

Abstract
Potassium channels are widespread in living cells and are involved in many diseases. The scorpion toxin α-KTx12.1 interacts with various K+ channels, suggesting its capacity to match diverse channel pores. It is recognized that tissue injuries may affect the pH at toxins site of action, thereby modulating both protein conformation and activity. To better understand its molecular mechanism of action, we studied α-KTx12.1 using pH as a tool to explore its plasticity and NMR in combination with MD calculations to detect it. The toxin solution structure consists of an α-helix and a triple-stranded β-sheet stabilized by four disulfide bridges. The NMR results show, in addition, that His28 possesses an unusually low pKa of 5.2. The best set of protein conformers is obtained at pH 4.5, while at pH 7.0, the reduced number of NOEs resulting from a faster hydrogen exchange does not allow to reach a good structural convergence. Nonetheless, MD calculations show that the toxin structure does not vary significantly in that pH range, while conformational changes and modifications of the surface charge distribution occur when His28 is fully protonated. Moreover, essential dynamics analysis reveals variations in the toxin’s coherent motions. In conclusion, His28, with its low pKa value, provides α-KTx12.1 with the ability to preserve its active conformation over a wide pH interval, thus expanding the range of cellular conditions where the toxin can fully exhibit its activity. Overall, the results further underline the role of histidine as a natural controller of proteins’ functionality.
Keywords: molecular dynamics, nuclear magnetic resonance, potassium channel, scorpion toxin, essential dynamics analysis
Animal venoms are constantly being studied in search of new pharmacologically active molecules that can be used for therapeutic purposes. In this respect, scorpion venom is an incredible resource of molecules, most of which are peptides and small proteins characterized by a variety of biological activities. A good number of them are able to bind specifically to ion channels, producing mainly neurotoxic effects.
On the other hand, it is worth remembering that from the point of view of scorpions, spiders, and snakes, these toxins are essential tools for their survival. As a result, they must be quite robust in order to reach their targets and exert their functions. In fact, they have to resist and overcome the defensive systems of the hosts such as, for instance, proteases and/or significant local pH variations that may result from inflammation states (Steen et al. 1996; Waldmann and Lasdunski 1998) induced by the animal bite itself.
Potassium channels are widely spread in the living kingdom and are part of a large variety of biological processes. In addition, it is now well recognized that they are involved in an increasing number of human pathologies (Ashcroft 2000; Shieh et al. 2000). Thus, the perception that potassium channels are important therapeutic targets explains the constantly growing efforts to comprehend their three-dimensional structure and mechanism of action.
Scorpion toxins selective for K+ channels (KTx) are short-chain peptides (23–43 amino acids) cross-linked by three or four disulfide bridges. They have been classified into α-, β-, and γ- toxins (Tytgat et al. 1999; Corona et al. 2002). The α-KTx family is the best studied to date. At first, it was divided into 12 subfamilies comprising a total of 49 members (Tytgat et al. 1999). More recently, six new subfamilies have been added (Batista et al. 2002; Goudet et al. 2002). Figure 1A ▶ shows the sequence alignment of a selected number of α-KTx toxins. Their global fold displays the same α/β scaffold formed by an α-helix and a two- or three-stranded β-sheet linked by two disulfide bridges (Tenenholz et al. 2000). This α/β fold, however, is shared by a variety of polypeptides with diverse functions, including toxins active on Na+ channels (Cohen et al. 2004), some antimicrobial peptides such as defensins (Bauer et al. 2001; Sawai et al. 2001), γ-thionins (Bloch et al. 1998), and antifungal proteins (Fant et al. 1998), thus suggesting that the α/β fold does not correlate directly with a specific target.
Figure 1.

(A) Sequence alignment of selected members of the α-KTx family carried out using CLUSTAL W (Thompson et al. 1994) followed by a manual adjustment. Identical and highly conserved residues are indicated by stars and by colons, respectively. (B) Pattern of disulfide pairing for the α-KTx12.1 sequence.
Overall, despite their similar architecture, α-KTx toxins exhibit distinct selectivity and affinity for a variety of K+ channels (Rodriguez de la Vega et al. 2003), thus posing interesting questions concerning the molecular factors that influence their diversified K+ channel blocker activity.
α-KTx12.1 is a four disulfide-bridged neurotoxin (Fig. 1B ▶), first isolated from the venom of the Brazilian scorpion Tityus serrulatus. Due to the lack of sequence homology with any other toxin belonging to the α-KTx family, it has been considered the first member of the subfamily 12 (Tytgat et al. 1999). Originally, it was named TsTX-IV (Tityus toxin IV) (Arantes et al. 1989) and described as a 41-residue protein (Novello et al. 1999). However, according to the results reported here and in agreement with data recently published by another group (Pimenta et al. 2003), it turns out that the polypeptide sequence lacks the residue Asn41. Based on these findings, it follows that α-KTx12.1 and the 40-residue-long butantoxin (BuTX) originally isolated from the venom of Tityus serrulatus and subsequently from T. bahiensis and T. stigmurus (Holaday et al. 2000) are the same molecule. Finally, a peptide recently purified from the venom of the Argentinean scorpion Tityus trivittatus, despite presenting the same primary sequence as α-KTx12.1, has been classified as α-KTx12.2 because of the different geographic origin (Coronas et al. 2003).
As for its biological activity, α-KTx12.1, at nM concentration, was shown to block the high-conductance Ca2+-activated K+ channels in Leydig cells (Novello et al. 1999). Furthermore, it was reported that α-KTx12.1, while it does not compete with 125I-apamin, a very specific ligand of the low-conductance activated K+ channels, is able to inhibit the binding of 125I-kaliotoxin to its receptor in rat brain synaptosomal preparations, with an IC50 of 46 nM, thus suggesting an affinity also for the Kv1.3 K+ channel (Pimenta et al. 2003). Finally, α-KTx12.2 was shown to block, reversibly, the Shaker B K+-channels, with a Kd of ~660 nM (Coronas et al. 2003). In summary, this toxin appears to be able to interact with a variety of K+ channels, exhibiting a variable affinity for each of them. Such behavior suggests that, despite the four disulfide bridges, the toxin may be characterized by a conformational flexibility that allows its structure to fit into the pore region of diverse K+ channels.
The current models describing the functionality of toxins assume that the type and position of the amino acids exposed at the molecular surface are critical in determining the biological functions and binding properties of each particular toxin. In fact, these models suggest that the inhibition of the K+ conductance results from the ability of the toxin to occlude the pore of the channel at the extracellular opening. As for the binding process, it is described as a reversible, bimolecular reaction which is governed by electrostatic interactions between negatively charged residues in the channel and positively charged residues in the toxin. Consequently, the binding is expected to be affected by both the pH and the ionic strength of the external media (Ellis et al. 2001; Garcia et al. 2001).
In this work, aiming to disclose how α-KTx12.1 succeeds to actively interact with various types of K+ channels and knowing that the binding is influenced by the structural features the toxin possesses just before interacting with the channel, we investigated the conformational properties of the toxin at that particular step. Recognizing that the toxin may experience environments where the pH varies significantly, and that the protein ionization state can affect its structural stability and flexibility (Fersht 1998; Kipping et al. 2001; Hu et al. 2003), we combined NMR spectroscopy and molecular dynamics (MD) simulations to probe the structural and dynamic features of α-KTx12.1 at both acidic and neutral pH.
Results
Purification and mass spectrometry
The chromatographic “Fraction X” of Tityus serrulatus venom accounts for 4%–6% of the soluble proteins present in it. Among the four subfractions obtained when “Fraction X” is passed through a reverse-phase HPLC, α-KTx12.1 elutes as subfraction X-3 (Supplemental Material, Fig. 1 ▶), which represents 46%–48% of “Fraction X.” The presence of a single band in the PAGE electrophoresis (Supplemental Material, inset of Fig. 1 ▶, lane 4) confirms the purity of the toxin.
Accurate mass measurement was achieved on desalted and purified α-KTx12.1 by electron spray ionization (ESI) analysis. The molecular weight calculated for the toxin according to the sequence deposited in the Swiss-Prot Data Base (SCK4_TITSE) is 4628 Da. The measured average molecular weight was 4505.8 ± 0.5 Da (Supplemental Material, Fig. 2A ▶), in agreement with the lack of residue Asn41 (Δm = −114 Da) (Pimenta et al. 2003) and the presence of four disulfide bridges (Δm = −8 Da). Considering that α-KTx12.1 contains two cysteines in its N terminus (Fig. 1 ▶) that cannot be aligned with any of the other α-KTx toxins isolated from scorpion venom (Tytgat et al. 1999), it turned out to be important to chemically verify the disulfide bridge pattern.
Figure 2.

Sequence of α-KTx12.1 with the sequential and medium-range NOE pattern at pH 4.5 (top) and pH 6.0 (bottom) taken from 150-msec NOESY spectra (35°C). For sequential connectivities, the thickness of the bars indicates NOE intensities; medium-range NOEs are identified by lines connecting the two coupled residues. The secondary structure was obtained from the global result of a PROCHECK-NMR (Laskowski et al. 1996) analysis of the final ensemble of NMR conformers at pH 4.5. Amino acids whose backbone amide proton resonances were not assigned at pH 6.0 are set in boxes.
In contrast to an earlier work (Pimenta et al. 2003), we selected pepsin proteolysis for two main reasons: (1) the acidic conditions that exclude disulfide reshuffling, and (2) the low residue specificity of this proteolytic enzyme that is expected to favor a higher level of fractioning. The enzyme treatment time course of the native α-KTx12.1 was followed by MALDI-TOF mass spectrometry.
Even after an extended proteolysis, only two peptide signals, with m/z values of 3587 and 3605, respectively, were obtained (Supplemental Material, Fig. 2B ▶). The lighter molecular mass corresponds to the fragment of α-KTx12.1 encompassing residues 9–40 and comprising three disulfide bridges (expected mass = 3586 Da). As for the heavier peptic fragment, on the basis of the mass difference of 18 Da, we assume it can be related to the same toxin core structure with a proteolytic nick, namely a cleavage in the primary structure which does not generate two fragments due to the presence of the disulfide bridge connection. Inspection of MALDI-TOF spectra at low mass range could not detect the N-terminal fragment, residues 1–8. However, on the basis of these results, the unique disulfide Cys2-Cys5 is unequivocally inferred. The other three bridges, Cys10-Cys31, Cys16-Cys36, and Cys20-Cys38 correspond to the equivalent positions in the known three disulfide bridges containing toxins (Fig. 1A ▶).
Sequential NMR resonance assignments at different pH values
The sequence-specific resonance assignments of α-KTx12.1 at pH 4.5, 6.0, and 7.0 were carried out according to the classical sequential assignment procedure (Wüthrich 1986). A combined analysis of DQF-COSY and TOCSY spectra at 35°C was used to identify the 1H spin systems, while the sequential connectivities were obtained from NOESY spectra. Experiments recorded at lower temperatures were also used to confirm assignments in cases of peak overlap or proximity to the water resonance. The 1H chemical shift values were deposited in the BioMagResBank database (http://www.bmrb.wisc.edu) under accession number 6428.
Unlike a previous study of BuTX at pH 5.0 and 20°C (Holaday et al. 2000), where a number of assignments were missing, the resonances of α-KTx12.1 at pH 4.5 were completely assigned with the exception of the aliphatic protons of Trp1. On the other hand, at both pH 6.0 and 7.0, the spin systems of Cys2, Ser3, Ser13, and Arg14 could not be identified. The protein N terminus seems to be sensitive to pH variation, as suggested by the chemical shift change of Thr4 and Cys5 amide protons. Additional pH-sensitive amide proton resonances include Lys22, Phe24, Ala27, Gly29, and Thr40. While Gly29 appears to sense the change in ionization state of the neighboring His28 ring, a rearrangement of some aromatic side chains is likely to affect the chemical shift values of the other aforementioned residues (see next section).
The analysis of the pH dependence of the His28 imidazole ring Hɛ1 resonance, monitored by 2D 1H-NMR spectra, provided an unusually low pKa of 5.2 (Supplemental Material, Fig. 3 ▶).
Figure 3.
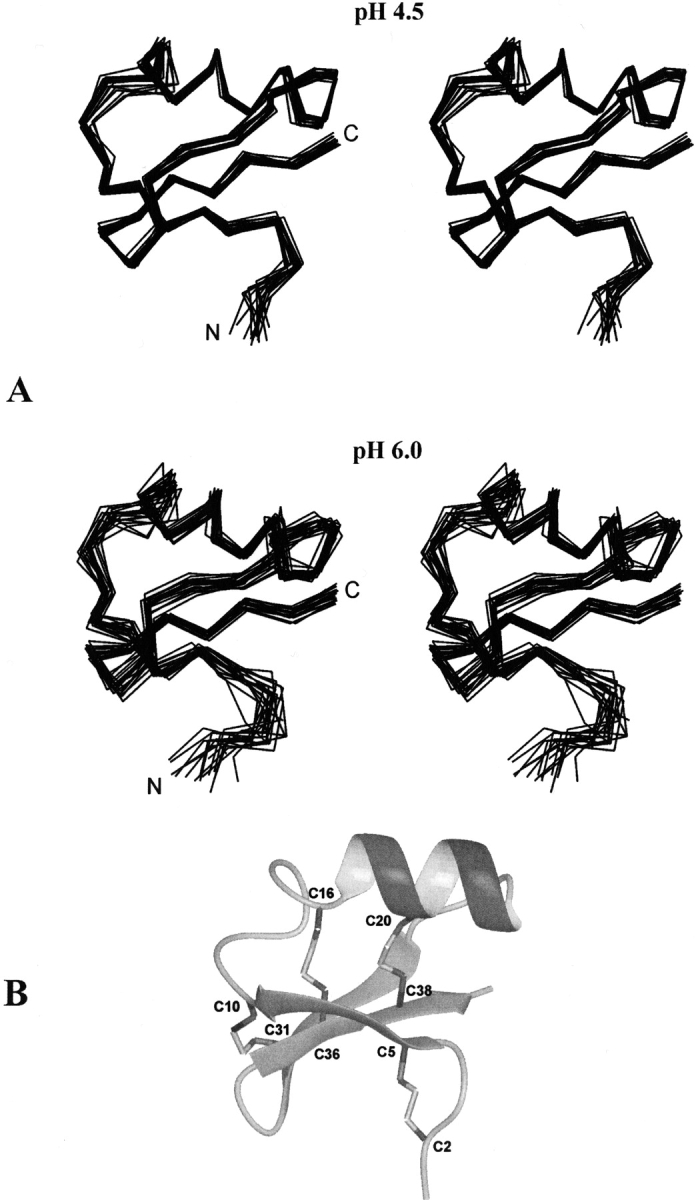
(A) Stereo view of the solution structure ensembles (as Cα-traces) of α-KTx12.1 at pH 4.5 and pH 6.0. The ensemble at pH 6.0 displays a higher backbone disorder. (B) Ribbon diagram of the best NMR conformer at pH 4.5. The disulfide bond connections are indicated.
Solution structure determination and analysis
At pH 4.5 the nuclear Overhauser effect (NOE) pattern identifies an α-helix in the central region of the toxin’s sequence and three short antiparallel β-strands, the first of which precedes the α-helix (Fig. 2 ▶, top). By increasing the pH to 6.0, we observed a significant reduction of the number of NOE signals throughout the protein sequence (Fig. 2 ▶, bottom; Table 1). In particular, focusing on the domains of regular secondary structure, we observed the disappearance of several NOEs defining the α-helix (Fig. 2 ▶, bottom) as well as of five interstrand connectivities (data not shown). At pH 7.0, the total number of NOEs is further reduced, but the most significant difference is found for the intraresidual NOEs rather than for those defining the protein fold (Table 1). It is worth mentioning that an analysis of the Hα resonance deviations based on the chemical shift index procedure (Wishart et al. 1992) did not indicate any relevant changes in the toxin’s secondary structure at the three pH values, as also confirmed by circular dichroism measurements (data not shown).
Table 1.
Structural statistics of the solution structure ensembles of α-KTx12.1
| pH 4.5 | pH 6.0 | pH 7.0 | |
| Restraint statistics | |||
| Total number of meaningful distance restraints | 397 | 245 | 212 |
| Intraresidual (i = j) | 89 | 66 | 44 |
| Sequential (|i − j|= 1) | 114 | 62 | 59 |
| Medium-range (1 < |i − j|≤4) | 75 | 44 | 39 |
| Long-range (|i − j|> 4) | 119 | 73 | 70 |
| NOE violations after energy minimization | |||
| 0.20 Å – 0.25 Å | 8 | 6 | |
| 0.26 Å – 0.35 Å | 2 | 5 | |
| >0.35 Å | 0 | 1 | |
| Structural precision, RMSD (Å) | 18 structures | 18 structures | |
| Backbone atomsa (residues 2–39) | 0.65 ± 0.19 | 1.19 ± 0.24 | |
| All heavy atoms (residues 2–39) | 1.31 ± 0.17 | 1.97 ± 0.24 | |
| Ramachandran plot analysis (%) | |||
| Residues in most favored regions | 70.6 | 68.7 | |
| Residues in additionally allowed regions | 28.3 | 27.6 | |
| Residues in generously allowed regions | 1.1 | 2.6 | |
| Residues in disallowed regions | 0.0 | 1.1 | |
a Backbone atoms always refer to (N, Cα, C′).
The three-dimensional structures of α-KTx12.1 at each pH were computed and subsequently refined by using, in the last run of calculations, the NOE-derived nonredundant distance restraints listed in Table 1. Figure 3A ▶ shows the final ensembles that were selected to represent the solution structure of the toxin at pH 4.5 and 6.0, each containing 18 energy-minimized conformers. The ensemble at pH 7.0 is not shown, because the reduced number of experimental data did not allow a good structure convergence. The structural statistics information is summarized in Table 1. As for the models obtained at pH 4.5 and 6.0, the absence of significant violations of single distance restraints indicate a good agreement between the NOE data and the calculated conformers. The structural quality of the models was examined with the PROCHECK-NMR program (Laskowski et al. 1996). Analysis of the Ramachandran plot showed that 100% and 98.9% of all nonglycine/nonproline residues fall within the allowed regions of conformational space at pH values 4.5 and 6.0, respectively (Table 1).
The results obtained at pH 4.5, where we had the most complete set of NMR data, indicate that the three β-strands span residues Cys5-Ala9, His28-Met32, and Lys35-Tyr39, while the β-helix comprises the segment Tyr17-Phe24 (Fig. 2 ▶, top). The first and the second β-strand both run anti-parallel to the third one, thus giving a +2x,-1 β-sheet topology: Figure 3B ▶ shows the ribbon diagram of the toxin solution structure. The first β-strand, which includes a bulge at Asp7 with the lack of the typical NOEs, is connected to the β-helix by a seven-residue-long, solvent-exposed loop. Finally, a type-I′β-turn is connecting the second and third β-strands to form a β-hairpin.
A comparison with the secondary structure reported for BuTX at almost identical pH but lower temperature (Holaday et al. 2000) revealed generally good agreement. In BuTX, two of the β-strands are just one residue shorter, spanning residues 29–32 and 35–38, while the helical segment appears to be longer. In fact, it is described as an α-helix from Glu15 to Ala23, possibly preceded by a 310 helical turn (Ala12-Arg14) that we did not observe. Within our ensemble of β-KTx12.1 models, the helix could be extended from Tyr17 to Ser13 in only three of 18 models.
The structure of β-KTx12.1 is very well defined at acidic pH (Fig. 3A ▶, top) with a backbone root mean square deviation (RMSD) of 0.65 ± 0.19 Å. Aside from Trp1 and Thr40, only the region Gly11-Arg14, which is part of the loop connecting the first β-strand with the α-helix, displays a certain degree of backbone disorder. The models of the toxin at pH 6.0 superpose to the ones at pH 4.5 (Fig. 3A ▶, bottom vs. top) with an average RMSD between the two ensembles of 1.26 ± 0.21 Å for the backbone atoms and 2.09 ± 0.18 Å for all heavy atoms (residues 2–39). The most apparent difference between the two families of conformers is the general higher backbone disorder observed at pH 6.0. However, a lower structure definition at this pH was expected, because of the reduced number of NOE-derived distance constraints (Table 1).
The side-chain orientations of residues belonging to the regions of regular secondary structure are rather well defined at pH 4.5, resulting in relatively small circular variances of the dihedral angles χ1 and χ2; exceptions are Cys5 and Lys30. Other residues displaying a rather high degree of side-chain disorder are Trp1, Cys2, Ser3, Cys16, and Thr40. At pH 6.0, these residues do not increase their circular variances any further, while a higher disorder is observed for the side chains of Thr4, Asp7, Ser 13, Arg14, Arg26, Asn33, and Tyr39. As for Tyr39, its side chain takes on a different orientation at the two pH values, with the phenol ring located much closer to the Gly29 Cα protons at acidic pH. This side-chain reorientation might be responsible for both the very unusual Gly29 Hα chemical shift value of 4.87 ppm, observed at pH 4.5, and the significant shift detected at higher pH. The different orientation of the Tyr39 ring at the two pHs is very likely responsible also for the shift of the Thr40 HN resonance. Similarly, the pH-dependent changes in the amide resonances of Lys22, Phe24, and Ala27 may be caused by spatial rearrangements of other nearby aromatic side chains, such as the ones of Tyr17, Phe21, and Phe24.
Unrestrained molecular dynamics calculations and essential dynamics analysis
As fewer NOE-derived distance constraints were available at neutral pH, it did not seem appropriate to directly compare the experimental structures at low and neutral pH. Therefore, we chose to perform MD simulations at pH 7.0 and 4.0 as representatives of neutral and acidic conditions; the corresponding theoretical ionization states of the toxin (Table 2) were defined according to the consistent valence force field (CVFF).
Table 2.
Ionization state of α-KTx12.1 at pH values of 4.0 and 7.0

The RMSD of the backbone atoms during 10 nsec of unrestrained MD simulation was calculated with respect to the well defined NMR solution structure of lowest energy at pH 4.5. The simulation at pH 7.0 shows that the toxin retains the experimental structure all the way through the simulation (Fig. 4 ▶). At pH 4.0, instead, a significant deviation from the NMR structure is detected; in fact, the toxin undergoes a number of conformational displacements, and only after 5 nsec it stabilizes in a new structural state, as can be seen in Figure 4 ▶.
Figure 4.

RMSD of α-KTx12.1 backbone atoms at pH 4.0 (black) and 7.0 (gray), during 10 nsec of unrestrained MD simulation. RMSDs are calculated relative to the lowest-energy NMR-derived solution structure at pH 4.5.
With the aim to eliminate a possible bias introduced by the use of the NMR structure as a reference, we performed a pairwise backbone RMSD analysis of the MD models collected every 10 psec. The matrix obtained at pH 4.0 confirms a conformational fluctuation during the initial 5 nsec of simulation, while at pH 7.0 the toxin backbone reaches a relatively stable state already after the first 2 nsec with small structural modifications (Supplemental Material, Fig. 4 ▶).
Inspection of the protein’s secondary structure elements during the last 5 nsec of the MD simulation shows that, while at pH 7.0 they are rather stable, at pH 4.0 the first β-strand and the N-terminal portion of the α-helix are clearly destabilized, with the β-strand being practically disrupted (Fig. 5 ▶).
Figure 5.

Behavior of the secondary structure elements of α-KTx12.1 during 10 nsec of unrestrained MD simulation at pH 7.0 (A) and 4.0 (B).
To gain some insight into the toxin’s backbone dynamics, we first evaluated the root mean square fluctuation (RMSF). Throughout the entire protein sequence, similar behavior was observed at both pH values, with the residues outside the regions of regular secondary structure exhibiting higher fluctuation (Supplemental Material, Fig. 5 ▶). Nonetheless, knowing that essential dynamics (ED) analysis allows us to distinguish concerted molecular motions from irrelevant fluctuations (van Aalten et al. 1996), we performed a comparative principal component analysis of the toxin’s Cα atoms.
Figure 6A ▶ shows the eigenvalue spectrum of the diagonalized covariance matrices for the last 5 nsec of unrestrained MD simulation at both pH values. Only the first 10 eigenvectors are plotted, as they already represent the core of the toxin’s degrees of freedom. To probe in more detail the existence of concerted fluctuations, we calculated the atomic displacements along the first three eigenvectors on a per-residue basis (Fig. 6B ▶). The fact that the first three eigenvectors account for only ~40% of the main molecular concerted motions can be interpreted as an indication of a general random flexibility of the backbone. Nonetheless, the plots provide useful information (Fig. 6B ▶). At pH 4.0 we can observe concerted motions involving a number of protein domains: the N-terminal segment, residues Trp1-Thr4, the loop connecting the α-helix with the second β-strand, residues Phe24-Ala27 (eigenvector 1); the N- and C-terminal region of the α-helix, residues Asp18-Pro19 and Lys22-Phe24, respectively, (eigenvector 2); the regions involving residues Ala12-Arg14 and Ala23-Arg26 (eigenvector 3), which encompass a portion of the loop connecting the first β-strand with the α-helix and a region including the C terminus of the α-helix plus a portion of the subsequent loop, respectively. At pH 7.0, besides a concerted motion in the N terminus, residues Trp1-Asp7, the eigenvectors are mainly dominated by the motion of the loop connecting the second with the third β-strand, residues Met32-Lys35, and by minor fluctuations involving a portion of the loop connecting the first β-strand with the α-helix, residues Arg14-Cys16, and the C-terminal half of the helix, residues Phe21-Ala23. Figure 7 ▶ gives a pictorial representation of these coherent fluctuations.
Figure 6.
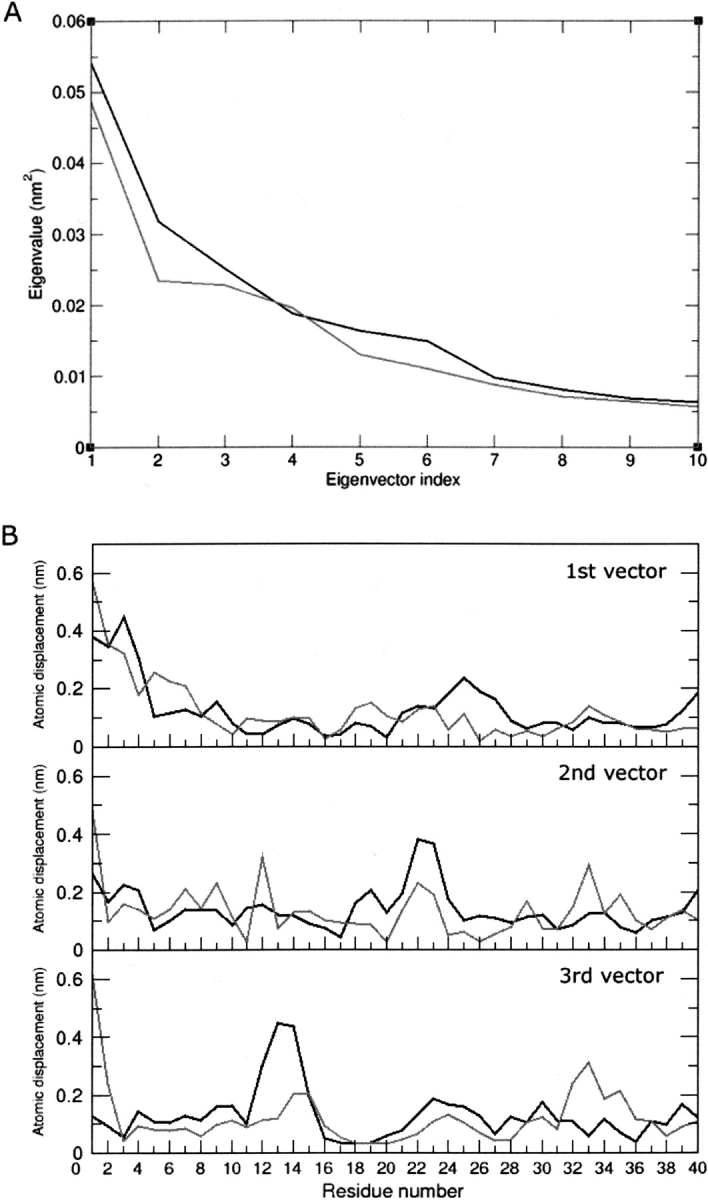
(A) Eigenvector spectra for the last 5 nsec of each 10-nsec unrestrained MD simulation at pH 4.0 (black) and 7.0 (gray). (B) Atomic displacements (nm) along the first three eigenvectors for each trajectory.
Figure 7.

α-KTx12.1 structures representing the Cα traces of filtered trajectories from the last 5 nsec of MD simulation that were carried out at pH 4.0 (A) and 7.0 (B). The trace radius is proportional to the atomic displacements according to the first three eigenvectors (labeled “Eig”).
Overall these results indicate that the change in ionization state affects the structural organization of the toxin and alters some of its coherent intramolecular motions. Indeed, if we observe the projection of the trajectories of the unrestrained simulation in the plane defined by the first two eigenvectors, it is possible to recognize that the protein is fluctuating in a more disperse conformational space at pH 4.0 than at pH 7.0 (Fig. 8 ▶). More interestingly, it is evident that the NMR solution structure obtained at pH 4.5 falls into the conformational space sampled by the unrestrained MD simulation carried out at neutral pH.
Figure 8.

Projections of the trajectories from the last 5 nsec of MD simulation at pH 4.0 (A) and 7.0 (B) onto the planes defined by the first two corresponding eigenvectors. The square corresponds to the projection of the initial conformation, i.e., the NMR-derived structure of lowest energy at pH 4.5.
In order to understand the possible reasons that could account for the conformational displacement that takes place moving from the pH range 7.0–4.5 toward a more acidic pH, we analyzed the two MD simulations (Fig. 4 ▶) looking for changes in hydrogen bond patterns or in some interresidue distances. We focused our attention on residues Glu15 and His28, since they have different ionization states in the two simulations (Table 2) and therefore their interactions with the surroundings may be expected to change. In the case of Glu15, no meaningful differences could be found. As for His28, in contrast, we found that only in the simulation at pH 4.0 a salt bridge is formed between the protonated His28 Nɛ2 atom and the C-terminal carboxylate group of Thr40 (Fig. 9 ▶, panels A,D). The onset of this interaction, well preserved throughout the entire calculation, initiates a series of significant structural changes such as the disruption of the hydrogen bond between Thr4 CO and Cys38 NH (Fig. 9 ▶, panels B,E) that most likely contributes to the destabilization of the first β-strand (Fig. 5B ▶), and a conformational displacement that involves mainly the N-terminal portion of the toxin (Trp1-Thr4). This latter structural modification brings the side-chain amino group of Lys35 and the Trp1 indole ring at a distance compatible with the onset of a π-interaction (Fig. 9 ▶, panels C,F).
Figure 9.

Distances between specific residues during the MD simulations at pH 7.0 (A–C) and 4.0 (D–F). The abbreviations used for specific atoms are: NE2, histidine imidazole nitrogen; O2, terminal carboxylate oxygen; CO, backbone carbonyl oxygen; NH, backbone amide nitrogen; OD2, aspartate side-chain oxygen; NZ, lysine side-chain nitrogen; CM, center of mass of the tryptophan indole ring.
Discussion
The biological functionality of macromolecules, in particular proteins, depends upon the presence of both specific structural determinants and dynamics features that result from either incoherent or concerted molecular motions. In scorpion toxins it is known that some structural requirements are conserved, e.g., the α-type toxins where a lysine residue must have a specific orientation to be able to insert into the K+ channel pore (Rodriguez de la Vega et al. 2003). Interestingly, toxin activity is also pH-dependent, a finding that is generally attributed to the modulation of electrostatic attraction and repulsion between the protein and its target (Meunier 1993; Dyke et al. 1996; Frémont et al. 1997; Thompson and Begenisich 2000; Ellis et al. 2001; Everart et al. 2004).
The α-KTx12.1 NMR solution structure at pH 4.5 has a fold typical for the α-KTx toxin family (Bontems et al. 1991; Fernández et al. 1994; Krezel et al. 1995; Wang et al. 2002). When the pH is increased to values closer to physiological conditions (pH 6.0 and 7.0), even though the structures do not show significant conformational differences, they exhibit a higher backbone disorder (Fig. 3 ▶) due to the diminished number of measurable NOEs. To assess the origin of such a reduction of NOEs (a faster NH exchange rate induced by the higher pH or a higher molecular flexibility), and recognizing that MD simulations can produce results consistent with NMR measurements (Hu et al. 2003; Sforça et al. 2004) as well as qualitatively reproduce the kinetics (Wu and Brooks 2004) and folding (Gnanakaran et al. 2003) of a peptide system, we complemented the NMR data with an ensemble of MD calculations carried out in explicit solvent.
The RMSF obtained analyzing the traces of the last 5 nsec of the MD simulations showed that the molecular flexibility of the toxin does not vary significantly in the considered pH range (Supplemental Material, Fig. 5 ▶), thus pointing to a faster hydrogen exchange as the cause of the reduced number of NOEs. Nonetheless, the ED analysis revealed that, upon pH variation, significant changes take place with respect to the nature of the intramolecular coherent motions (Fig. 7 ▶).
Overall, the MD results at pH 7.0 show similarities to some dynamic features reported for the toxin Tc1 (Grottesi and Sansom 2003), while they differ from the results obtained for kaliotoxin (Canto et al. 1999), since they do not show any correlated motion between the α-helix and the β-sheet.
The MD simulations also indicated important structural changes that are expected to affect the toxin’s functionality. In fact, the full protonation of His28 initiates a cascade of alterations of selected intramolecular interactions that lead to the destabilization of the first β-strand and to the conformational displacement of the N terminus. This last event places the side chain of Trp1 at a distance to the Lys35 side chain compatible with the formation of a π-interaction (Fig. 9 ▶, panels C,F), thus less available to act as a first anchoring point in the process of target selection (Pimenta et al. 2003).
Interestingly, the presence of a His residue with an anomalous low pKa was reported also for other toxins in the α-KTx12.1 family. In addition, a significant structural modification, brought about by the change in the His ionization state, was observed also in the case of kaliotoxin (Fernández et al. 1994; Gairí et al. 1997).
The toxins’ surface charge distribution is recognized as a key factor in the control of their ability to bind to receptors. The strategic location of charged groups modulates also the selectivity exhibited by different channel blockers (Meunier et al. 1993; Frémont et al. 1997). The charge distribution in the models of α-KTx12.1 obtained by the MD simulations is shown in Figure 10 ▶. Besides a change in the overall molecular shape, at pH 7.0 we observed a patch of positive charge centered at the site of the key residue Lys30, while at pH 4.0 the toxin surface that is expected to face the channel mouth is characterized by a diffuse positive potential. As a result, we may envisage that only when His28 is deprotonated, the protein surface exhibits selected regions characterized by positive charges able to assist the correct approach of the toxin to the channel pore.
Figure 10.

Accessible surfaces of the last structures from each MD simulation at pH 7.0 (A,B) and 4.0 (C,D). Surfaces represented in A and C are colored according to the electrostatic potential. In the transparent molecule representations (B,D), residues His28 and Lys30 are highlighted in yellow.
In conclusion, these results indicate that the presence of a His residue characterized by an unusually low pKa (5.2) allows α-KTx12.1 to maintain the structural and dynamics features that define its active state in a sufficiently wide pH range. Although this study does not completely describe the origins of the broad spectrum of action exhibited by α-KTx12.1, it reveals the existence of pH-dependent structural and dynamics changes that are regulated by the ionization state of a single amino acid and that may be associated with toxin functionality.
Materials and methods
Toxin isolation, purification, and analysis
Tityus serrulatus venom was extracted and fractionated by chromatography as previously described (Arantes et al. 1989). Briefly, the clear extract of desiccated venom (200 mg) was first passed through a 2.2 × 67.0 cm column of CM-cellulose-52 (Whatman) which was equilibrated and initially eluted with 0.01 M ammonium bicarbonate buffer, pH 7.8, up to 540 mL effluent, when a convex concentration gradient was started from 0.01 to 1.00 M buffer. “Fraction X” (2 mg) from the above fractionation was submitted to a reverse-phase chromatography performed in an HPLC system (Shimadzu) using a 4.6 mm × 12 cm column (C-18, Shimpack CLC-ODS, Shimadzu) initially equilibrated with 5% (v/v) acetonitrile in 0.1% (v/v) trifluoroacetic acid (TFA) (solvent A). Elution was performed with a 5%–80% (v/v) acetonitrile linear gradient in 0.1% (v/v) TFA at a flow rate of 1.0 mL/min. Absorbance was monitored at 214 nm. Solvent B was 80% (v/v) acetonitrile in 0.1% (v/v) TFA.
PAGE electrophoresis was run as previously described (Arantes et al. 1989).
Mass spectrometry
α-KTx12.1 was desalted and further purified using a LabService Analytica HPLC system equipped with a UV detector and connected to a mass spectrometer by a T-splitter. An aliquot (4 μL) was loaded on a C8 column (Vydac, 21 × 150 mm, 5 μm) and subsequently eluted at a flow rate of 0.15 mL/min, with a linear gradient of acetonitrile containing 0.2% (v/v) TFA. Mass spectra were performed on-line on 1/3 eluent, using an LCQ ion trap mass spectrometer (ThermoFinnigan) equipped with an electrospray ionization source. The MS spray voltage was 5.15 kV, and the heated capillary was maintained at 260°C. Deconvolution of averaged mass spectra was automatically performed by the software provided with the instrument. Fractions were collected manually, according to the UV trace, and finally lyophilized.
Native proteolysis
Purified α-KTx12.1 was dissolved in 0.1% TFA and submitted to proteolysis without disulfide reduction by addition of 2 μg of pepsin (Sigma-Aldrich). Five identical aliquots were taken at different times (0 min, 5 min, 30 min, 60 min and overnight), mixed immediately (1:1 v/v) with a saturated hydroxycinnamic acid solution (Sigma-Aldrich), and deposited on a stainless steel target. MALDI-TOF analyses were performed on a Voyager DE mass spectrometer (Applera) equipped with a nitrogen laser (337 nm). Data were acquired over 250 shots; an external calibration was used for the molecular mass determinations (Sequazime Peptide Mass Standards kit).
NMR data collection and processing
The samples were prepared in buffered solutions (90% H2O/10% D2O) at three different pH values, namely 4.5, 6.0, and 7.0; the protein concentration was in the range 0.5–0.7 mM. NMR spectra were collected at different temperatures (15°, 25°, and 35°C) on either a Bruker DMX or a Varian Inova spectrometer, both operating at 600 MHz as the proton Larmor frequency.
The two-dimensional homonuclear experiments included DQF-COSY (Rance et al. 1983), TOCSY (Davis and Bax 1985), and NOESY (Kumar et al. 1980), which were all performed using standard pulse sequences. Water suppression was achieved either by pulsed field gradients employing the WATERGATE scheme (Piotto et al. 1992) or by selective low-power irradiation of the water signal during the relaxation delay. The TOCSY spectra were acquired using both MLEV-17 (Bax and Davis 1985) and clean-DIPSI (Griesinger et al. 1988) spin-lock sequences with isotropic mixing periods of either 70 msec or 5.5 msec (in order to obtain COSY-type information). The NOESY spectra were recorded with mixing times of 150 msec and 200 msec. All spectra were collected in phase-sensitive mode with time-proportional phase incrementation of the initial pulse. Quadrature detection was used in both dimensions, with the carrier placed in the center of the spectrum on the water resonance. The spectral width in both dimensions was set to either 10 ppm or 12 ppm; typically, 512 t1 increments, each containing 2 K complex data points, were collected with a variable number of transients. The proton chemical shifts were referenced to the water signal at 4.81 ppm (25°C) and 4.69 ppm (35°C).
The spectral data were processed on a Silicon Graphics O2 workstation using either the XWIN-NMR 1.3 (Bruker) or Felix 97 (Accelrys) software package. Sine-squared window functions, phase shifted by 60°–90°, were applied in both dimensions prior to Fourier transformation. Baseline correction was applied in both dimensions wherever necessary.
The pKa of His28 was determined from a series of 2D 1H-NMR spectra carried out in the pH range 3.0–7.5. The pKa value was obtained from a least-squares fit of the data to the Henderson-Hasselbalch equation.
Distance constraints and structure calculations
Automated assignments of the NOEs were obtained with the home-written program nmr2st (Pristovšek et al. 2000). An internal calibration, based on the intensity of the well defined geminal Pro19 Hβ2-Hβ3 NOE connectivity, was used to set the upper distance limits. The integrated peak volumes from the NOESY spectra (mixing time 150 msec, 35°C) were converted into approximate interproton distances by normalizing them against the calibrated volume. The upper distance bounds were subsequently divided into four categories corresponding to 2.5, 3.2, 4.0, and 5.0 Å; lower bounds were taken as the sum of the van der Waals radii (1.9 Å) of the interacting protons.
The structures were calculated on a Silicon Graphics O2 workstation with the program DYANA (Güntert et al. 1997), which uses a simulated annealing algorithm combined with torsion angle dynamics. Starting ab initio, 100 conformers were calculated in 6000 annealing steps each. Initial structures were calculated by using the unambiguously assigned NOE-derived distance restraints and typical disulfide bond restraints. Subsequently, an iterative strategy was used for the structure refinement. In each round, the newly computed NMR structures were employed to additionally assign ambiguous NOE cross-peaks, to correct erroneous assignments, and to loosen the NOE distance bounds if spectral overlap was deduced.
Stereospecific assignments were obtained for 19 prochiral methylene and the two isopropyl groups using the program GLOMSA (Güntert et al. 1991). Pseudo-atom correction was applied in cases of magnetically equivalent protons as well as unknown stereospecific assignments, as proposed by Wüthrich (1986).
In the final calculation run, the structures were computed using meaningful distance restraints as listed in Table 1. The 18 best DYANA conformers were selected according to their standard target function values (Güntert et al. 1997) and subjected to restrained energy minimization by means of the DISCOVER module of the INSIGHT 97 software package (Accelrys). The consistent valence force field (CVFF) (Dauber-Osguthorpe et al. 1988) was used with a dielectric constant equal to r (distance in Å). A force constant of 20 kcal Å−2 mol−1 was applied in the NOE restraint term. The quality of the structures was determined using PROCHECK-NMR (Laskowski et al. 1996).
Molecular dynamics simulations
Energy minimization calculations, molecular dynamics simulations, and trajectory analysis were performed using the GROMACS program (Lindahl et al. 2001) running on a Compaq AlphaServer ES40 machine. Molecular visualization was done in the INSIGHT II graphical environment (Accelrys) operating on an Octane2 workstation (Silicon Graphics). The BIOPOLYMER module implemented in INSIGHT II was used to generate the starting conformations for the simulations.
System setup
Among the ensemble of 18 NMR-derived conformers of α-KTx12.1 at pH 4.5, the one with lowest energy was used to generate the starting coordinates for the simulations. Based on the tabulated pKa values of the CVFF implemented in the INSIGHT II package, the protonation state of the toxin residues at pH 4.0 and 7.0 were manually adjusted to the expected values, resulting in a peptide net charge of +5 and +3, respectively (Table 2). The N and C termini were treated as charged ammonium and carboxylate groups. Each model was neutralized with the necessary number of chloride counter ions and immersed in a rhombic dodecahedron box filled with 2244 and 2246 simple point charge (SPC) water molecules (Berendsen et al. 1981) for the simulation at pH 4.0 and 7.0, respectively.
Simulation parameters
The calculations were performed using the GROMACS all-hydrogen force field. The molecular dynamics simulations of both models were performed using periodic boundary conditions with a cutoff radius of 0.8 nm for short-range interactions and updating the neighbor pair list every 10 steps. Long-range electrostatic interactions were treated with the Particle Mesh Ewald method (Darden et al. 1993; Cheatham et al. 1995). The LINCS algorithm (Hess et al. 1997) was used to constrain all bond lengths, and SETTLE (Miyamoto and Kollman 1992) was employed to constrain water geometries. A time step of 2 fsec was chosen for integrating the equations of motion.
Simulation protocol
Initially, relaxation of solvent and ions was performed through 500 steps of the steepest descent energy minimization algorithm, while keeping the toxin backbone fixed. After removing any positional restraints, the system was submitted to another short minimization step that included NMR-derived distance restraints applied with a force constant of 10,000 kJ/mol/nm2. Thereafter, the system was equilibrated during a 200-psec step of molecular dynamics (MD) simulation with identical distance restraints at 308 K followed by another 800-psec step using a reduced force constant (2000 kJ/mol/nm2).
After equilibration, two consecutive MD simulations were carried out. In the first 1-nsec MD simulation, the NMR-derived restrains were applied with a force constant of 2000 kJ/mol/nm2. The second MD simulation was carried out without any experimental restraints for 10 nsec. The structures were collected every 10 psec during the equilibration step and every 0.1 psec during the remainder of the simulation.
Simulation analysis
The root mean square deviation (RMSD) was calculated for each structure collected during the simulations with respect to the selected NMR-derived conformer of lowest energy at pH 4.5. A pairwise RMSD for the backbone atoms was also determined, where each structure in the trajectory was compared with respect to all other structures. Finally, the root mean square fluctuation (RMSF) of the backbone atoms was computed on a per residue basis.
Essential dynamics analysis was carried out to obtain a quantitative characterization of the dynamic properties of both systems (van Aalten et al. 1996). The covariance matrices describing the positional fluctuations of the Cα atoms were diagonalized, resulting in sets of eigenvectors and corresponding eigenvalues. The eigenvectors (or principal modes) represent the concerted fluctuations of the Cα atoms in a 3N dimensional space (with N = number of atoms). The eigenvalue of each eigenvector represents the total mean square displacement of the atoms. The existence of concerted intramolecular motions within the subspace was investigated by projecting the trajectories onto the individual eigenvectors.
Acknowledgments
We thank Dr. Andrea Amadei for his critical and precious suggestions regarding the ED analysis. This work was partially supported by MIUR (Italy) and FAPESP (Brazil). The Interfaculty Center for Measurements (CIM) of the University of Parma, Italy; the CeBiME-LNLS, Campinas, Brazil; the Slovenian NMR Center in Ljubljana; and the European Large Scale Facility for Biomolecular NMR at the University of Frankfurt (UNIFRANMR), Germany are acknowledged for the use of the CD, NMR, and computing facilities. P.P. acknowledges financial support from the Ministry of Education, Science and Sport of Slovenia; T.A.P. is the recipient of a FAPESP (Brazil) Career Award Fellowship.
Abbreviations
BuTX, butantoxin
CVFF, consistent valence force field
DQF-COSY, double quantum filtered correlated spectroscopy
ED, essential dynamics
ESI, electron spray ionization
LCMS, liquid chromatography mass spectrometry
MALDI-TOF, matrix-assisted laser desorption ionization–time of flight
MD, molecular dynamics
NOE, nuclear Over-hauser effect
NOESY, nuclear Overhauser and exchange spectroscopy
PAGE, polyacrylamide gel electrophoresis
RMSD, root mean square deviation
RMSF, root mean square fluctuation
TFA, trifluoroacetic acid
TFE, trifluoroethanol
TOCSY, total correlation spectroscopy
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041131205.
Supplemental material: see www.proteinscience.org
References
- Arantes, E.C., Prado, W.A., Sampaio, S.V., and Giglio, J.R. 1989. A simplified procedure for the fractionation of Tityus serrulatus venom: Isolation and partial characterization of TsTX-IV, a new neurotoxin. Toxicon 27 907–916. [DOI] [PubMed] [Google Scholar]
- Ashcroft, F.M. 2000. Ion channels and disease. Academic Press, London.
- Batista, C.V.F., Gomez-Lagunas, F., Rodriguez de la Vega, R.C., Hajdu, P., Panyi, G., Gaspar, R., and Possani, L.D. 2002. Two novel toxins from the Amazonian scorpion Tityus cambridgei that block Kv1.3 and Shaker B K+-channels with distinctly different affinities. Biochim. Biophys. Acta 1601 123–131. [DOI] [PubMed] [Google Scholar]
- Bauer, F., Schweimer, K., Kluver, E., Conejo-Garcia, J.R., Forssmann, W.G., Rosch, P., Adermann, K., and Sticht, H. 2001. Structure determination of human and murine β-defensins reveals structural conservation in the absence of significant sequence similarity. Protein Sci. 10 2470–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bax, A. and Davis, D.G. 1985. MLEV-17-based two-dimensional homonuclear magnetization transfer spectroscopy. J. Magn. Reson. 65 355–360. [Google Scholar]
- Berendsen, H.J.C., Postma, J.P.M., van Gunsteren, W.F., and Hermans, J. 1981. Interaction models for water in relation to protein hydration. In Intermolecular forces (ed. B. Pullman), pp. 331–342. D. Reidel Publishing Co., Dordrecht, The Netherlands.
- Bloch Jr., C., Patel, S.U., Baud, F., Zvelebil, M.J., Carr, M.D., Sadler, P.J., and Thornton, J.M. 1998. 1H NMR structure of an antifungal γ-thionin protein SIα1: Similarity to scorpion toxins. Proteins 15 334–349. [DOI] [PubMed] [Google Scholar]
- Bontems, F., Roumestand, C., Boyot, P., Gilquim, B., Doljansky, Y., Menez, A., and Toma, F. 1991. Three-dimensional structure of natural charybdotoxyn in aqueous solution by 1H-NMR. Charybdotoxyn possesses a structural motif found in other scorpion toxins. Eur. J. Biochem. 196 19–28. [DOI] [PubMed] [Google Scholar]
- Canto, J., Fernandez, Y., Pons, M., Giralt, M., and Perez, J.J. 1999. Molecular dynamics study of Kaliotoxin in water. J. Biol. Macromol. 24 1–9. [DOI] [PubMed] [Google Scholar]
- Cheatham III, T.E., Miller, J.L., Fox, T., Darden, T.A., and Kollman, P.A. 1995. Molecular dynamics simulations on solvated biomolecular systems: The Particle Mesh Ewald method leads to stable trajectories of DNA, RNA, and proteins. J. Amer. Chem. Soc. 117 4193–4194. [Google Scholar]
- Cohen, L., Karbat, I., Gilles, N., Froy, O., Corzo, G., Angelovici, R., Gordon, D., and Gurevitz, M. 2004. Dissection of the functional surface of an anti-insect excitatory toxin illuminates a putative “hot spot” common to all scorpion β-toxins affecting Na+ channels. J. Biol. Chem. 279 8206–8211. [DOI] [PubMed] [Google Scholar]
- Corona, M., Gurrola, G.B., Merino, E., Cassulini, R.R., Valdez-Cruz, N.A., García, B., Ramírez-Domínguez, M.E., Coronas, F.I.V., Zamudio, F.Z., Wanke, E., et al. 2002. A large number of novel Ergtoxin-like genes and ERG K+-channels blocking peptides from scorpions of the genus Centruroides. FEBS Lett. 532 121–126. [DOI] [PubMed] [Google Scholar]
- Coronas, F.V., de Roodt, A.R., Olamendi-Portugal, T., Zamudio, F.Z., Batista, C.V.F., Gomez-Lagunas, F., and Possani, L.D. 2003. Disulfide bridges and blockage of Shaker B K+-channels by another butantoxin peptide purified from the Argentinean scorpion Tityus trivittatus. Toxicon 41 173–179. [DOI] [PubMed] [Google Scholar]
- Darden, T., York, D., and Pedersen, L. 1993. Particle Mesh Ewald: An N-log(N) method for Ewald sums in large systems. J. Chem. Phys. 98 10089–10092. [Google Scholar]
- Dauber-Osguthorpe, P., Roberts, P., Osguthorpe, V.A., Wolff, D.J., Genest, M., and Hagler, A.T. 1988. Structure and energetics of ligand binding to proteins: E. coli dihydropholate reductase-tremethoprim, a drug-receptor system. Proteins 4 31–47. [DOI] [PubMed] [Google Scholar]
- Davis, D.G. and Bax, A. 1985. Assignment of complex proton NMR spectra via two-dimensional homonuclear Hartmann-Hahn spectroscopy. J. Am. Chem. Soc. 107 2820–2821. [Google Scholar]
- Dyke, T.R., Duggan, B.M., Pennington, M.W., Byrnes, M.E., Kem, W.R., and Norton, R.S. 1996. Synthesis and structural characterization of analogues of the potassium channel blocker charybdotoxin. Biochim. Biophys. Acta 1292 31–38. [DOI] [PubMed] [Google Scholar]
- Ellis, K.C., Tenenholz, T.C., Jerng, H., Hayhurst, M., Dudlak, C.S., Gilly, W.F., Blaustein, M.P., and Weber, D.J. 2001. Interaction of a toxin from the scorpion Tityus serrulatus with a cloned K+ channel from squid (sqKv1A). Biochemistry 40 5942–5953. [DOI] [PubMed] [Google Scholar]
- Everhart, D., Cartier, E.G., Malhotra, A., Gomes, A.V., McIntosh, J.M., and Luetje, C.W. 2004. Determinants of potency on a β-conotoxin MII, a peptide antagonist of neuronal nicotinic receptors. Biochemistry 43 2732–2737. [DOI] [PubMed] [Google Scholar]
- Fant, F., Vranken, W., Brekaert, W., and Borremans, F. 1998. Determination of the three-dimensional solution structure of Raphanus sativus antifungal protein 1 by 1H NMR. J. Mol. Biol. 279 257–270. [DOI] [PubMed] [Google Scholar]
- Fernández, I., Romi, R., Szendeffy, S., Martin-Eauclaire, M.F., Rochat, H., Van Rietschoten, J., Pons, M., and Giralt, E. 1994. Kaliotoxin (1–37) shows structural differences with related potassium channel blockers. Biochemistry 33 14256–14263. [DOI] [PubMed] [Google Scholar]
- Fersht, A. 1998. Structure and mechanism in protein science. W.H. Freeman and Co., New York.
- Frémont, V., Blanc, E., Crest, M., Martin-Eauclaire, M., Gola, M., Darbon, H., and van Rietschoten, J. 1997. Dipole moments of scorpion toxins direct the interaction towards small- or large-conductance Ca+2-activated K+ channels. Lett. Pept. Sci. 4 305–312. [Google Scholar]
- Gairí, M., Romi, R., Fernandez, I., Rochat, H., Martin-Eauclaire, M.F., Van Rietschoten, J., Pons, M., and Giralt, E. 1997. 3D Structure of Kaliotoxin: Is residue 34 a key for channel selectivity? J. Pept. Sci. 3 314–319. [DOI] [PubMed] [Google Scholar]
- Garcia, M.L., Gao, Y.D., McManus, O.B., and Kaczorowski, G.J. 2001. Potassium channels: From scorpion venoms to high-resolution structure. Toxicon 39 739–748. [DOI] [PubMed] [Google Scholar]
- Gnanakaran, S., Nymeyer, H., Portman, J., Sanbonmatsu, K.Y., and Garcia, A.E. 2003. Peptide folding simulations. Curr. Opin. Struct. Biol. 13 168–174. [DOI] [PubMed] [Google Scholar]
- Goudet, C., Chi, C.W., and Tytgat, J. 2002. An overview of toxins and genes from the venom of the Asian scorpion Buthus martensi Karsch. Toxicon 40 1239–1258. [DOI] [PubMed] [Google Scholar]
- Griesinger, C., Otting, G., Wüthrich, K., and Ernst, R.R. 1988. Clean TOCSY for 1H spin-system identification in macromolecules. J. Am. Chem. Soc. 110 7870–7872. [Google Scholar]
- Grottesi, A. and Sansom, M.S.P. 2003. Molecular dynamics simulation of a K+ channel blocker: Tc1 toxin from Tityus cambridgei. FEBS Lett. 535 29–33. [DOI] [PubMed] [Google Scholar]
- Güntert, P., Braun, W., and Wüthrich, K. 1991. Efficient computation of three-dimensional protein structures in solution from nuclear magnetic resonance data using the program DIANA and the supporting programs CALIBA, HABAS and GLOMSA. J. Mol. Biol. 217 517–530. [DOI] [PubMed] [Google Scholar]
- Güntert, P., Mumenthaler, C., and Wüthrich, K. 1997. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273 283–298. [DOI] [PubMed] [Google Scholar]
- Hess, B., Bekker, H., Berendsen, H.J.C., and Fraaije, J.G.E.M. 1997. LINCS: A linear constraint solver for molecular simulations. J. Comp. Chem. 18 1463–1472. [Google Scholar]
- Holaday Jr., S.K., Martin, B.M., Fletcher Jr., P.L., and Krishna, N.R. 2000. NMR solution structure of butantoxin. Arch. Biochem. Biophys. 379 18–27. [DOI] [PubMed] [Google Scholar]
- Hu, H., Clarkson, M.W., Hermans, J., and Lee, A.L. 2003. Increased rigidity of eglin c at acidic pH: Evidence from NMR spin relaxation and MD simulations. Biochemistry 42 13856–13868. [DOI] [PubMed] [Google Scholar]
- Kipping, M., Zarnt, T., Steffen, K., Reimer, U., Fischer, G., and Bayer, P. 2001. Increased backbone flexibility in threonine45-phosphorylated hirudin upon pH change. Biochemistry 40 7957–7963. [DOI] [PubMed] [Google Scholar]
- Krezel, A.M., Kasibhatla, C., Hidalgo, P., MacKinnon, R., and Wagner, G. 1995. Solution structure of the potassium channel inhibitor agitoxin 2: Caliper for probing channel geometry. Protein Sci. 4 1478–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, A., Ernst, R.R., and Wüthrich, K. 1980. A two-dimensional nuclear Overhauser enhancement (2D NOE) experiment for the elucidation of complete proton-proton cross-relaxation networks in biological macromolecules. Biochem. Biophys. Res. Commun. 95 1–6. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., Rullmann, J.A., MacArthur, M.W., Kaptein, R., and Thornton, J.M. 1996. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8 477–486. [DOI] [PubMed] [Google Scholar]
- Lindahl, E., Hess, B., and van der Spoel, D. 2001. Gromacs 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Mod. 7 306–317. [Google Scholar]
- Meunier, S., Bernassau, J.-M., Sabastier, M.F., Martin-Eauclaire, M.F., van Rietschoten, J., Cambillau, C., and Darbon, H. 1993. Solution structure of P05-NH2, a scorpion toxin analog with high affinity for apamin-sensitive potassium channels. Biochemistry 32 11969–11976. [DOI] [PubMed] [Google Scholar]
- Miyamoto, S. and Kollman, P.A. 1992. SETTLE: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comp. Chem. 13 952–962. [Google Scholar]
- Novello, J.C., Arantes, E.C., Varanda, W.A., Oliveira, B., Giglio, J.R., and Marangoni, S. 1999. TsTX-IV, a short chain four-disulfide-bridged neurotoxin from Tityus serrulatus venom which acts on Ca2+-activated K+ channels. Toxicon 37 651–660. [DOI] [PubMed] [Google Scholar]
- Pimenta, A.M.C., Mansuelle, P., Diniz, C.R., and Martin-Eauclaire, M.F. 2003. Covalent structure and some pharmacological features of native and cleaved α-KTx12.1, a four disulfide-bridged toxin from Tityus serrulatus venom. J. Pept. Sci. 9 132–140. [DOI] [PubMed] [Google Scholar]
- Piotto, M., Saudek, V., and Sklenár, V. 1992. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2 661–665. [DOI] [PubMed] [Google Scholar]
- Pristovšek, P., Lücke, C., Reincke, B., Ludwig, B., and Rüterjans, H. 2000. Solution structure of the functional domain of Paracoccus denitrificans cytochrome c552 in the reduced state. Eur. J. Biochem. 267 4205–4212. [DOI] [PubMed] [Google Scholar]
- Rance, M., Sørensen, O.W., Bodenhausen, G., Wagner, G., Ernst, R.R., and Wüthrich, K. 1983. Improved spectral resolution in COSY 1H NMR spectra of proteins via double quantum filtering. Biochem. Biophys. Res. Commun. 117 479–485. [DOI] [PubMed] [Google Scholar]
- Rodriguez de la Vega, R.C., Merino, E., Becerril, B., and Possani, L.D. 2003. Novel interactions between K+ channels and scorpion toxins. Trends Pharmacol. Sci. 24 222–227. [DOI] [PubMed] [Google Scholar]
- Sawai, M.V., Jia, P.H., Liu, L., Aseyev, V., Wiencek, J.M., McCray Jr., P.B., Ganz, T., Kearney, W.R., and Tack, B.F. 2001. The NMR structure of human β-defensin-2 reveals a novel α-helical segment. Biochemistry 40 3810–3816. [DOI] [PubMed] [Google Scholar]
- Sforça, M.L., Oyama Jr., S., Canduri, F., Lorenzi, C.C.B., Pertinhez, T.A., Konno, K., Souza, M.B, Palma, M.S., Neto, J.R., Azevedo Jr., W.F., et al. 2004. How C-terminal carboxyamidation alters the biological activity of peptides from the venom of the eumenine solitary wasp. Biochemistry 43 5608–5617. [DOI] [PubMed] [Google Scholar]
- Shieh, C.C., Coghlan, M., Sullivan, J.P., and Gopalakrishnan, M. 2000. Potassium channels: Molecular defects, diseases, and therapeutic opportunities. Pharmacol. Rev. 52 557–593. [PubMed] [Google Scholar]
- Steen, K.H., Steen, A.E., Kreysel, H-W., and Reeh, P.W. 1996. Inflammatory mediators potentiate pain induced by experimental tissue acidosis. Pain 66 163–170. [DOI] [PubMed] [Google Scholar]
- Tenenholz, T.C., Klenk, K.C., Matteson, D.R., Blaustein, M.P., and Weber, D.J. 2000. Structural determinants of scorpion affinity: The charybdotoxin (α-KTX) family of K+-channel blocking peptides. Rev. Physiol. Biochem. Pharmacol. 140 135–185. [DOI] [PubMed] [Google Scholar]
- Thompson, J. and Begenisich, T. 2000. Electrostatic interaction between charybdotoxin and a tetrameric mutant of Shaker K+ channels. Biophys. J. 78 2382–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tytgat, J., Chandy, K.G., Garcia, M.L., Gutman, G.A., Martin-Eauclaire, M.F., van der Walt, J.J., and Possani, L.D. 1999. A unified nomenclature for short-chain peptides isolated from scorpion venoms: α-KTx molecular subfamilies. Trends Pharmacol. Sci. 20 444–447. [DOI] [PubMed] [Google Scholar]
- van Aalten, D.M.F., de Groot, B.L., Findlay, J.B.C., Berendsen, H.J.C., and Amadei, A. 1996. A comparison of techniques for calculating protein essential dynamics. J. Comp. Chem. 18 169–181. [Google Scholar]
- Waldmann, R. and Lasdunski, M. 1998. H+-gated cation channels: Neuronal acid sensors in the NaC/DEG family of ion channels. Curr. Opin. Neurol. 8 418–424. [DOI] [PubMed] [Google Scholar]
- Wang, I., Wu, S.-H., Chang, H.-K., Shieh, R.-C., Yu, H.-M., and Chen, C. 2002. Solution structure of a K+-channel blocker from the scorpion Tityus cambridgei. Protein Sci. 11 390–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart, D.S., Sykes, B.D., and Richards, F.M. 1992. The Chemical Shift Index: A fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry 31 1647–1651. [DOI] [PubMed] [Google Scholar]
- Wu, X. and Brooks, B.R. 2004. β-Hairpin folding mechanism of a nine-residue peptide revealed from molecular dynamics simulations in explicit water. Biophys. J. 86 1946–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wüthrich, K. 1986. NMR of proteins and nucleic acids. Wiley Interscience, New York.