

Abstract
Both NMR and IR studies of carbonyl (13C′) isotopomers of designed helices can provide residue-level details regarding the fractional occurrence and melting behavior of helical φ/ψ angles along the sequence of helical peptides, details that cannot be obtained from CD or 1H-NMR studies. We have studied a classic series of helical models, Ac-YGG-(KAXAA)3K-NH2 (X=A,V), in both aqueous and helix-favoring media containing fluoroalcohol cosolvents, including a solvent system allowing the observation of cold denaturation. These studies confirmed the strong N-capping associated with this sequence and revealed more extensive C-terminal fraying than that calculated using current helicity prediction algorithms. In the X=A series, the central residues are somewhat resistant to thermal melting; it instead occurs predominantly at the frayable C terminus. For the X=V series under cold-denaturing conditions, the temperature of maximal helicity is not uniform along the sequence and both solvated and nonsolvated helical alanine sites (13C=O stretches at 1592 cm−1 and 1615 cm−1, respectively) are apparent. Correlation between the two spectroscopies employed yielded the intriguing observation that the valine side chain is able to desolvate the i − 4 amide in short monomeric helices. In addition, we report further measurements of the temperature dependence of alanine statistical coil chemical shifts, the temperature dependence of the 13C chemical shift of urea (employed as chemical shift reference), and a useful formula for converting 13C′ shifts into fractional helicities.
Keywords: helix, chemical shift deviation, carbonyl stretch frequencies, backbone solvation, cold denaturation
Circular dichroism (CD) spectroscopy has proven to be an extremely useful method for quantifying the helical character of peptides and has been the primary source for data used in the calibration of current theoretical and empirical models for estimating the extent and distribution of helicity in peptide sequences. Unfortunately, there is a basic disconnect between the prediction algorithms and this experimental calibration method: the former operates on a per-residue basis, while the CD data are a summation over the complete sequence for all conformers present. The comparison of mutants in host/guest systems can provide parameters for the complete library of amino acids, but CD measurements cannot (in the absence of a model) define the distribution of helicity or extent of fraying across a single sequence. In order to parameterize helix/coil models to provide more accurate, finer-grained assessments of helicity, a different probe is required.
NMR data can provide residue-level measures of helicity. The secondary structure of a polypeptide chain is reflected in the chemical shifts of backbone atoms. We have recently applied chemical shift deviations (CSDs) to the study of short peptide hairpins (Andersen et al. 2004; Dyer et al. 2005). In designed helices (Scholtz and Baldwin 1992), the repetitive sequences result in significant signal overlap by proton NMR. With regard to 1H CSDs, the structuring shifts associated with secondary structure (local φ/ψ torsion angles) and backbone amide H-bonding status are rather small (~0.2–1 ppm for Hα and HN). Structuring shifts of this magnitude can also result from diamagnetic anisotropy contributions due to nearby side-chain functions, particularly from ring current effects of aryl groups. Studies focusing on specifically labeled 13C compounds, however, overcome these issues by providing a manageable number of signals for observation, and prior studies (Shalongo et al. 1994a; Park et al. 1998) have revealed larger structuring shifts associated with helix formation. The backbone carbons are also, due to their location, less likely to be influenced by ring current effects.
The application of 13C=O NMR to the study of alanine- rich helices was pioneered in the Stellwagen lab as a method for monitoring thermal transitions (Shalongo et al. 1994a), measuring helix distribution (Shalongo et al. 1994b), and analyzing N-capping (Park et al. 1998). CSDs were calculated as the change in chemical shift between the folded and the heat-denatured states. For alanine, CSDs on the order of 2.75 ppm were considered appropriate for centrally located positions in a helix. Conversion to fraction-folded values was accomplished with a correction for the temperature dependence of both the helical- and coil-state chemical shift expectation values.
Part of the interest in monitoring the chemical shift of 13Cα and 13C=O sites stems from model chemical shift calculations which indicate that ring current shifts for buried heavy atom backbone sites are much smaller than the secondary structure induced shifts. This differs from 1H chemical shift measures, for which ring current effects can often be as large or larger than those due to helix, strand, or turn formation. For helical systems, the carbonyl appeared particularly interesting, as the structuring transition represents not only a change of the neighboring φ and ψ dihedral angles, but also a change in hydrogen bond state for structured carbonyls. Furthermore, 13C′ labeling provides access to residue-specific carbonyl stretching frequencies (Decatur and Antonic 1999; Silva et al. 2000; Huang et al. 2002, 2004). The amide-I band for 13C-labeled carbonyls is shifted 39 cm−1 lower in frequency in comparison to 12C amide units, which is essentially the same as the ~37 cm−1 expected due to the isotopic mass effect (Tadesse et al. 1991; Werner et al. 2002). This shift in frequency allows labeled sites to be probed individually. The amide-I band has been employed to measure the kinetics of helix formation in both proteins and peptide helices (Huang et al. 2001, 2002; Werner et al. 2002). In this study, we employ 13C=O isotopomers of previously studied helical peptide sequences and controls to obtain a more precise estimate of the CSD associated with helix formation and to explore sequence effects on the cross-correlation of IR, CD, and NMR measures of helicity, the magnitude of end fraying effects, and helix melting behavior.
Materials and methods
Peptide synthesis and purification
Peptides were synthesized on an Applied Biosystem 433A synthesizer employing standard FMOC solid-state peptide synthesis methods and were acetylated while still on-resin. Rink resin provided an amidated C terminus upon cleaving using 95% trifluoroacetic acid, with 2.5% triisopropylsilane and 2.5% water. The cleavage product was then purified using reverse-phase HPLC on a Varian C18 preparatory-scale column using gradients of water and acetonitrile spiked with 0.1% and 0.085% trifluoroacetic acid, respectively. Collected fractions were lyophilized and the product molecular weight was confirmed on a Bruker Esquire ion trap mass spectrometer, from the (M + H)+1 peak. 13C′-labeled (99%) alanine was purchased from Cambridge Isotope Laboratories, Inc., and FMOC-protected by reacting with 1 equivalent of FMOC-OSuccinate (from Novabiochem) overnight in 1:1 acetone/water with 2% NaHCO3 by weight. The product (>90% yield) was isolated by ethyl acetate extraction after removal of the organic cosolvent.
Cosolvents
Hexafluorisopropanol (HFIP) and trifluoroethanol (TFE) were employed throughout our experiments in order to enhance folding and, in the case of HFIP, provide access to the cold-unfolding limb (Andersen et al. 1996a, 1999) of peptide helix thermostability curves. Cosolvent additions are expressed as final volume-% of cosolvent.
CD methods
Circular dichroism stock solutions were prepared by dissolving weighed amounts of peptide in 20 mM aqueous (pH 7.0) phosphate buffer to make solutions with an ~700-μM peptide concentration. The concentration of stock solutions of peptides containing a tyrosine residue was determined by UV based on the absorptivity coefficient, ɛ =1420 M−1 cm−1 at 276 nm. CD samples were diluted appropriately with buffer to obtain 35-μM peptide solutions. Fluoroalcohols were added as needed by gas-tight microsyringes.
Spectra were recorded on a Jasco J720 spectropolarimeter using 0.10-cm pathlength cells. The calibration of the wavelength and degree ellipticity scales and spectrum accumulation procedures have been described previously (Andersen et al. 1996b, 2002). For melting experiments, the temperature was increased from ~275K to 370K in 5° to 15° increments, depending on the degree of resolution required to elucidate the response. Each step was equilibrated at the target temperature for at least 5 min before data were acquired; larger temperature changes were provided additional time. CD data are reported in residue-molar ellipticity units (deg cm2 · residue · dmol−1).
NMR methods
All NMR experiments were collected on a Bruker DRX- 500 spectrometer. The samples consisted of ~2 mM peptide in 20 mM phosphate buffer (pH 7.0) with 10% D2O. All samples also contained 0.005 mM 13C-labeled urea as a chemical shift reference. The fully deuterated forms of HFIP and TFE were added in the appropriate amounts by gas-tight syringe. Sodium 2,2-dimethyl-2- silapentane-5-sulfonate (DSS) was used as the direct or indirect (below) internal chemical shift reference and set to 0 ppm for all conditions independent of temperature, pH, and cosolvent concentration.
Calibration of 13C′ shifts and calculation of chemical shift deviations
We employed 13C-labeled urea, which has a carbon shift in the same region as the peptide backbone amides, as the internal reference in hopes of increasing the precision of our shift data: Putting the reference signal in close proximity to the signals of interest allowed for a much narrower sweep width (32 ppm), yielding a resolution of 4.9 × 10−4 ppm/point. Because urea is not a commonly used reference standard, we measured its chemical shift (vs. DSS and dioxane) over a range of temperatures in all relevant solvent mixtures. The temperature response curves are displayed in the (Table 1S). The modest temperature dependence of the urea shift does, however, not affect our CSD calculations, as these are the difference between two signals (that of sites in the helical peptide and the comparable site in a “coil” reference peptide), both calibrated to urea. All chemical shifts were back calculated using the temperature dependence of urea versus DSS.
Calculation of residue-specific fractional helicities
In determining fH values, the chemical shifts obtained from the short (presumably statistical coil) reference peptides under matching conditions are assumed to represent fH=0 in water and in 9% HFIP. The previously determined (Andersen et al. 1999) equation for the 13C′ Δδ value for 100% helicity (taken from comparisons of 13C′, proton CSDs, and CD data for several peptides), ΔδHelix=3.040 + 0.00715T(°C), is used. Fractional helicity values were therefore calculated as
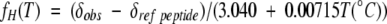 |
(1) |
Further validation of this equation is provided herein. The methodology is less reliable at high-percent fluoroalcohol. The statistical coil peptides used to provide sequence- and solvent-matched δref could display some helical character under highly structure-favoring conditions. Since the reference peptides lack a UV chromophore, we estimate helicity based on the R1 value, the ratio of the maximum CD signal between 190 and 195 nm versus the minimum signal between 195 and 210 nm (Bruch et al. 1991; Munoz et al. 1995), using the equation of Munoz et al.:
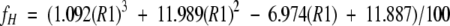 |
(2) |
Infrared spectroscopy
Peptide solutions were prepared from peptide samples lyophilized from TFA-containing aqueous solutions. Repeated lyophilization with D2O deuterated the sites with exchangeable hydrogens, but did not completely remove the TFA impurity. The solid peptides were dissolved in 99.9% D2O and appropriate fluoroalcohols to yield a concentration on the order of 2–4 mg/mL.
Equilibrium FTIR spectra were collected on a Bio-Rad FTS-40 interferometer using a temperature-controlled IR cell. The cell contained both the sample and a D2O reference solution between CaF2 windows with a 100-μm spacer. The cell is translated laterally under computer control to acquire matching sample and reference single beam spectra, and the protein absorption spectrum is computed as −log(Isamp/Iref).
Results
The YGG-3X model helices from the Baldwin laboratory (Marqusee and Baldwin 1987), which contained three repeats of a KAXAA sequence where X was either alanine or valine, have been very informative in prior studies of helix cold denaturation (Andersen et al. 1996a, 1999) and folding dynamics (Werner et al. 2002). Multiple sequences were synthesized with varying 13C′ labeling strategies, resulting in the sampling of 13C′ labels shown in Table 1.
Table 1.
YGG-3X and coil reference peptides: Sequence and 13C′ label positions (underlined)
| All 13C′ positions studied | |
| YGG-3A | Ac-YGGKAAAAKAAAAKAAAAK-NH2 |
| YGG-3V | Ac-YGGKAVAAKAVAAKAVAAK-NH2 |
| Helix Nos.a | 8 7 6 5 4 3 2 1 1 2 3 4 5 6 7 8 |
| Specific isotopomers of YGG-3V | |
| 3V(10,12) | Ac- YGGKAVAAKAVAAKAVAAK-NH2 |
| 3V(7,12) | Ac- YGGKAVAAKAVAAKAVAAK-NH2 |
| 3V(8,12,13) | Ac- YGGKAVAAKAVAAKAVAAK-NH2 |
| Coil reference peptides | |
| Coil-A | Ac- GKAAAKG- NH2 |
| Coil-V | Ac- GKAVAAK- NH2 |
a Helix positions are numbered from the center of the helix, which spans from K4 to K19 under conditions favoring helix formation; the central residues are numbered as −1 and +1, with larger negative and positive numbers on progressing to the N and C terminus, respectively.
In order to calculate the CSDs associated with structuring and fractional helicities (fH), the statistical coil and, for fH, helical expectation shifts must be known. Because 13C′ coil shifts exhibit nearest-neighbor effects (Wishart et al. 1995; Schwarzinger et al. 2001; Wang and Jardetzky 2002) and reference values are not available at all temperatures and solvent conditions, the “coil shifts” are most readily obtained using reference peptides. Two labeled reference peptides were constructed—coil-A and coil-V (Table 1)—that reproduced each of the five nearest- neighbor environments available to 13C′-labeled alanines in the YGG-3X peptides (KAA, AAA, AAK, KAV, VAA). The range of values due to environment was 0.57 ppm at 275K. A table of observed coil chemical shifts appears in (Table 2S). While the coil peptides appeared to be appropriately nonhelical in aqueous solution and in solutions containing only moderate amounts of FA cosolvent, similar systems display CD melting curves (Luo and Baldwin 1997) in 30%–40% vol-% TFE that indicate helicity loss on warming. If the coil-A and -V reference peptides are partially helical at high FA levels, this would result in underestimates of fractional helicity for test compounds under these conditions when Equation 1 is employed.
The helical models were studied in three solvents: aqueous, 30% TFE, and 9% HFIP. Thirty percent of TFE provides a helix-favoring environment for species normally only able to populate helical states to a moderate extent outside of a protein context. Nine percent of HFIP is a unique environment for this series of helical peptides, as it both stabilizes the helical structures to some extent and maximizes the cold-unfolding effect observed for certain systems (Andersen et al. 1996a, 1999). For our two model systems, only the valine-containing sequence displays cold unfolding, though both sequences show significant stabilization from FA addition (Fig. 1 ▶; Supplemental Material, Fig. 1S).
Figure 1.
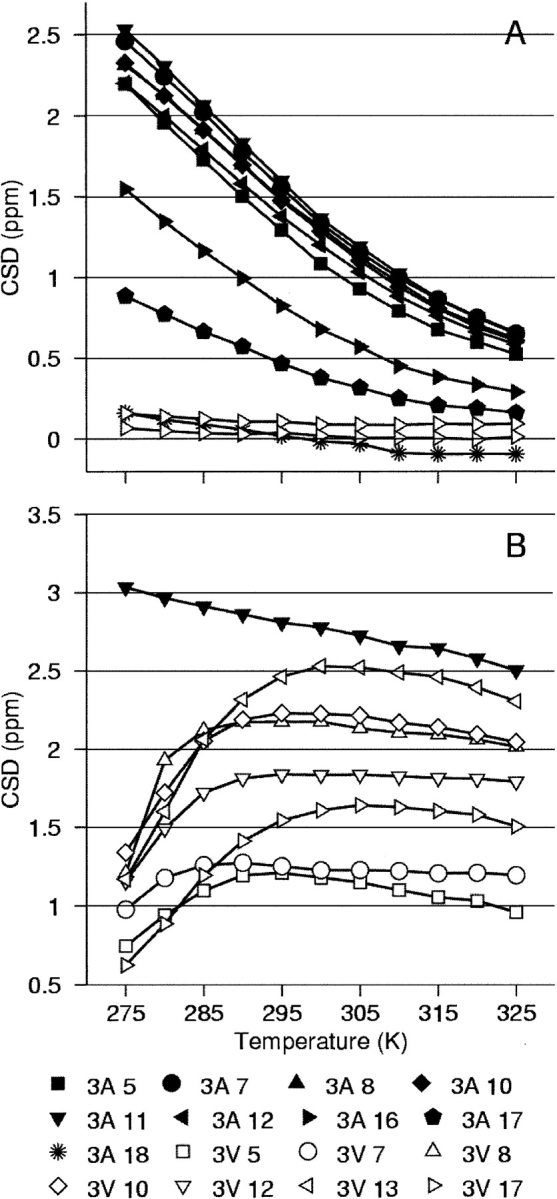
Panels A and B plot the temperature dependence of representative 13C′ CSDs for YGG-3A and −3V in aqueous and 9% HFIP solvents, respectively. Panel A includes representative traces for YGG- 3V; the CSDs were similarly small at all monitored positions. Panel B only includes the most-shifted position observed for YGG-3A. All YGG-3A positions display a similar monotonic temperature response, though the magnitude of the CSD varies with position.
Under all conditions, YGG-3A shows a monotonic loss of structuring shifts with increasing temperature at each labeled site. The resulting melting curves indicate a more stable, higher melting helix in the fluoroalcohol-containing media. In contrast, YGG-3V displays little or no helicity in aqueous solution, significant helicity (and steady loss with increasing temperature) in 30%TFE (see Supplemental Material), and a nonmonotonic response curve in 9% HFIP3, with a temperature of maximal structuring and helicity loss on both warming and cooling relative to this temperature. Dramatic cold denaturation in 6%–9% HFIP has previously been observed for YGG-3V by CD, and the thermodynamic parameters for the folding transition, as well as the temperature dependence of the 100% helix and statistical coil CD signals, have been determined (Andersen et al. 1996a, 1999).
Similar temperature responses have been observed for the amide-I bands in equilibrium IR spectra (see Fig. 1 ▶ in Werner et al. 2002) of unlabeled YGG-3X peptides (absorbance change at 1627 cm−1). While the IR melting curves show the same trends previously observed by CD and NMR, in the present study they do not provide a precise match to the NMR melting curves based on central 13C′ shift deviations. As in other spectroscopic melting studies, this can occur when the signals for the folded and unfolded states display intrinsic temperature dependencies and the temperature gradients are not the same for the different spectroscopic probes.
The 13C′ shift deviations can be brought into agreement with CD melt data by assuming that the CSD representing 100% conversion of a coil state alanine to a helical configuration increases by 0.24%/°C (as in Equation 1). Compared with the reconciled CD and NMR data, the IR tends to overestimate the extent of melting. This likely reflects the broadening of the amide-I band on warming.
We have previously reported IR difference spectra for some of the 13C′-isotopomers prepared for this NMR study (Andersen et al. 1999) and assigned the observed IR signals (Werner et al. 2002). Centrally labeled YGG-3A (positions 10 and 12) displayed 12C′ (1629 cm−1) and 13C′ (1590 cm−1) helical amide-I′ bands that responded monotonically on warming, helicity being lost as the temperature increased. Based on other literature reports (Manas et al. 2000; Walsh et al. 2003), these correspond to a solvated helical C=O band. The similarly labeled YGG-3V, isotopomer 3V(10,12), displayed the same two bands (1627 and 1590 cm−1) with an additional 13C=O band that appeared as a shoulder near 1615 cm−1 (Fig. 2 ▶). The two readily quantitated IR signals reached maximal values at ~24°C and decreased both on warming and cooling. The extent of cold- versus heat-induced unfolding observed at the two bands was comparable.
Figure 2.

Difference IR spectra for the amide-I′ region of YGG-3V with centrally located 13C′ labels (residue numbers 10 and 12) in 10% HFIP. The upper series of traces (Y-axis on right) cover the cold-unfolding limb (4°–21°C) of the peptide’s thermal transition; increasing signals at 1590 cm−1 and 1627 cm−1 indicate increasing helicity on warming over this range. A shoulder at ~1615 cm−1 is also visible. The lower series (Y-axis on left) similarly display the loss of helicity with continued temperature increase.
The additional band, evident as a shoulder in Figure 2 ▶, was assigned to a “nonsolvated” helix C=O. Based on the mass effect on a pure C=O stretch, this would place the corresponding 12C=O at 1652 cm−1, the position at which buried helices in proteins are observed. In support of this rationale, DeGrado and coworkers have reported similar frequencies for sequestered (13C′-Leu at 1607 cm−1) and exposed (13C′-Ala at 1587 cm−1) positions in a dimeric α-helical coiled-coil (Manas et al. 2000; Walsh et al. 2003). Although other rationales for backbone shielding in helices have been reported (vide infra), the sequestered (nonsolvated) band observed for YGG-3V is clearly absent in YGG-3A, and thus, evidently, due to the valine side chains. It was also clear that the nonsolvated amide-I′ band of isotopomer 3V(10,12) displayed a different temperature dependence in 10% HFIP: The ~1615 cm−1 shoulder continues to increase in intensity until a higher temperature is reached (~36°C rather than 24°C). Reference to panel B of Figure 1 ▶ shows that the maximal 13C-NMR measure of helicity can occur anywhere from 17° to 32°C, depending on the location of 13C′ label.
In order to explore this and to assign the solvated versus nonsolvated 13C′ sites, additional YGG-3V isotopomers were examined by IR. We hypothesized that the different label-related helical bands observed were due to the positions of the labels relative to the valines. Through specific label placement, we attempted to create an isotopomer that provided only a single 13C-amide-I′ absorbance. Due to the size of the ~1615 cm−1 band and its position relative to the much larger 1627 cm−1, our attempt focused on trying to minimize the size of the 1590 cm−1 signal using labels that could be verified by 13C NMR to be in helical regions: This was realized with the Ala7,12–13C′, the 1590 cm−1 feature reduced to <5% of 12C helix band at 1627 cm−1 over the melting limb (Fig. 3 ▶, lower panel). This isotopomer also displayed essentially no melting limb in the 13C′ shift melting curves (Fig. 1B ▶). The ~1615 cm−1 shoulder seen in Figure 2 ▶ is also seen in the 3V(7,12) melt and becomes an extremum in temperature difference spectra (upper series) over the cold-denaturation limb.
Figure 3.

Difference IR spectra for the amide-I′ region of isotopomer 3V(7,12) in 10% HFIP. The data presentation in Figure 2 ▶ is employed such that positive peaks in the top panel reflect IR changes associated with increasing helicity on warming from 4° to 25°C. The 1615 cm−1 peak increases throughout the 4°–25°C range, while the 1630 cm−1 peak in the difference spectra reaches its maximal value at 15°C. The lower panel reflects changes on further warming beyond 25°C. Placement of labels at residues 7 and 12 severely reduces the size of the 1590 cm−1 band in the melting limb relative to that observed for 3V(10,12), shown in Figure 2 ▶.
Discussion
The YGG-3A peptide was the only sequence studied that was helical under purely aqueous conditions (Fig. 1 ▶). By 13C NMR at 275K, the peptide displayed significant helicity in the central portion of the sequence, with continued helicity through residue 5 (position – 7 relative to the center of the helix), near the N terminus. Residues 16, 17, and 18 (helix positions +5, +6, +7) displayed less helicity than the central residues: The deviations decreased linearly, the three residues having 65.3%, 37.3%, and 6.7%, respectively, of the average deviation observed for central residues 10, 11, and 12 (positions −2, −1, +1). Residue 5, by comparison, displayed 92.6% of the average central-residue deviation. A similar observation, of significant C-terminal fraying along a designed poly-Ala helix, has been made by FTIR (Huang et al. 2002).
The thermal melts of the YGG-3A peptide indicated that the C-terminal fraying observed at the low-temperature limit increased with temperature. By 325K, residues 16 and 17 only displayed 48.3% and 26.9%, respectively, of the deviation observed for the central residues. The N terminus also lost helicity relative to the central positions with increasing temperature, though the effect was less pronounced: At 325K, residue 5 displayed 87.4% of the central residues’ average deviation. These observations support a structure ensemble in which the helical state is not only sampled less frequently with increasing temperature, but the average length of the sampled helical state also decreases; the majority of the decrease in length appears to be due to fraying at the C terminus (see Fig. 4B ▶).
Figure 4.
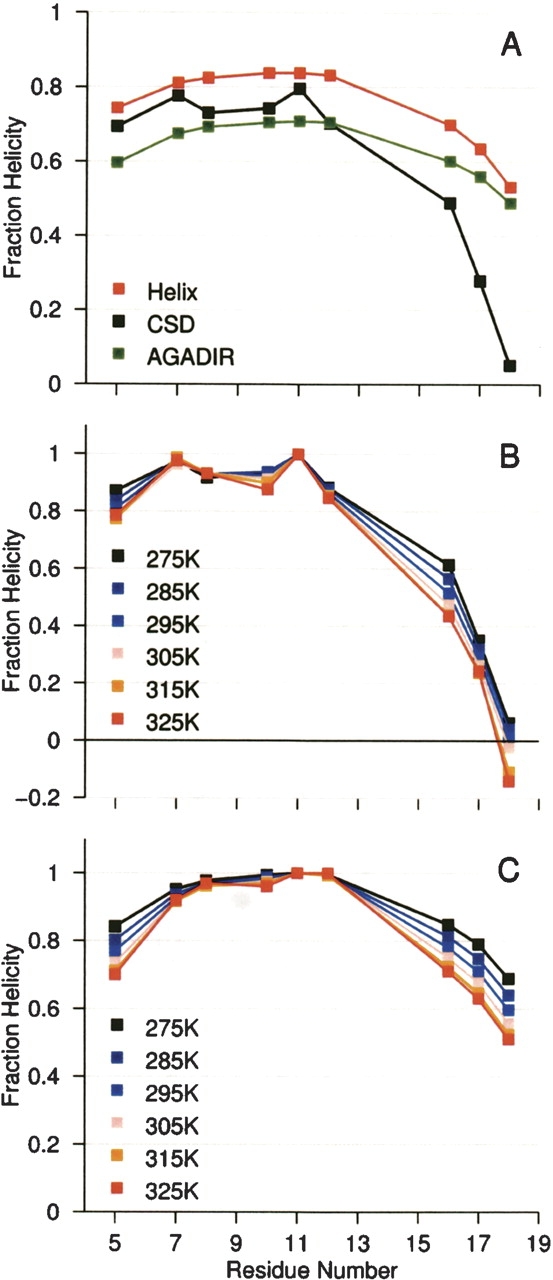
A comparison of observed and calculated helicity values for YGG-3A in aqueous solution. Panel A compares residue-level helicity estimations as determined by 13C′ labels, AGADIR, and Helix. The experimental and AGADIR values are at 275K. Panels B and C display changes in helicity values as a function of temperature by dividing all positions by the fH at the most-helical position at a given temperature. Panel B displays the experimental values determined from 13C′ CSDs. Panel C contains values calculated using AGADIR.
Turning to the temperature dependence of 13C′ CSDs in 9% HFIP, YGG-3A displays uniform melting but YGG-3V displayed cold denaturation with notable melting curve differences for specific sites (Fig. 1B ▶). The data from the 3V(10,12) isotopomer are noteworthy: The 13C′ CSD at residue 12 is smaller and its temperature response flatter than neighboring sites. Residue 7 is similar; these two sites display the least CSD melting. (Differences in the thermal profile of residues 7 and 12 relative to other sites is even more distinct in CSD data collected in 30% TFE [see Supplemental Material, Fig. 1S].) The common feature of these two positions, not shared with any other positions examined, is their placement i − 4 to a valine side chain.
Isotopomer 3V(10,12) was also studied by FTIR. In 10% HFIP, the peptide displayed separate bands assigned to solvent-exposed (1590 cm−1) and solvent-sequestered (1615 cm−1) helical sites, the latter being a surprising observation for a monomeric 19-residue peptide. The solvent-sequestered band was not observed for any YGG-3A isotopomer. Models of ideal helices suggested that the labeled amide unit of a residue i with a 13C′ substitution would be shielded by the side chain of a valine placed at position i + 4. The choice of isotopomer 3V(7,12) reflected this hypothesis, with both labels located four residues before a valine. In comparison to difference spectra of 3V(10,12) and 3V(8,12,13), both of which displayed significant 13C′ solvated helix bands (1590 cm−1), the same band for 3V(7,12) was greatly diminished, particularly over the extensively studied melting limb. The sequestered 13C=O band appeared as a 1615 cm−1extremum in difference spectra over the cold denaturing limb. The changes on warming from 4° to 25°C deserve more detailed comment. From 4° to 11°C, the solvent-sequestered 13C′ and solvated 12C′ bands indicate increasing helicity (Fig. 3 ▶). As further warming- induced helicity appears, the 1615 cm−1 band dominates, in part reflecting the conversion of some solvated 13C=O species to a sequestered state: A negative difference spectral feature appears at 1592 cm−1. This can be rationalized as the result of cold denaturation producing shorter helices lacking the turns that would afford protection for H-bonding interactions to backbone amides. Warming allows the helices to lengthen and backbone 13C=O positions to become sequestered. The change in the relative magnitudes of the 1615 and 1630 cm−1 features over the cold limb of this experiment reflects position-specific differences in melting behavior. For 3V(10,12) (Fig. 2 ▶), we also observed that the 1625–1630 cm−1 feature begins to melt at lower temperatures than the 1615 cm−1 feature.
To compare our experimental results with theoretical models of helicity, it is essential to convert the CSDs to residue-specific fractional helicities (fH). The statistical coil expectation value for chemical shift is set by the values obtained from our short, nonhelical peptides. These values have an inherent correction for the temperature dependence of the coil state; the five different positions have, in aqueous conditions, a slope of −0.00982 ppm/°C with a standard deviation of 0.00082. An expectation CSD value for 100% helicity is obtained using Equation 1, utilizing a Δδ(0%–100% helix) of 3.04 ppm that increases by 0.00715 ppm/°C on warming (mostly reflecting the negative slope of the coil state reference value). Our helicity Δδ is, as it should be, somewhat larger than the values (2.46–2.83 ppm) observed at central alanine positions by Shalongo et al. (1994b): even central helical positions do not reach 100% helicity. The Stellwagen laboratory has also reported temperature-dependence values for alanine in the coil and the helix states (Park et al. 1998) that, when combined, provide a temperature-dependence value for the CSD due to helix formation: 0.0053 ppm/°C, 0.0059 ppm/°C in an earlier report (Shalongo et al. 1994a), both values somewhat smaller than the 0.00715 ppm/°C value employed in this study.
Conversion of the CSDs to fH is straightforward for data obtained in aqueous and 9% HFIP solutions and correlated to values obtained by other methods. Centrally labeled YGG-3A (positions −2 and +1) was previously reported (Andersen et al. 1999) as losing 12% of its 1589 cm−1 IR band between 3° and 29°C. The band corresponds to the helicity at the labeled sites; over the similar range of 275–300 K, the average fH loss for these residues by 13C′ CSD measures was 13%. Obtaining fH for the experiments in 30%TFE is compromised by partial structuring of the coil reference peptides. The reference chemical- shift values observed in 9% HFIP at 275K were only slightly more downfield than those observed with no fluoroalcohol: An average difference of 0.02 ± 0.03 ppm was observed over the five positions that were labeled. By comparison, in 30% TFE, the chemical shifts moved, on average, 0.29 ± 0.15 ppm downfield. By 325K, this difference had been reduced to 0.14 ± 0.09 ppm. This implies helicity in this medium at the lower temperatures. Coil-V, with the largest shift changes upon transport to 30%TFE, was also examined by CD (in 20% HFIP, a helicity-supporting environment similar to 30% TFE). Helicity was quantified based on the R1 value (Bruch et al. 1991); Equation 2 (Munoz et al. 1995) indicated a fH of 0.20 at 295K. The ellipticity at 221 nm was essentially constant over the temperature range observed (274K–314K) but the maximum near 187 nm and the minimum near 201 nm decreased in absolute intensity with temperature, indicative of helix melting. Returning to 13C′ CSDs, if coil-V in 30% TFE is referenced to the coil values obtained in 9% HFIP, the two monitored positions have fH values of 0.08. The discrepancy between the two measures (0.12 in fH units or twofold in relative helicity) may come as a result of each method detecting different structural features. Circular dichroism measures the cumulative effect of a specific orientation of amide planes (dependent on an extensive string of φ/ψ values), while the 13C′ shift depends predominantly on local φ/ψ values and, presumably, the H-bonding status of the C=O (Xu and Case 2002). If the H-bonding effect (vide infra) is a significant component of the total CSD ascribed to helicity, then the helicity of short peptides would consistently be underestimated by 13C′ NMR, as even central residues may lack a reliable hydrogen bond partner. The absence of a H-bonding contribution could also result in smaller 13C′ CSDs in the C-terminal turn of a helix.
The stability of the YGG-3A helix in aqueous solution allowed for a comparison between NMR- and theory-derived measures of fractional helicity (Fig. 4 ▶). The 275K values for YGG-3A were compared with residue-level helicity values arrived at using Helix v.1.5 (Andersen and Tong 1997) and AGADIR (Munoz and Serrano 1995a,b, 1997; Lacroix et al. 1998). In general, the theory-based estimates of helicity match the experimental helicity distribution in the N-terminal and central portions. Both Helix and AGADIR indicated a single helix with a most-structured point near residue 11 (helix position −1), the center of the helix-favoring portion of the sequence. In this region, however, where the theoretical methods describe a smooth distribution of helical character, the NMR data suggest a more varied structural landscape. The variability is similar to that observed for another alanine-rich, helical peptide, Ac-(AAQAA)3- NH2 (Shalongo et al. 1994b).
In addition to differences in the central portion of the helix, the C-terminal end fraying observed by NMR does not match that estimated by current helicity-estimation methods (Fig. 4 ▶). The 13C′ CSDs toward the C terminus provide much smaller fH estimates than those expected based on both the Helix and AGADIR algorithms, with the experimental value falling to an fH of 0.10 at the penultimate residue. Some of the difference may be attributable to an H-bonding contribution to the 13C′ CSD. Chemical shift calculations based on density functional theory have previously estimated the contribution of hydrogen bond formation to the 13C′ CSD observed for a central helical residue to be as much as 1.4 ppm (Xu and Case 2002); thus, perhaps as much as 46% of the experimental CSD may be due to H-bonding. The chemical-shift-derived fH value calculated for our C-terminal 13C′ positions would therefore underestimate the degree of folding, as the residues are not capable of forming intramolecular, i/i + 4 hydrogen bonds. Even so, the experimental data still require more C-terminal fraying than that calculated by either helicity prediction method. Consider that if residue 18 were indeed 51% helical (the average of the Helix and AGADIR values), the expectation CSD after removing the 0.7 ppm H-bond effect would still be 0.84 ppm. Instead, a CSD of 0.16 ppm is observed.
The overestimation of C-terminal helicity by AGADIR is consistent across the temperature range studied. The comparisons appear in Figure 4, B and C ▶. Though the estimation of absolute helicity is inconsistent in some regions, the AGADIR predictions and experimental 13C′ CSD values indicate equivalent central helical melting, while distal positions have steeper thermal gradients.
The drop in fH observed for the C terminus of YGG-3A under aqueous conditions is mirrored under other conditions. At 325K in 30% TFE, YGG-3A displays a central fH (residue 11) of 0.65. The C-terminal residues, 16–18, have fH values of 0.45, 0.28, and 0.10, respectively. These terminal values are a close match to those of YGG-3A at 275K without FA addition (0.49, 0.28, 0.05). Under the latter conditions, however, residue 11 attained a fH of 0.80. This result is congruent with helix stabilization by fluoroalcohol resulting in folded ensembles with helices longer than those found under aqueous conditions. Indeed, at 295K, when the fH of residue 11 in 30% TFE nearly matches the aqueous value (0.79), the C-terminal fractional helicities have risen to 0.63, 0.39, and 0.15. The previously mentioned underestimation of helix formation at C-terminal sites due to the lack of backbone hydrogen bond formation does not affect this comparison.
The N terminus of the YGG-3A helix, by comparison, shows little difference between the thermal fraying predicted by AGADIR and that observed using NMR. Both profiles show some additional loss of helicity at the N terminus, with AGADIR predicting somewhat greater N-terminal fraying than the experimental shift data.
Concluding remarks
There is substantial agreement between theory and experiment regarding the degree of helicity observed at central helical residues. The 13C′ CSDs indicate some heterogeneity in central residue helicity; our observations mimic those of the Stellwagen group (Shalongo et al. 1994b). It should also be noted that a significant H-bonding component in the 13C′ shift associated with helicity could imply that this heterogeneity corresponds to changes in H-bonding distances over this region. Our experimental data confirm the N-capping effect in the YGG-3X peptides and indicate substantial fraying in the C-terminal segment of these helices. For both cold denaturation and helicity loss upon warming, the resulting helical ensembles appear to contain shorter helices, with most of the loss occurring at the frayable C terminus. Computational models of helicity do not reproduce this observation. Studies currently under way will extend this method to helices with C-capping motifs and should provide additional models to test the validity of Equation 1 for deriving fractional helicities from 13C′ shifts.
The most surprising observation in this study was the observation of high-frequency (~1615 cm−1) 13C=O bands for simple monomeric helices. This frequency is essentially that observed for the carbonyls of buried helices in proteins and also has been observed at buried interfacial sites in coiled coils (Manas et al. 2000; Walsh et al. 2003). The “shielding” of specific amide carbonyls from solvation has been predicted by Gnanakaran et al. from simulations of the IR spectrum of AA(AA KAA)3AAY (Gnanakaran et al. 2004). Replica exchange molecular dynamics simulations of the effects on the IR spectrum of conformational variability and H-bonding (internal and with water) led to the prediction that the Lys side chain shields the carbonyl group at i − 4, leading to an increase in the vibrational frequency. Furthermore, this shielding effect has been recently reported by the Decatur group on the basis of the FTIR spectrum of labeled helical peptides (Starzyk et al. 2005). Desolvation (upon TFE addition) of the residue at i − 4 from a Lys in a helical (AAKAA)n peptide was demonstrated using specific isotope labeling of the backbone carbonyls. No evidence for this specific effect of a lysine side chain was observed for the present set of isotopomers; rather, the correlations observed between IR and NMR data provided independent corroboration of the desolvation of helical alanine backbone sites by valine side chains located at the i + 4 position. A similar effect has been noted for an i + 4 Thr side chain (Walsh et al. 2003). The latter effect is due to intramolecular H-bonding; the Val effect is presumably strictly the result of steric inhibition of backbone solvation. Correlations between IR and NMR studies of 13C isotopomers should also prove useful in the study of β-hairpin peptides, a topic we plan to explore in future publications.
Acknowledgments
Support for this work was derived from NSF grant CHE0315361 (N.H.A.) and NIH grantGM053640 (R.B.D.).
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051510705.
Supplemental material: see www.proteinscience.org
Footnotes
The cold-denaturation effect in nonamphiphilic helices with limited HFIP addition has been most pronounced when valine is included in the sequence. This residue is also the only one that displayed a uniform increase in the Zimm-Bragg s-value in early host–guest studies of helix propensities (Scheraga et al. 2002).
References
- Andersen, N.H. and Tong, H. 1997. Empirical parameterization of a model for predicting peptide helix/coil equilibrium populations. Protein Sci. 6 1920–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen, N.H., Cort, J.R., Liu, Z., Sjoberg, S.J., and Tong, H. 1996a. Cold denaturation of monomeric peptide helices. J. Am. Chem. Soc. 118 10309–10310. [Google Scholar]
- Andersen, N.H., Liu, Z., and Prickett, K.S. 1996b. Efforts toward deriving the CD spectrum of a 3(10) helix in aqueous medium. FEBS Lett. 399 47–52. [DOI] [PubMed] [Google Scholar]
- Andersen, N.H., Dyer, R.B., Fesinmeyer, R.M., Gai, F., Liu, Z., Neidigh, J.W., and Tong, H. 1999. Effect of hexafluoroisopropanol on the thermodynamics of peptide secondary structure formation. J. Am. Chem. Soc. 121 9879–9880. [Google Scholar]
- Andersen, N.H., Brodsky, Y., Neidigh, J.W., and Prickett, K.S. 2002. Medium-dependence of the secondary structure of exendin-4 and glucagon-like-peptide-1. Bioorg. Med. Chem. 10 79–85. [DOI] [PubMed] [Google Scholar]
- Andersen, N.H., Fesinmeyer, R.M., and Hudson, F.M. 2004. Analysis of β peptides using chemical shift deviations. In Peptide revolution: Genomics, proteomics and therapeutics (eds. M. Chorev and T.K. Sawyer), pp. 462–463. American Chemical Society, Cardiff, CA.
- Bruch, M.D., Dhingra, M.M., and Gierasch, L.M. 1991. Side chain-backbone hydrogen bonding contributes to helix stability in peptides derived from an α-helical region of carboxypeptidase A. Proteins 10 130–139. [DOI] [PubMed] [Google Scholar]
- Decatur, S.M. and Antonic, J. 1999. Isotope-edited infrared spectroscopy of helical peptides. J. Am. Chem. Soc. 121 11914–11915. [Google Scholar]
- Dyer, R.B., Maness, S.J., Franzen, S., Fesinmeyer, R.M., Olsen, K.A., and Andersen, N.H. 2005. Hairpin folding dynamics: The cold-denatured state is predisposed for rapid refolding. Biochemistry 44 10406–10415. [DOI] [PubMed] [Google Scholar]
- Gnanakaran, S., Hochstrasser, R.M., and Garcia, A.E. 2004. Nature of structural inhomogeneities on folding a helix and their influence on spectral measurements. Proc. Natl. Acad. Sci. 101 9229–9234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, C.Y., Getahun, Z., Wang, T., DeGrado, W.F., and Gai, F. 2001. Time-resolved infrared study of the helix-coil transition using (13)C-labeled helical peptides. J. Am. Chem. Soc. 123 12111–12112. [DOI] [PubMed] [Google Scholar]
- Huang, C.Y., Getahun, Z., Zhu, Y., Klemke, J.W., DeGrado, W.F., and Gai, F. 2002. Helix formation via conformation diffusion search. Proc. Natl. Acad. Sci. 99 2788–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, R., Kubelka, J., Barber-Armstrong, W., Silva, R.A., Decatur, S.M., and Keiderling, T.A. 2004. Nature of vibrational coupling in helical peptides: An isotopic labeling study. J. Am. Chem. Soc. 126 2346–2354. [DOI] [PubMed] [Google Scholar]
- Lacroix, E., Viguera, A.R., and Serrano, L. 1998. Elucidating the folding problem of α-helices: Local motifs, long-range electrostatics, ionic-strength dependence and prediction of NMR parameters. J. Mol. Biol. 284 173–191. [DOI] [PubMed] [Google Scholar]
- Luo, P. and Baldwin, R.L. 1997. Mechanism of helix induction by trifluoro-ethanol: A framework for extrapolating the helix-forming properties of peptides from trifluoroethanol/water mixtures back to water. Biochemistry 36 8413–8421. [DOI] [PubMed] [Google Scholar]
- Manas, E.S., Getahun, Z., Wright, W.W., and DeGrado, W.F. 2000. Infrared spectra of amide groups in α-helical proteins: Evidence for hydrogen bonding between helices and water. J. Am. Chem. Soc. 122 9883–9890. [Google Scholar]
- Marqusee, S. and Baldwin, R.L. 1987. Helix stabilization by Glu-...Lys + salt bridges in short peptides of de novo design. Proc. Natl. Acad. Sci. 84 8898–8902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz, V. and Serrano, L. 1995a. Elucidating the folding problem of helical peptides using empirical parameters. II. Helix macrodipole effects and rational modification of the helical content of natural peptides. J. Mol. Biol. 245 275–296. [DOI] [PubMed] [Google Scholar]
- ———. 1995b. Elucidating the folding problem of helical peptides using empirical parameters. III. Temperature and pH dependence. J. Mol. Biol. 245 297–308. [DOI] [PubMed] [Google Scholar]
- ———. 1997. Development of the multiple sequence approximation within the AGADIR model of α-helix formation: Comparison with Zimm- Bragg and Lifson-Roig formalisms. Biopolymers 41 495–509. [DOI] [PubMed] [Google Scholar]
- Munoz, V., Serrano, L., Jimenez, M.A., and Rico, M. 1995. Structural analysis of peptides encompassing all α-helices of three α/β parallel proteins: Che-Y, flavodoxin and P21-ras: Implications for α-helix stability and the folding of α/β parallel proteins. J.Mol. Biol. 247 648–669. [DOI] [PubMed] [Google Scholar]
- Park, S.H., Shalongo, W., and Stellwagen, E. 1998. Analysis of N-terminal capping using carbonyl-carbon chemical shift measurements. Proteins 33 167–176. [DOI] [PubMed] [Google Scholar]
- Scheraga, H.A., Vila, J.A., and Ripoll, D.R. 2002. Helix-coil transitions revisited. Biophys. Chem. 101–102 255–265. [DOI] [PubMed] [Google Scholar]
- Scholtz, J.M. and Baldwin, R.L. 1992. The mechanism of α-helix formation by peptides. Annu. Rev. Biophys. Biomol. Struct. 21 95–118. [DOI] [PubMed] [Google Scholar]
- Schwarzinger, S., Kroon, G.J., Foss, T.R., Chung, J., Wright, P.E., and Dyson, H.J. 2001. Sequence-dependent correction of random coil NMR chemical shifts. J. Am. Chem. Soc. 123 2970–2978. [DOI] [PubMed] [Google Scholar]
- Shalongo, W., Dugad, L., and Stellwagen, E. 1994a. Analysis of thermal transitions of a model helical peptide using 13C NMR. J. Am. Chem. Soc. 116 2500–2507. [Google Scholar]
- ———. 1994b. Distribution of helicity within the model peptide acetyl(AAQAA) 3amide. J. Am. Chem. Soc. 116 8288–8293. [Google Scholar]
- Silva, R.A., Kubelka, J., Bour, P., Decatur, S.M., and Keiderling, T.A. 2000. Site-specific conformational determination in thermal unfolding studies of helical peptides using vibrational circular dichroism with isotopic substitution. Proc. Natl. Acad. Sci. 97 8318–8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starzyk, A., Barber-Armstrong, W., Sridharan, M., and Decatur, S.M. 2005. Spectroscopic evidence for backbone desolvation of helical peptides by 2,2,2-trifluoroethanol: An isotope-edited FTIR study. Biochemistry 44 369–376. [DOI] [PubMed] [Google Scholar]
- Tadesse, L., Nazarbaghi, R., and Walters, L. 1991. Isotopically enhanced infrared spectrsocopy: A novel method for examining secondary structure at specific sites in conformationally heterogeneious peptides. J. Am. Chem. Soc. 113 7036–7037. [Google Scholar]
- Walsh, S.T.R., Cheng, R.P., Wright, W.W., Alonso, D.O.V., Daggett, V., Vanderkooi, J.M., and DeGrado, W.F. 2003. The hydration of amides in helices: A comprehensive picture from molecular dynamics, IR, and NMR. Protein Sci. 12 520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. and Jardetzky, O. 2002. Investigation of the neighboring residue effects on protein chemical shifts. J. Am. Chem. Soc. 124 14075–14084. [DOI] [PubMed] [Google Scholar]
- Werner, J.H., Dyer, R.B., Fesinmeyer, R.M., and Andersen, N.H. 2002. Dynamics of the primary processes of protein folding: Helix nucleation. J. Phys. Chem. B 106 487–494. [Google Scholar]
- Wishart, D.S., Bigam, C.G., Holm, A., Hodges, R.S., and Sykes, B.D. 1995. 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J. Biomol. NMR 5 67–81. [DOI] [PubMed] [Google Scholar]
- Xu, X.P. and Case, D.A. 2002. Probing multiple effects on 15N, 13Cα, 13Cβ, and 13C′ chemical shifts in peptides using density functional theory. Biopolymers 65 408–423. [DOI] [PubMed] [Google Scholar]