

Abstract
A recent paper in this journal has challenged the idea that complex adaptive features of proteins can be explained by known molecular, genetic, and evolutionary mechanisms. It is shown here that the conclusions of this prior work are an artifact of unwarranted biological assumptions, inappropriate mathematical modeling, and faulty logic. Numerous simple pathways exist by which adaptive multi-residue functions can evolve on time scales of a million years (or much less) in populations of only moderate size. Thus, the classical evolutionary trajectory of descent with modification is adequate to explain the diversification of protein functions.
Keywords: evolutionary theory, gene duplication, microevolutionary theory, multi-residue functions, mutation, neofunctionalization, population genetics, protein evolution, random genetic drift
A major achievement of 20th century biology was the integration of the machinery of mathematical population genetics into the life sciences. The resultant theory has withstood the test of time, and modern extensions have greatly advanced our understanding of the mechanisms of evolution at the molecular, genomic, cellular, and developmental levels, while also substantially influencing many areas of applied biology, including medicine, agriculture, and conservation biology. Occasional questions are still raised about the sufficiency of microevolutionary theory to explain certain complexities at the biochemical and/or phenotypic levels (e.g., Gould 1980), but such skepticism is usually attributable to a lack of familiarity with the mathematical and/or biological foundations of population genetics (as pointed out, for example, by Charlesworth et al. 1980).
In a recent paper in this journal, Behe and Snoke (2004) questioned whether the evolution of protein functions dependent on multiple amino acid residues can be explained in terms of Darwinian processes. Although an alternative mechanism for protein evolution was not provided, the authors are leading proponents of the idea that some sort of external force, unknown to today’s scientists, is necessary to explain the complexities of the natural world (Behe 1996; Snoke 2003). The following is a formal evaluation of their assertion that point-mutation processes are incapable of promoting the evolution of complex adaptations associated with protein sequences. It will be shown that the contrarian interpretations of Behe and Snoke are entirely an artifact of incorrect biological assumptions and unjustified mathematical oversimplification.
Before proceeding, a fundamental flaw in the argument of Behe and Snoke needs to be pointed out. Although the authors claim to be evaluating whether Darwinian processes are capable of yielding new multi-residue functions, the model that they present is non- Darwinian (King and Jukes 1969). Contrary to the principles espoused by Darwin, that is, that evolution generally proceeds via functional intermediate states, Behe and Snoke consider a situation in which the intermediate steps to a new protein are neutral and involve nonfunctional products. Although non-Darwinian mechanisms play an important role in contemporary evolutionary biology, there is no logical basis to the authors’ claim that observations from a non-Darwinian model provide a test of the feasibility of Darwinian processes. Moreover, given that the authors restricted their attention to one of the most difficult pathways to an adaptive product imaginable, it comes as no surprise that their efforts did not bear much fruit.
With a priority on being compatible with the conventional framework of population genetics, the following model is the closest possible Darwinian version of the Behe and Snoke model in that the intermediate states of protein evolution involve functional products (in accordance with Darwin) with no immediate positive effects on organismal fitness (consistent with the assumptions of Behe and Snoke). Using conservative biological assumptions, it is shown that the origins of new protein functions are easily explained in terms of well-understood population- genetic mechanisms.
The model
A major goal of evolutionary theory is to develop a mechanistic understanding of the observed features of molecules, individuals, and populations in terms of universally established principles of mutation, Darwinian selection and descent with modification, Mendelian segregation and recombination (in sexual species), and random genetic drift. To simplify the presentation as much as possible, the focus here is on a nonrecombining haploid genome (as assumed by Behe and Snoke), with the origin of a new adaptive function involving a two-residue interaction, for example, the disulfide bond between two cysteines. As in Behe and Snoke (2004), this adaptation is assumed to be acquired at the expense of an essential function of the ancestral protein, so that the new function can only be permanently established via gene duplication, with one of the copies maintaining the original function. The Behe-Snoke assumption that a selective advantage only results after both participating residues are in place is also adhered to. However, two significant deviations from the model of Behe and Snoke are incorporated.
First, Behe and Snoke start with a duplicate locus that has spread throughout the base population, although they also assume that most gene copies have actually been permanently silenced by previous mutational events, the number of functional copies actually varying arbitrarily among individuals (without upper bound) as a consequence of unknown deterministic mechanisms. In contrast, the model presented here starts with a more realistic base population harboring a single locus in all individuals. A duplicate gene then arises in a single random member of the population, as must always be the case with a mutational change. With many fewer initial targets for mutation, and the vast majority of new duplicates being rapidly lost by genetic drift, this starting condition imposes a much greater challenge for the evolution of a new gene function than that assumed by Behe and Snoke.
Second, Behe and Snoke assume that all mutational changes contributing to the origin of a new multi-residue function must arise after the duplication process. They justify this assumption by stating that the majority of nonneutral point mutations to a gene yield a nonfunctional protein. To stretch this statement to imply that all amino acid changes lead to nonfunctionalization is a gross mischaracterization of one of the major conclusions from studies on protein biology—most protein-coding genes are tolerant of a broad spectrum of amino acid substitutions (Kimura 1983; Taverna and Goldstein 2002a,b). For example, in a large mutagenesis screen, Suckow et al. (1996) found that >44% of amino acid positions in the Lac repressor of Escherichia coli are tolerant of replacement substitutions. Axe et al. (1998) found that only 14% of amino acid sites in a bacterial ribonuclease are subject to inactivation by some replacement substitutions, with only one site being entirely nonsubstitutable. For human 3-methyladenine DNA glycosylase, ~66% of single amino acid substitutions retain function (Guo et al. 2004). Even for the highly conserved catalytic core regions of proteins, approximately one-third of amino acid sites can tolerate substitutions (Materon and Palzkill 2001; Guo et al. 2004). Many other studies (e.g., Kim et al. 1998; Akanuma et al. 2002), including all of those cited by Behe and Snoke, have obtained results of this nature. A deeper understanding of the fraction of amino-acid-altering mutations that have mild enough effects to permit persistence in a population comes from observations on within- and between-species variation in protein sequences (Li 1997; Keightley and Eyre-Walker 2000; Fay and Wu 2003), which generally indicate that 10% to 50% of replacement mutations are capable of being maintained within populations at moderate frequencies by selection-mutation balance and/or going to fixation. Because there is strong heterogeneity of substitution rates among amino acid sites (Yang 1996), these average constraint levels should not be generalized across all sites, many of which evolve at rates close to neutrality. Thus, most proteins in all organisms harbor tens to hundreds of amino acid sites available for evolutionary modification prior to gene duplication.
Based on these observations and in contrast to Behe and Snoke, the following model assumes that the intermediate step toward a two-residue adaptation is nondebilitating with respect to the original function but also effectively neutral, with one caveat noted below. Under this assumption, the first step in the evolution of a two-residue function potentially resides at the ancestral locus, where two alternative classes of alleles may be present prior to duplication: those containing a key amino acid at one of the potentially participating sites (type 2), and those with none (type 1) (Fig. 1 ▶). Assuming that n amino acid sites can potentially participate in the origin of the new function, the expected frequencies of these two allelic classes are obtained by normalizing the first two terms of the Poisson distribution, as loss of the ancestral protein function renders inviable all single-copy alleles with two or more key residues. The expected frequencies of type-1 and type-2 alleles are then 20/(20+n) and n/(20+n), respectively, under the assumption that all 20 amino acids are equally substitutable in the intermediate neutral state. As a consequence of random genetic drift and mutation, very small populations will generally be monomorphic for one type of allele, with the nature of that allele varying stochastically over time according to the preceding probabilities. In contrast, larger populations will approach a drift-mutation equilibrium, with the two allelic classes following these respective frequencies. In either case, averaging over a long period of time, when a random gene duplicates, it will be of type 1 with probability 20/(20+n) and type 2 with probability n/(20+n). Thus, immediately following gene duplication, there is a single copy of either allelic type 3 or type 5 in the population (Fig. 1 ▶), with initial frequency equal to the reciprocal of the population size, 1/N.
Figure 1.
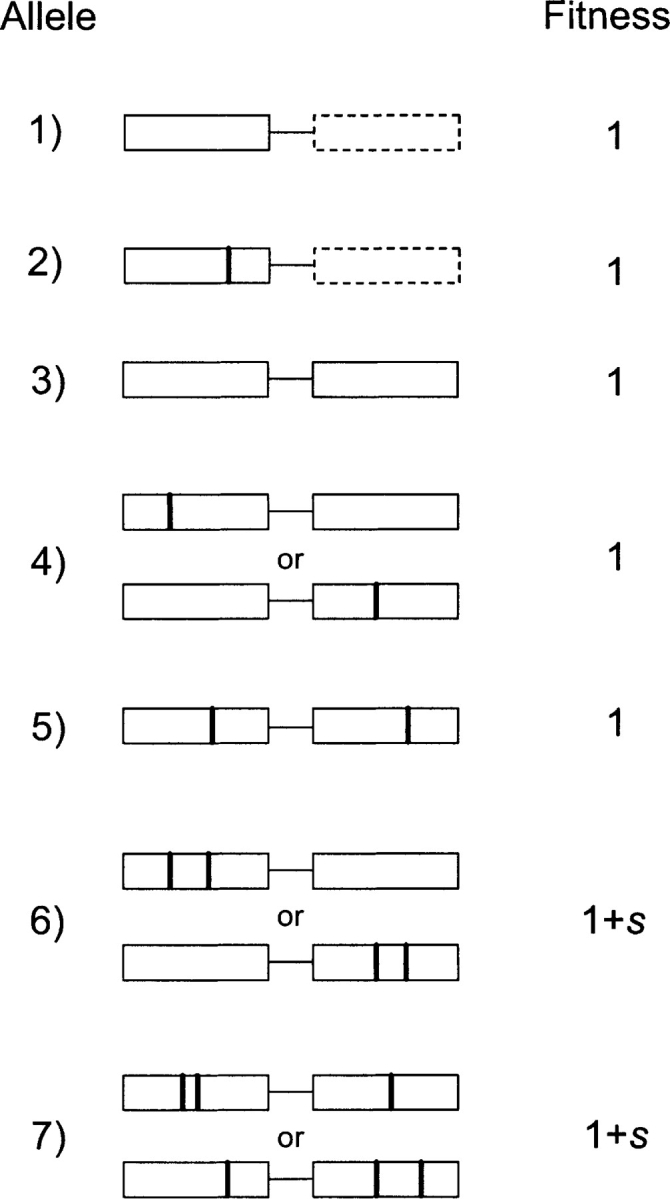
The seven possible classes of viable genotypes. The dashed boxes for allelic classes 1 and 2 denote “absentee” loci in the single-copy background. Dark vertical lines denote the presence of a key amino acid change that is essential for the production of the novel two-residue function; their location at various positions is meant to remind the reader that various pairs of amino acid sites may participate in the production of the new protein.
Successful establishment of the new function (neofunctionalization of one of the copies) requires the founding pair of linked gene duplicates to (1) initially attain a high frequency; (2) acquire the mutations essential to the expression of the new function (allelic types 6 or 7) while en route or subsequent to fixation; and (3) be preserved by positive selection subsequent to the origin of the new function. All three processes occur in parallel with a background production of null alleles. The two central issues to be resolved are then: (1) How frequently will a duplication event lead to neofunctionalization; and (2) How long will this take? Answers to these questions can be acquired by recursively following the population through the sequential steps of mutation, selection, and random sampling.
The mutation process can be summarized as follows. All gene copies mutate to defective nulls at rate μ (per gene per generation). Thus, one-copy alleles (types 1 and 2) mutate to an inviable state at rate μ, as do alleles of types 6 and 7 when the copy with the ancestral function is removed. In addition, two-copy alleles can be converted to functional one-copy alleles in several ways: 4 → 1, 4 → 2, 6 → 1, and 7 → 2 at rate μ; and 3 → 1 and 5 → 2 at rate 2μ. Letting v0 denote the rate of nonsynonymous mutation per codon (converting one amino acid to another), then the loss of a key amino acid residue causes the following allelic conversions: 2 → 1, 4 → 3, and 7 → 6 at rate v0; and 5 → 4, 6 → 4, and 7 → 5 at rate 2v0. Letting v1=v0n/19 be the rate of mutation to a gene carrying one key residue from a gene carrying none, and v2=v0(n−1)/19 be the rate of mutation to a gene carrying two key residues from a gene carrying one, the conversion to alleles with new key residues can be described as: 1 → 2, 4 → 5, and 6 → 7 at rate v1; 3 → 4 at rate 2v1; and 4 → 6 and 5 → 7 at rate 2v2. Finally, allelic types 2 and 7 are converted to nulls at rate v2 when the appearance of the new function in a one-residue allele results in the complete loss of ancestral function.
Each generation, after the production of mutant alleles by the preceding scheme, selection increases the frequencies of the neofunctionalized alleles (types 6 and 7) by a proportional amount s (the selection coefficient). Random sampling then yields the next generation of N individuals. This stochastic phase of random genetic drift ensures that various classes of alleles either become lost from the population or rise to fixation, eventually confining the system to one of two alternative stable states. If the population reaches a state in which all alleles are of type 1 and/or 2, then neofunctionalization is no longer possible without a new round of gene duplication. In contrast, once alleles of type 6 or 7 have attained a critical frequency, permanent neofunctionalization is essentially ensured. The concern here is with the frequency of duplication events that lead to the latter state, Pneo, and to this end the threshold allele frequency for neofunctionalization is defined by
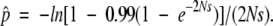 |
(1) |
This expression is obtained by rearranging the haploid version of Kimura’s (1962) diffusion equation for the fixation probability of a beneficial allele with initial frequency p. Here, p^ denotes the sum of frequencies of alleles of types 6 and 7 necessary for the probability of fixation of the new function to exceed 0.99.
The behavior of this model was evaluated by computer simulation using a range of parameter values justified by the following observations. Surveys of a wide variety of unicellular and multicellular species suggest that the genetic effective population sizes of species (N) range from a low of ~103 for some vertebrates and vascular plants to a high of ~109 for some prokaryotic species (Lynch and Conery 2003a). These numbers are, of course, substantially below the absolute numbers of individuals within species, but several factors including linkage, variance in family size, and population-size fluctuations conspire to reduce the genetic effective population size of a species below the actual number of adults (Caballero 1994; Gillespie 2001). The null mutation rate (μ) for protein-coding genes is generally on the order of 10−6 per generation (Drake et al. 1998). The per nucleotide mutation rate is generally in the range of 10−9 to 10−8 per generation or higher (Li 1997; Denver et al. 2004), and the latter is used as the total nonsynonymous mutation rate per codon, v0. It is uncertain how many potential amino acid sites can be used in the production of a new two-residue function, and a range of n=2–50 is considered, the lowest value matching the assumption of Behe and Snoke. The selection coefficient (s) will equal either 0.01 or 0.0001, covering the range of moderate to fairly weak selection intensities. Values of n and/ or s above these ranges, which are not out of the question, will result in higher rates of neofunctionalizaton than reported below.
It is useful to consider the probability of neofunctionalization per duplication event, Pneo, relative to the probability of fixation of a neutral mutation (1/N) by using the scaled parameter θ =NPneo. A value of θ =1 implies that neofunctionalization of a newly arisen duplicate gene occurs with the same probability as the fixation of a neutral mutation. Moreover, letting δ denote the rate of duplication per gene copy, then because δN is the rate of gene duplication at the population level, δθ is the rate of neofunctionalization at the population level, that is, the rate of establishment of the new selectable function.
The probabilities of neofunctionalization under this model increase with population size (N), the number of potential contributory sites to the new function (n), and the selective advantage of the new function (s) (Fig. 2 ▶). Increasing population size enhances both the efficiency of selection and the number of mutational targets for the production of neofunctionalized alleles, and depending on the nature of the determinants of the adaptation (s and n), θ increases linearly with population size above N ≃ 104 to 106. The results show that moderate levels of N, n, and s are sufficient to yield probabilities of neofunctionalization in excess of the neutral rate of fixation, that is, θ>1.
Figure 2.

The probability of neofunctionalization of a newly arisen duplicate gene scaled to the neutral expectation (θ), as a function of the effective population size (N), number of potentially participating amino acid sites in the new two-residue function (n), and the selective advantage of the new function (s). For the solid lines, s=0.01; for the dashed lines, s=0.0001. In all cases, the mutation rate to null alleles (μ) is 10−6, and the nonsynonymous mutation rate per codon (v0) is 10−8. Each data point is derived from 107 to 1010 stochastic simulations. The solid and dashed lines are the analytical approximations described in the text. The dotted line denotes the point at which the probability of neofunctionalization is equal to the fixation probability of a neutral mutation.
An analytical approximation
Insight into the mechanisms driving these patterns can be achieved with a population-genetic approximation. Although neofunctionalization can be precipitated by initial duplications of either type-1 or type-2 alleles, type-2 alleles are much more likely to be neofunctionalized because they have acquired one of the two key contributory residues prior to duplication. Thus, as a first approximation, the focus is only on the subset of initial duplications to type-5 alleles, which usually comprise a fraction n/(20+n) of initial duplication events, an exception noted below.
The fact that θ can greatly exceed 1 at large population sizes (Fig. 2 ▶) is revealing. If a type-5 allele were simply neutral with respect to the pre-existing single-copy alleles, then fixation would occur with probability 1/N, and θ would have a maximum value of n/(20+n)<1. Although duplicated genes are, indeed, assumed to be selectively neutral in the preceding simulations, two-copy alleles still have a slight intrinsic advantage over one-copy alleles in that the latter mutate to nulls at rate μ, whereas two-copy alleles must acquire two such mutations (one in each copy) to be inactivated. This weak mutational advantage acts like selection, yielding the probability of fixation of a newly arisen pair of linked duplicates:
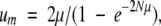 |
(2) |
which approaches a maximum of 2μ at large N (Lynch 2002). This expression also applies to founder events involving type-3 alleles.
Should fixation occur, then the ultimate fate of a two-copy allele will be determined by subsequent mutations. The next mutation to arise will be one of three types: (1) a reversion of a type-5 to a type-2 allele arises at rate 2μ, has a mutational disadvantage, and fixes with probability ud, defined by Equation 2 with −μ substituted for μ; (2) a conversion to a type-4 allele arises at rate 2v0, and fixes with the neutral probability 1/N; and (3) a conversion to a type-7 allele arises at rate 2v2, is beneficial, and fixes at rate:
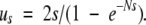 |
(3) |
Thus, assuming no selective interference between competing fixation events, the next mutation to fix results in neofunctionalization with probability:
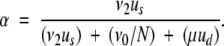 |
(4) |
Although there are additional paths to neofunctionalization (e.g., having mutated to a type-4 allele, a second mutation can resurrect a type-5 allele, which can then acquire a mutation to a type-7 allele), these indirect paths are of relatively low probability and can be ignored as a first approximation.
The preceding logic suggests that the scaled probability of neofunctionalization (θ) should be approximately equal to Numαn/(20+n), but if this were the only factor involved in the establishment of a two-copy allele, then θ would approach a maximum value of ~2Nμn/(20+n) at large N, as um → 2μ and α → 1. In contrast, θ attains values much greater than this prediction (Fig. 2 ▶). For example, with n=50, θ would be expected to approach (1.43 × 10−6)N at large N independent of s, whereas the values observed with s=0.01 are ~22.4 times higher and those observed with s=0.0001 are ~1.8 times higher.
The discrepancy is due to the chance occurrence of neofunctionalizing mutations during the initial phase of establishment of the duplicate. Without such mutations, a newly arisen type-5 allele would be destined to be lost by random genetic drift with probability 1−um. However, prior to loss, an approximately neutral allele destined to loss in a haploid population yields a cumulative average number of N descendant copies, each of which is subject to mutation. Should a two-copy allele en route to loss acquire a neofunctionalizing mutation prior to being silenced by a degenerative mutation, it will then have a boost in the probability of fixation defined by Equation 3. A simple expression for this rescue effect is not available, but a recursive approach developed in Lynch et al. (2001) is adapted for the purposes of this paper in the Appendix. Letting r denote the probability that a type-5 allele initially destined to loss is rescued by a neofunctionalizing mutation, then
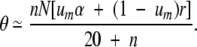 |
(5) |
The fit of this expression to the simulation data is quite good, except at very low n when selection is weak (s=0.0001) and the population size is large (Nμ>1) (Fig. 2 ▶). At large N and small s, violations of the assumption that no more than two alleles are simultaneously segregating may cause the breakdown in the mathematical approximations, which ignore the reduction in fixation probability resulting from selective interference.
One technical modification needs to be made to the above theory at very large N. Because they are one mutation removed from acquiring a two-residue function that eliminates the essential ancestral function, type- 2 alleles have a very weak mutational disadvantage. When the product of population size and the excess mutation rate to nulls is on the order of 1 or larger, this effect reduces the pre-duplication frequency of type-2 alleles to that expected under selection-mutation balance,  . With the parameters used in analyses herein, this condition was only approached in a few extreme situations, and in any event the deviation between these two results is not great unless n is large. For example, with n=2, both the preceding formula and n/(20+n) yield an expected frequency of type-2 alleles of 0.091, and with n=10, the respective frequencies are 0.333 and 0.282. Comparison of the simulated results including this modification with the analytical approximation just noted shows that this added complication at large N is of minor effect.
. With the parameters used in analyses herein, this condition was only approached in a few extreme situations, and in any event the deviation between these two results is not great unless n is large. For example, with n=2, both the preceding formula and n/(20+n) yield an expected frequency of type-2 alleles of 0.091, and with n=10, the respective frequencies are 0.333 and 0.282. Comparison of the simulated results including this modification with the analytical approximation just noted shows that this added complication at large N is of minor effect.
Time to neofunctionalization
Two temporal components contribute to the time to neofunctionalization: (1) the expected time of arrival of the first neofunctionalizing mutation that will go on to fixation (i.e., that satisfies Equation 1); and (2) the time span of the fixation process itself. The expected arrival time is equal to the reciprocal of the rate of origin of fixable neofunctionalizing mutations, 1/(δθ). Using Equation 5 to define θ, and noting that δ is generally at least 10−8 per gene copy per generation (Lynch and Conery 2000, 2003b), the average arrival times associated with the results in Figure 2 ▶ are provided in Figure 3 ▶. These times scale approximately inversely with population size, decrease with increasing n and s, and for a wide range of conditions are smaller than 107 generations. The mean fixation times, obtained directly from the simulations, were uniformly between 3 × 104 and 106 generations (data not shown). Thus, the arrival time is generally the limiting factor for populations of small to moderate size, while the fixation time can be the limiting factor in very large populations.
Figure 3.

Average arrival times for a neofunctionalized allele destined for fixation. The curves are based on the theory described in the text, and assume a rate of gene duplication of 10−8 per gene copy per year.
Discussion
To support their contention of the implausibility of adaptive protein evolution by Darwinian processes, Behe and Snoke started with an ad hoc non-Darwinian model with a highly restrictive and biologically unrealistic set of assumptions. Such extreme starting conditions guaranteed that the probability of neofunctionalization would be reduced to a minimal level. An alternative approach, adopted here, is to rely on a set of biologically justified premises and an explicit population-genetic framework. When this is done, contrary to the assertions of Behe and Snoke that neofunctionalization events involving multiple amino acid residues require 108 or more generations and population sizes in excess of 109 individuals, it is readily demonstrated that this process can go to completion with high probability on time scales of 106 yr or less in populations >106 in size. As is discussed below, this is a highly conservative conclusion with respect to both the time and population-size requirements. To put this into perspective, a span of 106 yr is small on the total evolutionary time scale (in years) of ~3.8 × 109 for all of life, ~2 × 109 for eukaryotes, ~7 × 108 for metazoans, ~4 × 108 for tetrapods and land plants, and ~2 × 108 for mammals (e.g., Knoll 2003). In addition, a population size of 106 is minuscule for most microbes (the species whose genome structure is most compatible with the Behe-Snoke model) (Finlay 2002).
It is difficult to pinpoint the source of the difference between the results of Behe and Snoke and those contained herein, as the latter authors do not explicitly model the evolutionary process, whereas the stochastic computer simulations presented here precisely track the joint dynamics of allele frequencies. In order to maintain a permanent reservoir of potentially neofunctionalizable duplicate genes, Behe and Snoke assume that a new gene copy is produced instantaneously every time an allele is thought to have been silenced. However, the further assumption that duplicate genes are entirely neutral until the final step in neofunctionalization has been achieved leads to a steady-state situation in which almost all alleles residing at the duplicate locus are “irrecoverably lost.” In contrast, the analyses presented here assume that a duplicate gene arises in a single individual in a population in which all other individuals carry a single copy of the gene. This difference in approach, as well as others, is actually expected to reduce the estimated time to neofunctionalization in the Behe-Snoke model relative to that presented here. For example, although Behe and Snoke focus on the time to the first appearance of a neofunctionalizing mutation, the focus here is on the necessarily longer time to achieve the first fixable mutation. In addition, although Behe and Snoke assume that the forward and backward point-mutation rates (per amino acid residue) are equal, it is assumed here that the former is just 1/19 of the latter, which necessarily reduces the likelihood of neofunctionalization.
Uncertainties on these issues aside, there are at least three reasons for the discrepancies between our results. First, for the most part, Behe and Snoke assume that the evolution of a multi-residue function requires the origin of a full set of mutations previously kept absent from the population as a consequence of their lethal pleiotropic effects on ancestral gene functions. However, if the intermediate steps toward the evolution of a selectable multi-residue function are entirely neutral after gene duplication, as Behe and Snoke assume, then there is no compelling reason that “one-off” (type-2) alleles should be absent from the population prior to duplication. In the context of a disulfide bond, for example, the Behe-Snoke assumption implies a situation in which no cysteine residue in an ancestral protein would be capable of participating in a cysteine–cysteine interaction in a subsequently modified copy, which seems highly implausible (e.g., Matsumura et al. 1989). An additional logical problem implicit in the Behe-Snoke model is how an organism producing 50% functional and 50% nonfunctional protein would avoid a reduction in fitness.
Second, Behe and Snoke assume that only two specific amino acid sites within a protein are capable of giving rise to a new selectable diresidue function. Given that the average protein in most organisms contains between ~300 and 600 amino acids, this assumption is also unrealistic. Increasing the number of participating amino acid sites from n=2 to just 10 can magnify the probability of neofunctionalization by more than 10- fold, but the results presented here also demonstrate that even with the restrictive Behe and Snoke assumption of n=2, the probability of neofunctionalization can exceed the neutral fixation probability for populations with effective sizes in excess of 106.
Third, although both of our analyses assume that the intermediate changes en route to a multi-residue function are selectively neutral with respect to protein function, Behe and Snoke failed to realize that a completely linked pair of duplicate genes has a mutational advantage equal to the mutation rate to null alleles (μ), owing to the fact that both members of a linked pair must be inactivated before the viability of the carrier is affected. This intrinsic mutational advantage, which is independent of the selectable multi-residue function, is of minor consequence when the power of random genetic drift exceeds that of mutation (N<1/μ), but when N is moderately large (2Nμ>1), the fixation probability approaches 2μ. Once fixed and maintained by positive selection, active two-copy alleles will not be the victims of the neutral accumulation of silencing mutations assumed by Behe and Snoke. This then essentially ensures that the new selectable function will evolve, provided the selective advantage of the new function exceeds the mutational advantage of maintaining two redundant ancestral copies. This argument applies to all potential founder duplicates, including those that are initially lacking in any contributory amino acid residues for the new function. Thus, although the preceding analytical approximation ignored events initiating with a type-3 allele, the actual computer simulations show that these events also lead to a high probability of neofunctionalization when N is large.
There are several reasons that the probabilities of neofunctionalization computed in this paper are likely to be underestimates. First, with respect to population structure, although the range of effective population sizes used in the simulations encompass the estimates available for a wide variety of species from measures of nucleotide variation at silent sites, the highest of these estimates may be biased downwardly (Lynch and Conery 2003a). That is, the upper limit to N is probably higher than the 109 used in this study. More importantly, molecular-based estimates of the long-term effective population size are equivalent to the harmonic means of the generation-specific measures, and hence much closer to population-size minima than maxima. Virtually all populations experience temporal changes in abundance, and there can be long phases in which the effective population size greatly exceeds that implied by long-term estimates. Given the near linear scaling of θ and N, these two complications alone could easily increase θ (and decrease the average arrival time) by more than an order of magnitude.
Second, although the simulations presented here rely on the assumption that the absolute and effective number of individuals in a population are equivalent, variance in family size resulting from spatial variation, selective sweeps associated with linkage, and other factors will generally result in the former being substantially greater than the latter. There is a simple reason why increased absolute population size can greatly magnify the likelihood of neofunctionalization. In the preceding analyses, it was assumed that the appearance of double mutations (e.g., the simultaneous origin of both cysteine residues contributing to a disulfide bond) is negligible, as this probability is just [n(n−1)/2](μ/19)2 per individual. Under the assumption that μ=10−8, the rate of origin of double mutants per gene copy is ~3 × 10−19 when n=2 and ~3 × 10−16 when n=50. For microbial species, the apparent focus of Behe and Snoke, whose total population densities can easily exceed 1016 (Finlay 2002), new double mutants can be expected to arise very frequently at the population level (reducing Behe and Snoke’s time to first appearance to just one generation or so).
Third, the key to neofunctionalization by gene duplication resides in the ability of the initial duplicate to rise to fixation and then remain in an active state. Although the model presented herein allows for the fixation of a pair of linked duplicates in large populations via its weak mutational advantage, there are more powerful mechanisms for the preservation of duplicate genes. In populations of moderate to large size, for example, neofunctionalizing mutations aside from those associated with a multi-residue function will be sufficient to preserve the duplicate copy in an active state. In populations of small to moderate size, neofunctionalizing mutations are less likely to arise before one or more degenerative mutations have gone to fixation, but the latter can also promote duplicate- gene preservation by subfunctionalization when each member of the pair partially or entirely loses a complementary ancestral subfunction (Force et al. 1999; Lynch et al. 2001; Prince and Pickett 2002). In either case, once a duplicate pair is stabilized in the population by selective forces, the mutations required for the origin of a multi-residue function are no longer competing for fixation with background silencing mutations. This substantially increases the likelihood of origin of a new multi-residue function by providing additional time and mutational targets for the process.
Fourth, although it has been assumed in both models that the intermediate steps toward neofunctionalization are selectively neutral, for multi-residue functions such as ligand-binding sites, it is likely that intermediate alleles will have some function. For example, substitutions of single amino acids may alter the three-dimensional structure of a protein in ways that facilitate the joint participation of other amino acids in a novel function. Selection on intermediate states of a magnitude si will cause a still further increase in θ by magnifying the initial fixation probability. Recalling Equation 2, the selective advantage would now be (μ+si) rather than μ. With μ=10−6, an si of just 10−5 to 10−4 would increase θ (and correspondingly decrease the mean arrival time) 10- to 100-fold at large N.
Fifth, with respect to the breeding system, it has been assumed that the population consists of haploid individuals with no recombination within or between genes. However, diploidy and recombination can greatly facilitate the probability of neofunctionalization by gene duplication. Suppose, for example, that prior to gene duplication an allele with the new function is lethal in the homozygous state, in accordance with the assumptions relied on above, but beneficial in heterozygotes containing both the ancestral and derived functions. The derived allele will then be maintained in the population at the single locus by balancing selection, poising the system for fixation of the neofunctionalized allele following gene duplication, with no requirement of a new mutation at all, provided there is recombination between the two loci (Spofford 1969; Lynch et al. 2001).
In summary, the conclusions derived from the current study are based on a model that is quite restrictive with respect to the requirements for the establishment of new protein functions, and this very likely has led to order-of-magnitude underestimates of the rate of origin of new gene functions following duplication. Yet, the probabilities of neofunctionalization reported here are already much greater than those suggested by Behe and Snoke. Thus, it is clear that conventional population-genetic principles embedded within a Darwinian framework of descent with modification are fully adequate to explain the origin of complex protein functions.
Acknowledgments
I am very grateful to B. Charlesworth, J. Dudycha, D. Fitch, A. Force, D. Scofield, and two anonymous reviewers for helpful comments. The work was supported in part by NSF grants EF- 0328516 and MCB-0342431 and NIH grant R01 GM36827.
Appendix
To estimate the probability that a type-5 allele en route to loss by random genetic drift will be rescued by acquiring a neofunctionalizing mutation prior to loss, which then propels it to fixation, it is necessary to know the probability that the original allele, initially present at frequency 1/N, is lost by time t, which is denoted here as uL(t). Starting with uL(0)=0 at the time of the original duplication event,
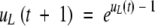 |
(Fisher 1922). From Lynch et al. (2001), conditional on the allele not yet having been lost, the expected number of copies in the population is
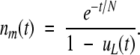 |
Accounting for the probability that the allele may have acquired a degenerative mutation in one copy and hence not be available for neofunctionalization, and conditional on the fate of the allele having not already been determined, the probability that a successful rescue occurs in generation t is
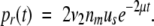 |
The total probability of rescue in generation t is then
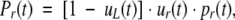 |
where ur(t) is the probability that the allele has not been rescued by time t:
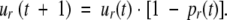 |
The total probability of a rescue (r) is equal to the sum of Pr(t) from t=0 to ∞.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041171805.
References
- Akanuma, S., Kigawa, T., and Yokoyama, S. 2002. Combinatorial mutagenesis to restrict amino acid usage in an enzyme to a reduced set. Proc. Natl. Acad. Sci. 99 13549–13553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axe, D.D., Foster, N.W., and Fersht, A.R. 1998. A search for single substitutions that eliminate enzymatic function in a bacterial ribonuclease. Biochemistry 37 7157–7166. [DOI] [PubMed] [Google Scholar]
- Behe, M.J. 1996. Darwin’s black box—The biochemical challenge to evolution. Free Press, New York.
- Behe, M.J. and Snoke, D.W. 2004. Simulating evolution by gene duplication of protein features that require multiple amino acid residues. Protein Sci. 13 2651–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero, A. 1994. Developments in the prediction of effective population size. Heredity 73 657–679. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B., Lande, R., and Slatkin, M. 1980. A neo-Darwinian commentary on macroevolution. Evolution 36 474–498. [DOI] [PubMed] [Google Scholar]
- Denver, D.R., Morris, K., Lynch, M., and Thomas, W.K. 2004. High mutation rate and predominance of insertions in the Caenorhabditis elegans nuclear genome. Nature 430 679–682. [DOI] [PubMed] [Google Scholar]
- Drake, J.W., Charlesworth, B., Charlesworth, D., and Crow, J.F. 1998. Rates of spontaneous mutation. Genetics 148 1667–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay, J.C. and Wu, C.-I. 2003. Sequence divergence, functional constraint, and selection in protein evolution. Annu. Rev. Genomics Hum. Genet. 4 213–235. [DOI] [PubMed] [Google Scholar]
- Finlay, B.J. 2002. Global dispersal of free-living microbial eukaryote species. Science 296 1061–1063. [DOI] [PubMed] [Google Scholar]
- Fisher, R.A. 1922. On the dominance ratio. Proc. Roy. Soc. Edinb. 52 399–433. [Google Scholar]
- Force, A., Lynch, M., Pickett, F.B., Amores, A., Yan, Y.-L., and Postlethwait, J. 1999. Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151 1531–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie, J.H. 2001. Is the population size of a species relevant to its evolution? Evolution 55 2161–2169. [DOI] [PubMed] [Google Scholar]
- Gould, S.J. 1980. Is a new and general theory of evolution emerging? Paleobiology 6 119–130. [Google Scholar]
- Guo, H.H., Choe, J., and Loeb, L.A. 2004. Protein tolerance to random amino acid change. Proc. Natl. Acad. Sci. 101 9205–9210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P.D. and Eyre-Walker, A. 2000. Deleterious mutations and the evolution of sex. Science 290 331–333. [DOI] [PubMed] [Google Scholar]
- Kim, D.E., Gu, H., and Baker, D. 1998. The sequences of small proteins are not extensively optimized for rapid folding by natural selection. Proc. Natl. Acad. Sci. 95 4982–4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M. 1962. The probability of fixation of mutant genes in a population. Genetics 47 713–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1983. The neutral theory of molecular evolution. Cambridge University Press, Cambridge, UK.
- King, J.L. and Jukes, T.H. 1969. Non-Darwinian evolution. Science 164 788–798. [DOI] [PubMed] [Google Scholar]
- Knoll, A.H. 2003. Life on a young planet: The first three billion years of evolution on earth. Princeton University Press, Princeton, NJ.
- Li, W.-H. 1997. Molecular evolution. Sinauer, Sunderland, MA.
- Lynch, M. 2002. Intron evolution as a population-genetic process. Proc. Natl. Acad. Sci. 99 6118–6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M. and Conery, J.S. 2000. The evolutionary fate and consequences of duplicate genes. Science 290 1151–1155. [DOI] [PubMed] [Google Scholar]
- ———. 2003a. The origins of genome complexity. Science 302 1401–1404. [DOI] [PubMed] [Google Scholar]
- ———. 2003b. The evolutionary demography of duplicate genes. J. Struct. Funct. Genomics 3 35–44. [PubMed] [Google Scholar]
- Lynch, M., O’Hely, M., Walsh, B., and Force, A. 2001. The probability of preservation of a newly arisen gene duplicate. Genetics 159 1789–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Materon, I.C. and Palzkill, T. 2001. Identification of residues critical for metallo-β-lactamase function by codon randomization and selection. Protein Sci. 10 2556–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura, M., Becktel, W.J., Levitt, M., and Matthews, B.W. 1989. Stabilization of phage T4 lysozyme by engineered disulfide bonds. Proc. Natl. Acad. Sci. 86 6562–6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince, V.E. and Pickett, F.B. 2002. Splitting pairs: The diverging fates of duplicated genes. Nat. Rev. Genet. 3 827–837. [DOI] [PubMed] [Google Scholar]
- Snoke, D. 2003. Natural philosophy: A survey of physics and Western thought. Access Research Network, Colorado Springs, CO.
- Spofford, J.B. 1969. Heterosis and the evolution of duplications. Amer. Natur. 103 407–432. [Google Scholar]
- Suckow, J., Markiewicz, P., Kleina, L.G., Miller, J., Kisters-Woike, B., and Muller-Hill, B. 1996. Genetic studies of the Lac repressor. XV: 4000 single amino acid substitutions and analysis of the resulting phenotypes on the basis of the protein structure. J. Mol. Biol. 261 509–523. [DOI] [PubMed] [Google Scholar]
- Taverna, D.M. and Goldstein, R.A. 2002a. Why are proteins so robust to site mutations? J. Mol. Biol. 315 479–484. [DOI] [PubMed] [Google Scholar]
- ———. 2002b. Why are proteins marginally stable? Proteins 46 105–109. [DOI] [PubMed] [Google Scholar]
- Yang, Z. 1996. Among-site rate variation and its impact on phylogenetic analyses. Trends Ecol. Evol. 11 367–372. [DOI] [PubMed] [Google Scholar]