; Patel and Borchardt 1990; Tyler-Cross and Schirch 1991). In the second solution, the iso-Asp form represents 45% of the deamidated protein and contains one high-affinity site (lgK = 8.0) and three lower affinity sites (average lgK = 6.3). This solution is less likely because fraction of iso-Asp is lower than generally obtained. It was not possible to fit the data with three or four strong sites in the iso-Asp form. Although there are two possible solutions, it is clear that the deamidated protein with iso-Asp binds Ca2+ in a sequential manner, and that deamidation affects more than one site. The two groups of sites differ by a factor of 50 in affinity. Figure 7F ▶ shows data that have been transformed to the number of bound Ca2+ ions versus free Ca2+ concentration, clearly illustrating the sequential binding to the deamidated protein.
Discussion
The results of the present study clearly show that post-translational modifications of calbindin have profound effects on its calcium-binding properties. One type of post-translational modification identified for calbindin in this study is the deamidation of Asn 203. Deamidation is a spontaneous nonenzymatic post-translational modification that converts asparagine to aspartate or glutamine to glutamate (Berson and Yalow 1966). Isopeptide linkages (β-peptide linkages) tend to dominate over normal α-peptide linkages at the site of deamidation. Deamidation of an Asn side chain primarily takes place via the formation of a cyclic imide intermediate (Fig. 2 ▶) that may open up in two different ways: either to give back the normal Asp-Gly linkage (α-peptide linkage) or to provide an isoaspartyl linkage (β-peptide linkage) (Bornstein 1970; Bornstein and Balian 1970). The deamidation process is influenced by several parameters, including the type of amino acid following Asn or Gln, temperature, and pH, with higher rate at elevated temperature and pH (for review, see Wright 1991; Bischoff and Kolbe 1994). Asn-Gly and Gln-Gly are the most frequently observed sequences in deamidation events. Glycine enhances dramatically the deamidation rate because of high local backbone flexibility and the lack of steric hindrance imposed by a side chain (Bischoff and Kolbe 1994), which favors the formation of the succinimide intermediate. The amino acid conversion leads to a mass increase of +1 Da per deamidation event, which can be accurately measured by MALDI-ToF MS on a peptide mixture using an internal reference.
Here, only one deamidation site was detected and only in peptides including Asn 203 and Asn 209. The localization of the deamidation site was performed by MS/MS sequencing of the A194-K216 peptide, showing unambiguously that Asn203 is the site of deamidation. Another regular deamidation sequences is found in calbindin at Gln 182-Gly 183. However, no mass change was detected in fragments encompassing Gln 182, which means that Gln 182 is not deamidated under the assessed conditions. However, deamidation of this residue could still occur at a very low rate. Asn 203 is located in the Ca2+-binding loop of EF-hand 5, which is one of four high-affinity Ca2+-binding sites of calbindin. The deamidation is observed only on the apo-form with a deamidation of 0.023 h−1 at 50°C. This rate implies a half-life of 1.4 d, in comparison with 0.9 d as reported for a GSNGG peptide at pH 7.4, 30°C (Robinson et al. 2001). Our value hence suggests considerable flexibility of EF-hand 5 in the apo form. Most likely, Ca2+-binding prevents deamidation by confining the backbone.
Deamidation introduces an extra charge. Therefore, two distinct bands are observed on agarose gel for the F456 and F56 fragments, which contain EF-hand 5 (Fig. 3 ▶). The side-chain oxygen of Asn 203 can be inferred as one of the Ca2+ ligands in EF-hand 5 because this is a regular EF hand. Using the ED-CCCCC mutant that corresponds to deamidated calbindin with the α-peptide linkage, we find that this form has slightly elevated Ca2+ affinity. In contrast, we find for the sample of deamidated wild type that the β-peptide linkage leads to significantly reduced Ca2+ affinity, not only for EF-hand 5 but also for one or two proximal site(s). Our findings agree with earlier reports on calbindin D9k, a small protein with two EF hands and a deamidation site at a Ca2+ ligand. For deamidated calbindin D9k, the α-form has marginally increased Ca2+ affinity, while the β-form displays reduced Ca2+ affinity for both sites (Chazin et al. 1989).
Recent work has shown that the function of calbindin as an activator of myo-inositol monophosphatase is dependent on the redox potential, and the presence of cysteine residues (Cedervall et al. 2005). For rat calbindin, one disulfide bond was identified between Cys 94 and Cys 100 (Johnson et al. 1999; Tao et al. 2002). The same bond was ruled out for human calbindin (Tao et al. 2002), which displays 98.5% sequence identity with the rat protein. The samples used in the present deamidation study allowed us to track the status of the cysteines in human calbindin. The results clearly demonstrate that cysteine 187, 219, and 257 are solvent-accessible and not present in any disulfide bonds (Table 1). On the contrary, our MS data show the presence of a disulfide bond between Cys 94 and Cys 100 (E1 and E2 fragments). The E1 fragment corresponds to the peptide I73-K105 with an intact Lys 98 peptide bond, hence the 94–100 disulfide bond is intramolecular. In fragment E2, the Lys 98 peptide bond is cleaved, and the disulfide bond could be intra- or intermolecular. The results of the ICAT approach show that the reduced and oxidized forms of Cys 100 coexist in human calbindin. This may explain why the Cys 94-Cys 100 bond was not detected in the earlier study (Tao et al. 2002).
Disulfide bond formation was found to be a Ca2+-dependent process. Ca2+ binding may stabilize a conformation of the linker between EF-hand 2 (no affinity for Ca2+) and EF-hand 3 (one of the four high-affinity Ca2+ sites) that favors the 94–100 disulfide formation. Disulfide bond formation has a significantly smaller effect on the Ca2+ binding properties compared to deamidation. The oxidized protein still binds four Ca2+ ions with high affinity and positive cooperativity, with only slightly reduced affinity.
It was recently found that the conformation and activity of calbindin is regulated by the redox potential, and data for a series of cysteine to serine mutants showed that Cys94 and Cys100 are responsible for this redox dependency (Cedervall et al. 2005). These and the present results indicate that the disulfide bond between Cysteine 94 and 100 in calbindin can exist under in vivo conditions and that its formation is controlled via the cellular redox potential.
Materials and methods
Materials
All chemicals were of analytical grade and purchased from Sigma Aldrich. The endoproteinase Lys C, was from Calbiochem, and chymotrypsin and Asp-N from Roche. Restriction enzymes were purchased from New England Biolabs. The ICAT reagent and phase Poros R2–50 μm were from Applied Biosystem. The PCR and sequencing primers were from DNA-technology, GFX plasmid and GFX PCR and gel band purification kits from Amersham Pharmacia Biotech, and DNA polymerase Pwo from Roche. BL21 (DE3) pLysS Star strain was from Invitrogen.
Calbindin mutant construction
The starting material for mutagenesis was the wild-type human calbindin D28k gene (wild-type calbindin) cloned in a modified Pet 3a plasmid (PetSac), which carries ampicillin resistance, a T7 RNA polymerase inducible by isopropyl-ß-D-thiogalactopyranoside (IPTG) (Studier and Moffatt 1986), and NdeI and SacI cloning sites (Thulin and Linse 1999). One calbindin mutant was constructed to provide protein with Gln182 and Asn203 deamidated to regular Glu and Asp, respectively. The mutant contains two amino acid substitutions, Q182E and N203D and is named ED-CCCCC (Fig. 1 ▶). The mutant was prepared using standard PCR methods and the sequences were verified using DNA sequencing (Cedervall et al. 2005). Five fragments of wild-type calbindin are constructed F12, F34, F56, F123, and F456 (Berggård et al. 2000b), including EF-hand 1–2, 3–4, 5–6, 1–3, and 4–6, respectively (Fig. 1 ▶).
Expression and purification
Wild-type calbindin and ED-CCCCC were expressed and purified as described (Thulin and Linse 1999), except that the Escherichia coli strain BL21 (DE3) pLysS Star strain was used. The EF-hands fragments were expressed and purified as reported (Berggård et al. 2000b). Note that the purification of proteins and fragments is performed at pH 5.6 to avoid deamidation, yielding up to 200 mg of pure protein per liter of culture.
Post-translational modifications
Deamidation of Gln and Asn residues, as well as the oxidation state of the five cysteine residues were assessed for apo- and Ca2+-loaded wild-type calbindin in reducing and nonreducing environment as described below. Ca2+-depleted protein (1 mM) was first dissolved in 50 mM ammonium acetate buffer with 10 mM DTT at pH 7.0. The apo and Ca2+-loaded forms were obtained by adding 10 mM EDTA or 10 mM CaCl2, respectively. Gel filtration of each sample was performed on a Sephadex 75 HR 10/30 column (in 50 mM ammonium acetate buffer, pH 7.0) to remove DTT and EDTA or excess Ca2+. The monomer fraction was collected and each sample was split in two parts. One part, samples referred to as nonreducing condition, were kept in neat buffer from this point an on, while the other part (reducing condition) was complemented with 1 mM DTT. Each part was divided into two tubes that were incubated at 37°C and 50°C, respectively. Deamidation and disulfide bond formation was followed over 1 wk with sampling every 24 h. Each day, 9 μg of protein was removed and submitted to complete proteolysis by endoproteinase Lys-C digestion for 3 h (protease/protein ratio = 1/50), or chymotrypsin (protease/protein ratio = 1/60), or Asp-N (protease/protein ratio = 1/250). The resulting peptide mixture was desalted and analyzed by MALDI-ToF, with and without adding DTT for disulfide reduction, and by ESI (electrospray ionization) mass spectrometry for MS-MS sequencing.
Labeling of free cysteine residues
The determination of the status of cysteines by the ICAT methodology (Gygi et al. 1999) was achieved as follows for wild-type calbindin. A protein aliquot (100 μg, 64 μM) in a 50 mM Tris/HCl buffer, pH 7.0, was denatured in the presence of 0.1% of SDS, 5 mM EDTA by heating up to 100 °C for 10 min. The ICAT alkylation (+ 442.225 Da) was achieved by transferring the sample to a vial containing the ICAT reagent. The molar ratio between ICAT and calbindin cysteines was 12. The mixture was incubated for 1 h at room temperature in the dark. Alkylated calbindin was then digested with the endo-Lys-C at pH 7.5. Peptides labeled by the ICAT reagent (fraction 1) were extracted from the peptide mixture on a streptavidin column, following the manufacturer’s protocol. The nonalkylated peptides were recovered, freeze-dried, and reconstituted with 20 mM ammonium acetate, prior to reduction with 1mM DTT, pH 7.0. After one hour incubation at 37 °C, the ICAT alkylation was repeated. Separation of alkylated peptides (fraction 2) from nonalkylated ones was carried out on a streptavidin column. The set of collected fractions were then analyzed by MALDI-ToF MS. Fraction 1 contains peptides with cysteines that were not involved in disulfide bonds and therefore accessible to alkylation in the nonreduced but denatured full-length protein, or in a peptide digest that had not been reduced. Fraction 2 contains peptides with cysteines that were disulfide bonded in the full-length protein.
Sample preparation for mass spectrometry
The peptide mixture was purified and concentrated on phase Poros R2–50 μm resin filled in a gel loader tip (Eppendorff). Peptides were washed with a solution of 5% formic acid and 5% methanol, and eluted with 80% methanol/5% formic acid. The MALDI-ToF MS analysis (see the following section for technical details) was performed with dihydroxy benzoic acid as a matrix (volume ratio of sample/matrix was 1/20). In order to identify peptides connected by a disulfide bridge, each sample was analyzed with and without adding 1 mM DTT. If reduction was suspected to be inefficient due to the formation of peptide aggregates, the peptide mix was subjected to sonication for 10 min, followed by reduction with 1 mM DTT. The resulting mixture was directly analyzed by MALDI-ToF MS.
Mass spectrometry analysis
Mass spectra for the proteolytic digests using three enzymes were acquired using a MALDI-ToF mass spectrometer (Voyager DE STR, Applied Biosystem), fitted with a pulsed nitrogen laser (337 nm). Mass spectra were acquired in the reflectron mode. The total acceleration voltage was set at 20 kV with a grid voltage of 65% and a delay extraction of 400 ns. External calibration was first realized using a standard peptide mix solution ranging from 573 to 3496 Da (LaserBio Labs). After assessment that no calbindin peptides would interfere with the calibrating peptides, this peptide mixture was used as internal reference and diluted to 1/300 in the calbindin peptide mixture. Mass measurement errors were <10 ppm. Samples were prepared in the DHB (DiHydroxyBenzoic acid) matrix at a final concentration of 20 mg/mL in acetonitrile/trifluoroacetic acid (70/0.1%) solution. The sample solution (1 μl) was then deposited on the MALDI target and allowed to dry. Peptide sequence identification was performed using the Cloe software (2.0, http://www.ibs.fr/software/). ESI mass spectra and tandem mass spectra were acquired in the positive ion mode using a QToF hybrid mass spectrometer (Qstar Pulsar I, Applied Biosystems), fitted with a nano-electrospray ionization source (Protana). The instrument was calibrated using a CsI solution. Prior to tandem mass spectrometry experiments, samples were desalted using a Poros reversed phase resin according to the procedure described above. Collision energy and the collision gas (N2) were optimized to obtain a good yield of precursor ion fragmentation. Collision energy was set between 45 and 110 eV and nitrogen was introduced in the collision cell at a final pressure ranging from 7.5 10−5 to 15 10−5 Torr.
Macroscopic Ca2+-binding constants
The macroscopic Ca2+-binding constants of calbindin D28k were assessed by Ca2+ titrations in the presence of the chromophoric chelator quin2, as described elsewhere (Linse 2002). In short, the absorbance at 263 nm is followed as a function of total Ca2+ concentration (added in small steps) and reports on Ca2+ binding to the chelator. However, the amount of Ca2+ bound to the chelator depends on the relative affinities for the protein and chelator and their respective concentrations. Each experiment was performed three times.
Parallel experiments were performed for nonreduced and reduced wild-type calbindin, for nonreduced and reduced ED-CCCCC mutant, and for a sample of reduced wild-type protein for which deamidation had been forced by 10-d incubation of the apo protein at pH 8.5 in 10 mM DTT in Ca2+-free buffer. The samples were subjected to gel filtration in a decalcified G25 column prior to the Ca2+-binding experiment to remove DTT. The Ca2+ titrations were performed in 2 mM Tris/HCl (tris(hydroxymethyl)aminomethane) pH 7.5 with no added salt, and 25–30 mM quin 2 and 20–25 mM protein. For reduced samples, the buffer contained 1 mM DTT. The quin 2 concentration was determined from the absorbance and the protein concentration was determined using amino acid analysis after acid hydrolysis. The residual calcium in the protein and buffer, and the calcium concentration of the stock, was determined by atomic absorption spectroscopy (purchased from SGA Analytica).
The procedure yields the macroscopic Ca2+-binding constants for the protein, which are extracted by computer fitting of Equation 1 directly to the measured absorbance using the CaLigator Software (Andre and Linse 2002).
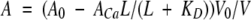 |
(1) |
A0 and ACa are the absorbances of apo and Ca2+-bound chelator, respectively, in the absence of dilution. KD is the dissociation constant for the Ca2+-chelator complex (fixed at 5.2 10−9 M−1). The ratio V0/V (initial over actual volume) gives the dilution effect on absorbance. The free Ca2+-concentration, L, was solved numerically from Equation 2 for nondeamidated samples, and from Equation 3 for deamidated samples.
 |
(2) |
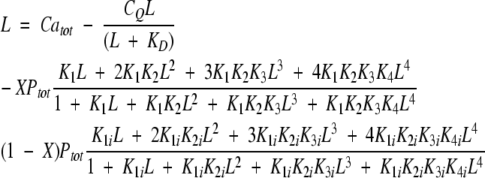 |
(3) |
Catot, CQ, and Ptot are the total Ca2+, chelator and protein concentrations, respectively, as corrected for dilution due to Ca2+ addition. K1, K2, K3, and K4 are the four macroscopic binding constants for the protein. In Equation 3, K1, K2, K3, and K4 are the four macroscopic binding constants for the deamidated protein with normal aspartic acid, and K1i, K2i, K3i, and K4i are the four macroscopic binding constants for the form with iso-aspartic acid. X is the fraction of deamidated protein with Asp, and 1-X the fraction with iso-Asp.
Acknowledgments
We thank Dr. Alain Brunelle for valuable and helpful scientific discussion, and Patrice Dosset for the development of the Cloe software. This work was supported by the Swedish Research Council and the Strategic Research Funds of Sweden (S.L.), and NSF Career Grant #9996074 and the Lise Meitner Foundation, Lund University (K.S.Å.).
Abbreviations
calbindin, calbindin D28k
quin 2, 2-[[2-[bis(carboxymethyl)amino]-5-methylphenoxy]methyl]-6-methoxy-8-[bis(carboxymethyl)amino]-quinoline
MS, mass spectrometry
EDTA, Ethylenediaminetetraacetic acid
F12, a fragment comprising residues 1–86 of human calbindin
F34, a fragment comprising residues 87–175 of human calbindin
F56, a fragment comprising residues 176–261 of human calbindin
F123, a fragment comprising residues 1–132 of human calbindin
F456, a fragment comprising residues 133–261 of human calbindin.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041157705.
References
- Airaksinen, M.S., Eilers, J., Garaschuk, O., Thoenen, H., Konnerth, A., and Meyer, M. 1997. Ataxia and altered dendritic calcium signaling in mice carrying a targeted null mutation of the calbindin D28k gene. Proc. Natl. Acad. Sci. 94 1488–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Åkerfeldt, K.S., Coyne, A.N., Wilk, R.R., Thulin, E., and Linse, S. 1996. Ca2+-binding stoichiometry of calbindin D28k as assessed by spectroscopic analyses of synthetic peptide fragments. Biochemistry 35 3662–3669. [DOI] [PubMed] [Google Scholar]
- Andre, I. and Linse, S. 2002. Measurement of Ca2+-binding constants of proteins and presentation of the CaLigator software. Anal. Biochem. 305 195–205. [DOI] [PubMed] [Google Scholar]
- Andressen, C., Blumcke, I., and Celio, M.R. 1993. Calcium-binding proteins: Selective markers of nerve cells. Cell Tissue Res. 271 181–208. [DOI] [PubMed] [Google Scholar]
- Arentz-Hansen, H., Korner, R., Molberg, O., Quarsten, H., Vader, W., Kooy, Y.M., Lundin, K.E., Koning, F., Roepstorff, P., Sollid, L.M., et al. 2000. The intestinal T cell response to α-gliadin in adult celiac disease is focused on a single deamidated glutamine targeted by tissue transglutaminase. J. Exp. Med. 191 603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellido, T., Huening, M., Raval-Pandya, M., Manolagas, S.C., and Christakos, S. 2000. Calbindin-D28k is expressed in osteoblastic cells and suppresses their apoptosis by inhibiting caspase-3 activity. J. Biol. Chem. 275 26328–26332. [DOI] [PubMed] [Google Scholar]
- Berggård, T., Silow, M., Thulin, E., and Linse, S. 2000a. Ca(2+)- and H(+)-dependent conformational changes of calbindin D(28k). Biochemistry 39 6864–6873. [DOI] [PubMed] [Google Scholar]
- Berggård, T., Thulin, E., Åkerfeldt, K.S., and Linse, S. 2000b. Fragment complementation of calbindin D28k. Protein Sci. 9 2094–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berggård, T., Miron, S., Önnerfjord, P., Thulin, E., Åkerfeldt, K.S., Enghild, J.J., Akke, M., and Linse, S. 2002a. Calbindin D28k exhibits properties characteristic of a Ca2+ sensor. J. Biol. Chem. 277 16662–16672. [DOI] [PubMed] [Google Scholar]
- Berggård, T., Szczepankiewicz, O., Thulin, E., and Linse, S. 2002b. myo-Inositol monophosphatase is an activated target of calbindin D28k. J. Biol. Chem. 9 9. [DOI] [PubMed] [Google Scholar]
- Berson, S.A. and Yalow, R.S. 1966. Deamidation of insulin during storage in frozen state. Diabetes 15 875–879. [DOI] [PubMed] [Google Scholar]
- Bischoff, R. and Kolbe, H.V. 1994. Deamidation of asparagine and glutamine residues in proteins and peptides: Structural determinants and analytical methodology. J. Chromatogr. B Biomed. Appl. 662 261–278. [DOI] [PubMed] [Google Scholar]
- Bornstein, P. 1970. Structure of α-1-CB8, a large cyanogen bromide produced fragment from the α-1 chain of rat collagen. The nature of a hydroxylamine-sensitive bond and composition of tryptic peptides. Biochemistry 9 2408–2421. [DOI] [PubMed] [Google Scholar]
- Bornstein, P. and Balian, G. 1970. The specific nonenzymatic cleavage of bovine ribonuclease with hydroxylamine. J. Biol. Chem. 245 4854–4856. [PubMed] [Google Scholar]
- Cedervall, T., Berggård, T., Borek, V., Thulin, E., Linse, S., and Åkerfeldt, KS. 2005. Redox sensitive cystein residues in calbindin D28k are structurally and functionally important. Biochemistry 44 684–693. [DOI] [PubMed] [Google Scholar]
- Chazin, W.J., Kordel, J., Thulin, E., Hofmann, T., Drakenberg, T., and Forsen, S. 1989. Identification of an isoaspartyl linkage formed upon deamidation of bovine calbindin D9k and structural characterization by 2D 1H NMR. Biochemistry 28 8646–8653. [DOI] [PubMed] [Google Scholar]
- Christakos, S., Gabrielides, C., and Rhoten, W.B. 1989. Vitamin D-dependent calcium binding proteins: Chemistry, distribution, functional considerations, and molecular biology. Endocr. Rev. 10 3–26. [DOI] [PubMed] [Google Scholar]
- Drakenberg, T., Fernlund, P., Roepstorff, P., and Stenflo, J. 1983. beta-Hydroxyaspartic acid in vitamin K-dependent protein C. Proc. Natl. Acad. Sci. 80 1802–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger, T. and Clarke, S. 1987. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J. Biol. Chem. 262 785–794. [PubMed] [Google Scholar]
- Gygi, S.P., Rist, B., Gerber, S.A., Turecek, F., Gelb, M.H., and Aebersold, R. 1999. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 17 994–999. [DOI] [PubMed] [Google Scholar]
- Helgstrand, M., Vanbelle, C., Thulin, E., Linse, S., and Akke, M. 2004. Sequential 1H, 15N and 13C NMR assignment of human calbindin D28k. J. Biomol. NMR 28 305–306. [DOI] [PubMed] [Google Scholar]
- Johansson, B.G. 1972. Agarose gel electrophoresis. Scand. J. Clin. Lab. Invest. Suppl. 124 7–19. [DOI] [PubMed] [Google Scholar]
- Johnson, K.L., Veenstra, T.D., Londowski, J.M., Tomlinson, A.J., Kumar, R., and Naylor, S. 1999. On-line sample clean-up and chromatography coupled with electrospray ionization mass spectrometry to characterize the primary sequence and disulfide bond content of recombinant calcium binding proteins. Biomed. Chromatogr. 13 37–45. [DOI] [PubMed] [Google Scholar]
- Kretsinger, R.H. and Nockolds, C.E. 1973. Carp muscle calcium-binding protein. II. Structure determination and general description. J. Biol. Chem. 248 3313–3326. [PubMed] [Google Scholar]
- Lerm, M., Pop, M., Fritz, G., Aktories, K., and Schmidt, G. 2002. Proteasomal degradation of cytotoxic necrotizing factor 1-activated rac. Infect. Immun. 70 4053–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linse, S. 2002. Calcium binding to proteins studied via competition with chromophoric chelators. Methods Mol. Biol. 173 15–24. [DOI] [PubMed] [Google Scholar]
- Luthra, M. and Balasubramanian, D. 1993. Nonenzymatic glycation alters protein structure and stability. A study of two eye lens crystallins. J. Biol. Chem. 268 18119–18127. [PubMed] [Google Scholar]
- Lutz, W., Frank, E.M., Craig, T.A., Thompson, R., Venters, R.A., Kojetin, D., Cavanagh, J., and Kumar, R. 2003. Calbindin D28K interacts with Ran-binding protein M: Identification of interacting domains by NMR spectroscopy. Biochem. Biophys. Res. Commun. 303 1186–1192. [DOI] [PubMed] [Google Scholar]
- McLachlin, D.T. and Chait, B.T. 2001. Analysis of phosphorylated proteins and peptides by mass spectrometry. Curr. Opin. Chem. Biol. 5 591–602. [DOI] [PubMed] [Google Scholar]
- Molberg, O., McAdam, S.N., Korner, R., Quarsten, H., Kristiansen, C., Madsen, L., Fugger, L., Scott, H., Noren, O., Roepstorff, P., et al. 1998. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat. Med. 4 713–717. [DOI] [PubMed] [Google Scholar]
- Ozaki, Y., Mizuno, A., Itoh, K., and Iriyama, K. 1987. Inter- and intramolecular disulfide bond formation and related structural changes in the lens proteins. A Raman spectroscopic study in vivo of lens aging. J. Biol. Chem. 262 15545–15551. [PubMed] [Google Scholar]
- Patel, K. and Borchardt, R.T. 1990. Chemical pathways of peptide degradation. II. Kinetics of deamidation of an asparaginyl residue in a model hexapeptide. Pharm. Res. 7 703–711. [DOI] [PubMed] [Google Scholar]
- Reissner, K.J. and Aswad, D.W. 2003. Deamidation and isoaspartate formation in proteins: Unwanted alterations or surreptitious signals? Cell Mol. Life Sci. 60 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, N.E., Robinson, A.B., and Merrifield, R.B. 2001. Mass spectrometric evaluation of synthetic peptides as primary structure models for peptide and protein deamidation. J. Pept. Res. 57 483–493. [DOI] [PubMed] [Google Scholar]
- Sen, A.C. and Chakrabarti, B. 1990. Effect of acetylation by aspirin on the thermodynamic stability of lens crystallins. Exp. Eye Res. 51 701–709. [DOI] [PubMed] [Google Scholar]
- Stenflo, J., Fernlund, P., Egan, W., and Roepstorff, P. 1974. Vitamin K dependent modifications of glutamic acid residues in prothrombin. Proc. Natl. Acad. Sci. 71 2730–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier, F.W. and Moffatt, B.A. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189 113–130. [DOI] [PubMed] [Google Scholar]
- Tao, L., Murphy, M.E., and English, A.M. 2002. S-nitrosation of Ca(2+)-loaded and Ca(2+)-free recombinant calbindin D(28K) from human brain. Biochemistry 41 6185–6192. [DOI] [PubMed] [Google Scholar]
- Thulin, E. and Linse, S. 1999. Expression and purification of human calbindin D28k. Protein Expr. Purif. 15 265–270. [DOI] [PubMed] [Google Scholar]
- Tyler-Cross, R. and Schirch, V. 1991. Effects of amino acid sequence, buffers, and ionic strength on the rate and mechanism of deamidation of asparagine residues in small peptides. J. Biol. Chem. 266 22549–22556. [PubMed] [Google Scholar]
- Wright, H.T. 1991. Sequence and structure determinants of the nonenzymatic deamidation of asparagine and glutamine residues in proteins. Protein Eng. 4 283–294. [DOI] [PubMed] [Google Scholar]