

Abstract
Combinatorial shotgun scanning mutagenesis was used to analyze two large, related protein binding sites to assess the specificity and importance of individual side chain contributions to binding affinity. The strategy allowed for cost-effective generation of a plethora of functional data. The ease of the technology promoted comprehensive investigations, in which the classic alanine-scanning approach was expanded with two additional strategies, serine- and homolog-scanning. Binding of human growth hormone (hGH) to the hGH receptor served as the model system. The entire high affinity receptor-binding sites (site 1) of wild-type hGH (hGHwt) and of an affinity-improved variant (hGHv) were investigated and the results were compared. The contributions that 35 residue positions make to binding were assessed on each hormone molecule by both serine- and homolog-scanning. The hormone molecules were displayed on the surfaces of bacteriophage, and the 35 positions were randomized simultaneously to allow equal starting frequencies of the wild-type residue and either serine or a homologous mutation in separate libraries. Functional selections for binding to the hGH receptor shifted the relative wild-type/mutant frequencies at each position to an extent characteristic of the functional importance of the side chain. Functional epitope maps were created and compared to previous maps obtained by alanine-scanning. Comparisons between the different scans provide insights into the affinity maturation process that produced hGHv. The serine and homolog-scanning results expand upon and complement the alanine-scanning results and provide additional data on the robustness of the high affinity receptor-binding site of hGH.
Keywords: shotgun phage display scanning, functional epitope, human growth hormone
Protein–protein interactions are at the heart of most biological processes, including metabolism, immune responses, signal transduction, and gene regulation (Pawson and Nash 2000; Yoshida et al. 2001; Warren 2002; Gascoigne and Zal 2004). Accordingly, an atomic-resolution exploration of the physiochemical principles that govern these interactions would have immense impact on our understanding of biology. In fact, one of the greatest challenges in structural biology is to develop approaches to quantify the driving forces that govern the formation and regulation of protein– protein interactions. The recent expansion of structural databases has been providing an increasing number of high-resolution structures of functionally relevant molecular complexes. This expanding data set represents an essential framework for revealing the fundamental structural organization of hydrogen-bonding, van der Waals, and electrostatic forces that are believed to underlie protein–protein interactions.
However, while we can attain a clearer picture by studying time-averaged representations of contact surfaces, or “structural epitopes,” these results cannot, by themselves, pinpoint contact residues that make significant energetic contributions to binding and define the “functional epitopes.” The crucial data that assess the magnitude and specificity of individual amino acid residue contributions to a protein–protein interaction are best obtained through carefully designed, structure-guided mutagenesis approaches.
The conventional method for mapping the energetic topography of functional epitopes is alanine-scanning mutagenesis. This site-directed approach involves the systematic removal of side chain atoms past the β-carbon of each targeted residue while limiting unwanted conformational and physicochemical disruptions (Cunningham and Wells 1989, 1993). In a sense, properties of alanine to effectively eliminate a side chain contact while restricting indirect effects gave it a “gold-standard” status among natural amino acid residues. However, we believe that other types of systematic scans can augment traditional alanine-scanning data. In this regard we have chosen two complementary scanning strategies, serine-scanning and homolog-scanning, which aim to further increase the resolution of energetic profiling of functional epitopes.
Serine-scanning is based on the logic that, in some cases, substitutions with the small, hydrophilic serine side chain may be less disruptive than with the more hydrophobic alanine side chain. Serine is a highly represented amino acid type in proteins, and alanine/serine substitute for each other with high frequency in homologous protein sequences as judged either by the PAM (percent accepted mutations) or BLOSUM (blocks substitution matrix) scoring schemes (Dayhoff et al. 1978; Henikoff and Henikoff 1992). In any case, systematic side-by-side comparisons between alanine and serine scans for statistically significant numbers of proteins should provide insight into the potential context dependence of interchangeability of these two amino acids and could identify types of environments where these scans provide parallel or different information.
The second type of scanning strategy is homolog-scanning. The logic behind homolog-scanning is to cause the least possible structural disruption upon side chain substitution and locate which residue contributions are the most specific for maintaining a function. The first homolog-scanning strategy applied mutations that were based on an alignment of related amino acid sequences from different species (Cunningham et al. 1989). The same term is used, however, for the more general approach, when amino acid residues are replaced with stereochemically similar residues, regardless of whether such replacements have actually been observed in nature (Vajdos et al. 2002; Sato et al. 2004). One issue regarding homolog-scanning that, to our knowledge, has not been systematically explored is what physicochemical characteristics best link two amino acid types. For instance, in general, which amino acid is the best homolog for asparagine: a glutamine that varies the length but not the charge, or aspartate, which varies the charge but not the length?
While it is apparent that serine- and homolog-scanning can provide additional substance for further characterizing functional epitopes, they are also arduous and have significant labor cost associated with them if conventional mutagenesis methods are used. To circumvent these practical barriers we employed a combinatorial approach: phage-displayed shotgun scanning mutagenesis that uses simple binding selection procedures and allows for the quantitative evaluation of the individual effects of many simultaneously introduced site-directed mutations (Weiss et al. 2000). The biophysical binding measurements are substituted for by a statistical analysis that uses wild-type/ mutant ratios determined at each of the targeted positions of DNA sequences from large numbers of binding-selected clones. These ratios have been shown to accurately recapitulate ΔΔGmut-wt values determined from traditional mutational scanning methods (Weiss et al. 2000).
To assess the information content extractable from serine- and homolog-scanning data, we chose the high affinity binding site (site 1) of human growth hormone (hGH) as a model system. Site 1 of hGH consists of 35 residues and binds to the extracellular domain of its receptor (hGHR) with nanomolar affinity (Cunningham and Wells 1989; de Vos et al. 1992). A series of previous studies on this site 1 interaction have been exceptionally fruitful in providing important insights into such general features as how binding energy is distributed among the component amino acid residues located at large protein– protein interfaces (Cunningham and Wells 1989, 1993; Clackson and Wells 1995). Determination of the energetic contribution of individual hGH residues in the site 1 interface by Wells and colleagues was the genesis of traditional alanine-scanning methodology, an approach that is now almost indispensable for structure-function analyses in protein science (Cunningham and Wells 1989).
The site 1 interaction of hGH with the hGH receptor (hGHR) was also among the first subjects for the use of phage display mutagenesis to affinity-mature a protein– protein interaction. It yielded a hormone variant (hGHv) that incorporates 15 mutations and binds two orders of magnitude tighter to the hGHR than does hGHwt (Lowman and Wells 1993). Furthermore, we previously described the use of phage-displayed protein libraries for the rapid mapping of functional epitopes using combinatorial shotgun alanine-scanning mutagenesis (Weiss et al. 2000; Vajdos et al. 2002; Pal et al. 2003). Finally, in a recent study we used this technology to perform a comprehensive shotgun alanine scan of the entire site 1 of hGHv and compared the functional epitope to that of hGHwt (Pal et al. 2003). We now present the results of serine and homolog scans that cover the entire site 1 receptor-binding interface comprising 35 amino acid residues on both hGHwt and hGHv. We demonstrate that, when combined with previous alanine-scanning results, these scans provide a more extensive and comprehensive functional description of the site 1 interface than alanine-scanning alone.
Results and Discussion
Display bias
The mutagenesis strategies for serine- and homolog-scanning are described in Table 1. As described in the Materials and Methods and indicated in Table 2, two separate binding selections were applied: binding to the receptor through the randomized site 1 region, and binding to a monoclonal antibody through a distinct region on hGH. Wild-type/mutant ratios determined by the statistical analysis of antibody-selected clones estimate display bias, the extent to which mutations affect display efficiency, which in turn is considered to correlate with folding efficiency and protein stability (Jin et al. 1992; Weiss et al. 2000). These data are generated to normalize data obtained from the receptor-binding selection. Wild-type/ mutant ratios in columns 6–9 of Table 2 that are larger or smaller than 1.0 suggest display biases corresponding to destabilizing or stabilizing effects, respectively. Large (over fourfold) deviations from unity are highlighted; of the 134 ratios, only 19 fell into this group, six for hGHwt and 13 for hGHv. For hGHwt there are two mutations (T60S and T67S) that indicate significant stabilizing effects, while for hGHv there are five such mutations (W14L, N21S, N167S, T175S, and R183S). Interestingly, all the display-enhancing or stabilizing effects correspond to mutations that conserve the general physicochemical character of the residue. Polar neutral residues (except R183) are mutated to the also polar neutral serine, while the nonpolar tryptophan is mutated to the nonpolar leucine. In contrast, most display-reducing or destabilizing effects correspond to mutations that change the charge and/or hydrophobic character of the mutated residue (such as M14S, H18D, Y164S, or E174S for hGHwt and Q29E, Y164S, N167D, K168S, F176S, and R178S for hGHv). Overall, the relatively small number of large deviations in both the hGHwt case and the affinity-improved hGHv case suggests that site 1 is robust in the sense that it tolerates extensive sequence perturbations without compromising the stability of the protein.
Table 1.
Shotgun-scanning codons
| Serine scanb | Homolog scanc | ||||
| Wild typea | Codond | m2 | m3 | Codond | Homo |
| D | KMC | A | Y | GAM | E |
| E | KMG | A | * | GAM | D |
| F | TYC | TWC | Y | ||
| H | MRC | N | R | MAC | N |
| I | AKC | RTT | V | ||
| K | ARM | N | R | ARG | R |
| L | TYG | MTC | I | ||
| M | AKS | I | R | MTG | L |
| N | ARC | RAC | D | ||
| P | YCT | SCA | A | ||
| Q | YMG | P | * | SAA | E |
| R | MGT | ARG | K | ||
| S | KCC | A | |||
| T | WCG | ASC | S | ||
| W | TSG | TKG | L | ||
| Y | TMC | TWC | F | ||
aAmino acids are represented by the single-letter amino acid code.
bThe serine-scanning codon for each amino acid ideally encodes only for the wild type or serine, but the degeneracy of the genetic code necessitates the occurrence of two other amino acids (m2 and m3) for some substitutions. Asterisks (*) indicate a stop codon.
c For homolog scanning, binary shotgun codons were designed to encode the wild type and a similar amino acid (Homo).
dEquimolar DNA degeneracies in shotgun codons are represented by the IUB code (K=G/T; M=A/C; R=A/G; S=G/C; W=A/T; Y=C/T).
Table 2.
Shotgun-scanning analysis of hGHwtand hGHv
| Wild–type/mutant ratiosb | ||||||||||||
| hGHR selection | Antibody selection | ΔΔGmut–wtc(kcal/mol) | ||||||||||
| hGHwt | hGHv | hGHwt | hGHv | hGHwt | hGHv | |||||||
| Residuea | Wt/Homo | Wt/Ser | Wt/Homo | Wt/Ser | Wt/Homo | Wt/Ser | Wt/Homo | Wt/Ser | Homo | Ser | Homo | Ser |
| M14W | 0.91 | 4.45 | 1.21 | 3.13 | 1.39 | 7.33 | 0.022 | 0.29 | −0.3 | −0.3 | 2.4 | 1.4 |
| H18D | 1.89 | 4.17 | 1.64 | 1.08 | 2.42 | 13.7 | 0.96 | 0.84 | −0.2 | −0.7 | 0.3 | 0.2 |
| H21N | 2.33 | 1.25 | 6.23 | 0.12 | 1.59 | 1.36 | 7.45 | 0.045 | 0.2 | −0.1 | −0.1 | 0.6 |
| Q22 | 1.42 | 0.37 | 2.88 | 1.64 | 0.69 | 0.43 | 1.85 | 1.85 | 0.4 | −0.1 | 0.3 | −0.1 |
| F25 | 1.02 | 1.31 | 2.17 | 1.31 | 1.71 | 1.49 | 1.50 | 1.24 | −0.3 | −0.1 | 0.2 | 0.0 |
| D26 | 0.70 | 3.63 | 1.04 | 1.04 | 1.22 | 0.94 | 0.92 | 1.92 | −0.3 | 0.8 | 0.1 | −0.4 |
| Q29 | 1.94 | 0.75 | 3.52 | 0.81 | 0.92 | 0.72 | 5.50 | 0.80 | 0.4 | 0.0 | −0.3 | 0.0 |
| K41I | 1.35 | 1.14 | 1.14 | 1.23 | 0.34 | 0.91 | 0.53 | 1.84 | 0.8 | 0.1 | 0.5 | −0.2 |
| Y42H | 1.04 | 0.77 | 1.21 | 1.22 | 0.47 | 0.90 | 1.09 | 2.50 | 0.5 | −0.1 | 0.1 | −0.4 |
| S43 | 0.84 | 1.21 | 1.53 | 0.94 | −0.4 | 0.0 | ||||||
| F44 | 1.57 | 2.80 | 0.92 | 2.20 | 2.79 | 2.03 | 1.09 | 0.90 | −0.3 | 0.2 | 0.2 | 0.5 |
| L45W | 3.52 | 1.26 | 1.24 | 1.71 | 2.00 | 1.94 | 0.70 | 0.54 | 0.4 | −0.3 | 0.3 | 0.7 |
| Q46W | 4.88 | 0.40 | 0.73 | 1.67 | 0.54 | 0.40 | 0.46 | 0.82 | 1.3 | 0.0 | 0.3 | 0.4 |
| N47 | 1.42 | 0.74 | 1.16 | 0.78 | 0.49 | 1.04 | 0.86 | 1.02 | 0.6 | −0.2 | 0.2 | −0.2 |
| P48 | 1.19 | 0.61 | 0.53 | 0.96 | 0.84 | 0.84 | 0.64 | 1.28 | 0.2 | −0.2 | −0.1 | −0.2 |
| T60 | 1.79 | 0.25 | 1.76 | 0.76 | 0.79 | 0.17 | 1.14 | 0.43 | 0.5 | 0.2 | 0.3 | 0.3 |
| P61 | 4.94 | 9.55 | 4.59 | 6.38 | 0.69 | 0.57 | 0.93 | 0.98 | 1.2 | 1.7 | 1.0 | 1.1 |
| S62 | 1.94 | 1.47 | 1.33 | 2.36 | 0.2 | −0.3 | ||||||
| N63 | 1.50 | 0.86 | 2.65 | 0.31 | 0.32 | 0.62 | 1.09 | 0.63 | 0.9 | 0.2 | 0.5 | −0.4 |
| R64K | 1.88 | 7.55 | 1.02 | 3.08 | 2.58 | 1.61 | 0.66 | 0.30 | −0.2 | 0.9 | 0.3 | 1.4 |
| E65 | 0.42 | 0.38 | 0.67 | 0.61 | 0.45 | 0.27 | 0.42 | 0.47 | 0.0 | 0.2 | 0.3 | 0.2 |
| E66 | 0.59 | 0.78 | 0.67 | 1.63 | 0.35 | 0.40 | 0.39 | 0.68 | 0.3 | 0.4 | 0.3 | 0.5 |
| T67 | 1.50 | 0.86 | 3.52 | 2.20 | 0.84 | 0.22 | 1.47 | 0.40 | 0.3 | 0.8 | 0.5 | 1.0 |
| Q68 | 1.79 | 0.86 | 2.65 | 1.50 | 1.27 | 0.42 | 1.20 | 0.40 | 0.2 | 0.4 | 0.5 | 0.8 |
| Y164 | 1.18 | 3.13 | 0.55 | 5.33 | 1.00 | 5.57 | 0.86 | 22.3 | 0.1 | −0.3 | −0.3 | −0.9 |
| R167N | 1.32 | 1.38 | 9.00 | 0.38 | 1.31 | 0.75 | 6.15 | 0.19 | 0.0 | 0.4 | 0.2 | 0.4 |
| K168 | 0.51 | 2.15 | 0.55 | 1.20 | 0.54 | 1.94 | 0.98 | 7.00 | 0.0 | 0.1 | −0.3 | −1.1 |
| D171S | 18.3 | 1.15 | 2.17 | 1.44 | 2.27 | 1.33 | 1.5 | −0.4 | 0.3 | |||
| K172 | 0.68 | 11.0 | 0.86 | 23.0 | 0.46 | 1.38 | 1.02 | 0.82 | 0.2 | 1.2 | −0.1 | 2.0 |
| E174S | 1.00 | 4.89 | 0.94 | 0.41 | 4.45 | 0.82 | 0.5 | 0.1 | 0.1 | |||
| T175 | 1.05 | 4.94 | 1.64 | 1.09 | 0.63 | 1.09 | 0.58 | 0.23 | 0.3 | 0.9 | 0.6 | 0.9 |
| F176Y | 2.25 | 46.5 | 0.79 | 46.0 | 2.32 | 3.50 | 0.52 | 6.15 | 0.0 | 1.5 | 0.3 | 1.2 |
| R178 | 2.37 | 3.32 | 1.26 | 17.8 | 2.57 | 0.93 | 2.58 | 4.35 | 0.0 | 0.8 | −0.4 | 0.8 |
| I179T | 0.43 | 3.23 | 2.24 | 3.04 | 0.56 | 1.00 | 2.00 | 0.71 | −0.2 | 0.7 | 0.1 | 0.9 |
| R183 | 0.69 | 4.53 | 9.56 | 9.33 | 1.00 | 1.00 | 6.15 | 0.034 | −0.2 | 0.9 | 0.3 | 3.3 |
aThe 35 residues included in the analysis are listed. The first letter denotes the hGHwt sequence in the single-letter amino acid code, followed by the position number in the hGH sequence. In positions where the hGHv sequence differs from that of hGHwt the letter following the position number denotes the hGHv sequence (e.g., M14W denotes position 14, which is methionine in hGHwt and tryptophan in hGHv).
b Wild-type/mutant ratios were calculated from ~100 sequences selected for binding to either the hGHR-ECD or an anti-hGH monoclonal antibody. Values significantly different from unity (≥4.0 or ≤0.25) are highlighted in bold.
cThe wild-type/mutant ratios were used to calculate the difference in binding free energy (ΔΔGmut–wt) corresponding to each serine or homolog mutation as described in Materials and Methods. Binding energy difference values indicating significant deleterious (≥0.4 kcal/mol) or beneficial (≤0.4 kcal/mol) effects are highlighted in bold.
Serine-scanning
The wild-type/mutant ratios obtained from receptor selected clones are affected by both display bias and the direct effect of the mutation on the molecular interaction with the receptor. The magnitude of the direct effect is expressed in terms of display bias–normalized changes in binding energy. These ΔΔGmut-wt values are listed in the last four columns of Table 2. The normalized values allow for many different types of comparisons to be made among the four data sets. In addition, these data can also be compared to previously published alanine-scanning data for the two proteins (Cunningham and Wells 1993; Pal et al. 2003). Data in Figures 1 ▶ and 2 ▶ are organized so as to facilitate these comparisons.
Figure 1.

ΔΔGmut-wt values measuring the effects of mutations on site 1 binding affinity for the hGHR. Data in (A) are for hGHwt and those in (B) are for hGHv. Data are shown for homolog (red bars), serine (white bars), or alanine (green bars) substitutions. The homolog- and serine-scanning data are from Table 2, alanine-scanning data for hGHwt are from Cunningham and Wells (1993), and shotgun alanine-scanning data for hGHv are from Pal et al. (2003). Asterisks (*) indicate positions for which alanine-scanning data are not available. The hGHwt and hGHv residues are denoted by the single-letter amino acid code followed by their position number in the sequence. The letter following the position number denotes the homolog substitution used at each position.
Figure 2.
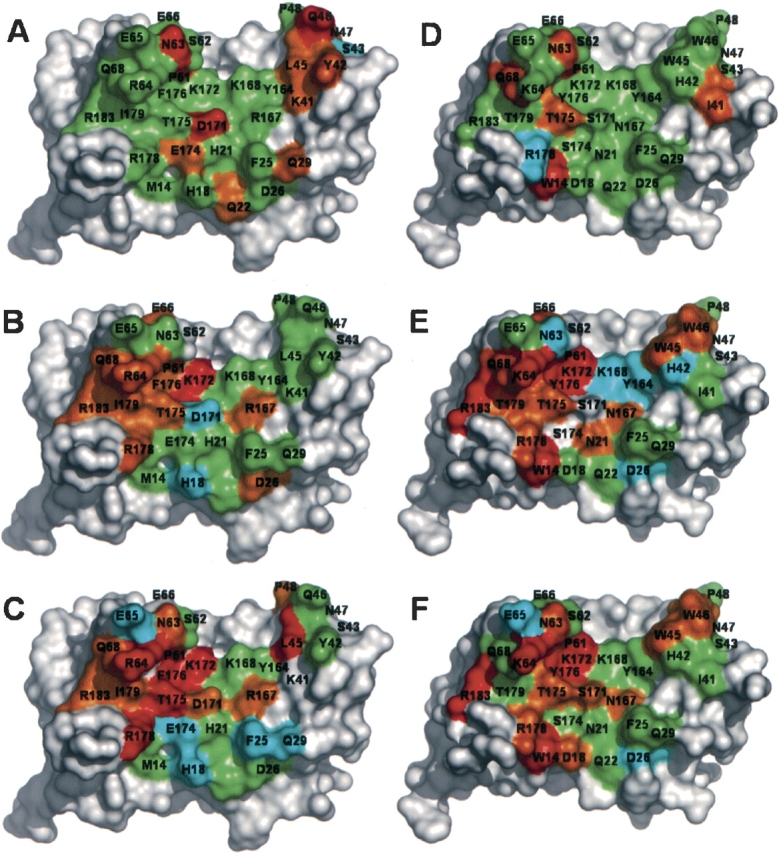
The effects of mutations on site 1 binding affinity for the hGHR mapped onto the structure of hGHwt (A–C) or hGHv (D–F). The maps show the effects of substitution by homologous residues (A,D), serine (B,E), or alanine (C,F). The residues are colored according to the ΔΔGmut-wt values shown in Table 2 and Figure 1 ▶, as follows: cyan< −0.4 kcal/mol; −0.4 kcal/mol ≤ green<0.4 kcal/mol; 0.4 kcal/ mol ≤ orange<1.0 kcal/mol; red ≥ 1.0 kcal/mol; gray, untested. The structures were derived from the 2:1 hGHR-ECD:hGH complexes for hGHwt (de Vos et al. 1992) and hGHv (Schiffer et al. 2002), and were rendered as molecular surfaces using Pymol (DeLano Scientific).
In general, the serine-scanning and alanine-scanning ΔΔGmut-wt values track together, but there are a few notable exceptions (>0.6 kcal/mol difference): D26, L45, D171, and E174 in hGHwt, and N63, E65, Y164, and K168 in hGHv. Interestingly, the E174A mutation substantially improves binding of hGHwt to the hGHR, but the E174S mutation is predicted to cause only a minor effect. This result is somewhat unexpected, because the E174S mutation was selected during the affinity maturation process that produced hGHv (Lowman and Wells 1993), and thus, one might expect the point mutation to improve the affinity of hGHwt for the hGHR. The fact that the serine scan of hGHwt does not predict an improvement suggests that the selection of the E174S mutation in hGHv was dependent upon some of the other mutations that were accumulated during the affinity maturation process, and consequently, the presence of any particular mutation in hGHv does not necessarily indicate that the single mutation alone would be beneficial.
The contact surfaces characteristic of most large protein– protein interfaces generally contain approximately equal amounts of hydrophobic and hydrophilic residues. Consequently, there may be many instances where the removal of a side chain causes less disruption if the substitute is a serine rather than an alanine. In fact, the versatility of serine to make a wide variety of packing interactions is implied by the observation that it is one of the most prevalent amino acids in antigen-binding sites (Mian et al. 1991; Padlan 1994; Davies and Cohen 1996; Fellouse et al. 2004, 2005). The overrepresentation of serine in the heavy chains of natural antibodies is due mostly to occurrence in the first and second complementarity determining regions (CDRs), which often interact with the antigen, but generally do not contribute to the overall affinity nearly as much as the third CDR does. Thus, serine is apparently versatile enough to pack against different types of surfaces, but it does so in ways that contribute little to the binding energy. However, it seems that these interactions do not hinder binding either. Therefore, serine appears to be functionally neutral in many cases, which makes it a valuable alternative to alanine for functional scanning strategies, as the goal of such strategies is to use substitutions that remove specific side chain interactions without introducing new interactions.
Homolog-scanning
Removal of a portion of a side chain through substitution with either alanine or serine can provide an overall measure of the energetic importance of a particular residue, but it provides little information as to how specific the side chain interaction is. Some side chain interactions might have absolute requirements for a particular geometry or functional group, while others might be adequately substituted for by a similar residue. Scanning by substitution with homologous residues aims to shed light on the level of specificity involved in the side chain interaction, which is an essential and characteristic element of most macromolecular recognition events.
Overall, homolog-scanning produced fewer and smaller deleterious effects than either serine-scanning or alanine-scanning (Figs. 1 ▶, 2 ▶), as most effects are in the ±0.4 kcal/ mol range. For simplicity, our homolog-scanning strategy used binary degenerate codons to include only one homologous residue in addition to the wild type. In some cases this strategy restricted the flexibility in choosing the best homolog. For instance, while many obvious homolog pairs could be generated (e.g. tyrosine/phenylalanine and threonine/serine), the best homolog substitution for tryptophan (phenylalanine or tyrosine) could not be introduced in a binomial format, and thus, leucine was used as the best available substitution (Table 1).
The majority of the mutations that caused large deleterious effects (Figs. 1 ▶,2 ▶) were those for which a highly similar homolog was not available. This is clearly true for the W14L mutation in hGHv, and also for the P61A mutation in both hGHv and hGHwt, since no appropriate homolog is available for proline among the natural amino acids. We also note that positions 14 and 61 are occupied by “framework” residues that influence binding by conferring conformational stability to the local polypeptide chain, but are not themselves involved directly in binding contacts.
One of the largest differences between the hGHwt and hGHv scans is seen at position 14, which is occupied by methionine in hGHwt and by tryptophan in hGHv. Position 14 is occupied by either a methionine or valine in almost all known growth hormone sequences. Therefore, the leucine substitution that was a poor compromise for the tryptophan in hGHv is actually the most appropriate one for the methionine in hGHwt. All three scans of this position in hGHwt show little functional difference upon substitution by alanine, serine, or leucine. In contrast, all three substitutions for the tryptophan at position 14 in hGHv result in significant decreases in binding affinity. From a structural standpoint, the side chain at this position functions as a framework residue and does not make direct contact with the hGHR. The implication is that the tryptophan at this framework position gained particular importance in the hGHv interaction, and substitution by leucine, a residue that is similar to the wild-type methionine, cannot rescue the new function.
Interestingly, some of the largest effects observed by homolog-scanning involve side chains that contain either carboxyl or amide groups. There is no general agreement in the literature with regards to the question of what is the best homologous substitution for such side chains. To our knowledge, no attempt was made to systematically evaluate the pros and cons of the possible homologous substitutions in these cases. In our homolog-scanning strategy, asparagine and glutamine were replaced by the acidic counterparts, aspartate and glutamate, respectively; this resulted in substitutions that retained the length of the wild-type side chain but introduced a negative charge. In contrast, the acidic side chains were substituted for each other, which conserved the negative charge but altered the side chain length. These are generally thought to be conservative mutations. However, the data clearly indicate that in some situations these so-called “homologous” substitutions have a much more deleterious effect on binding than truncating the side chain by substitution with alanine or serine. It is noteworthy that most of the large deleterious effects observed in the homolog-scanning data for hGHwt (Table 2) result from the replacement of side chain amide groups by carboxyl groups (Q22E, Q29E, Q46E, N47D, and N63D) or from alterations in the lengths of acidic side chains (D171E and E174D). For instance, the highly deleterious effect of the mutation Q46E is most likely due to the introduction of charge repulsion effects, since Q46 forms a hydrogen-bond with an acidic side chain of hGHR (E120) in the wild-type site 1 interaction. This interpretation is supported by the fact that the Q46S mutation does not affect binding. The D171E mutation lengthens the side chain but maintains the negative charge; the large deleterious effect of this mutation is presumably due to a mismatch in the geometry with its binding partners in the hGHR (R43 and W104). Interestingly, this is one of the positions where the alanine- and serine-scanning data differ (Fig. 1 ▶).
A general finding from the homolog-scanning data is that side chains involved in hydrogen-bonding interactions appear to be among the ones most sensitive to substitution. It is clear that both side chain length and charge effects can influence binding through homologous substitutions in the carboxyl-amide/carboxyl-acid side chain groupings. Structural analysis can help to rationalize the mutagenesis results, but nonetheless, the findings are descriptive rather than predictive, as it is not clear why substitutions of some hydrogen-bonding groups are detrimental while those of others are not.
It is somewhat surprising that, among all of the homologous side chain substitutions made in the hGHwt and hGHv site 1 interfaces, none are predicted to cause a substantial improvement in binding to the hGHR. This suggests that both naturally-evolved hGHwt and in vitro-evolved hGHv are already optimized within the scope of the geometrically and/or chemically similar sequence space. Indeed, we know from the affinity maturation studies that originally produced hGHv that substantial changes were needed to greatly increase the binding affinity of hGHwt (Lowman and Wells 1993; Pal et al. 2003). The important point is that the affinity maturation involved substitutions that significantly altered the chemical character and/or geometry of the wild-type residues they replaced. Thus, it appears that increasing the affinity of natural hormone binding surfaces requires more than just subtle tweaking; it apparently requires that new interactions be made by nonconservative substitutions with nonhomologous residues.
Conclusions
Alanine-scanning mutagenesis is a powerful method for systematically probing the contributions of individual protein side chains to binding and activity. However, in cases that involve the analysis of large protein–protein interfaces, a comprehensive scan is a quite laborious undertaking. Thus, making such a scan feasible often requires that the researcher make a guess as to which subset of residues would provide the most valuable information upon mutagenesis. In the absence of previous experimental information, it is a less than optimal strategy.
We believe that the recently developed shotgun-scanning mutagenesis approaches provide a viable alternative that is rapid and economical, and allows for parallel evaluation of many comparative scans that follow different logic and test different aspects of the interaction. Additionally, since protein binding surfaces are not rigid, effects due to single residue substitutions—at least in some cases—can be compensated for by local conformational rearrangements. As a result, the energetic effects that are measured for an alanine (or another) substitution would not account for the original contribution of the replaced side chain. In this regard, an important distinction between the classical site-directed scan compared to the shotgun scan is that the shotgun scan data are derived from a statistical average obtained from sequences that have multiple patterns of mutations over large numbers of scanned positions. Any localized compensatory effects for a mutation at any particular site are averaged out over the large set of protein variants used in the analysis. It is noteworthy, however, that if enough sequences are analyzed, highly correlated phenomena are not lost, but can actually be detected. We have shown that shotgun-scanning data are robust enough to enable the detection of even subtle pairwise interactions between residues which interact in a cooperative manner (Pal et al. 2005).
Materials and methods
Materials
Enzymes and M13-KO7 helper phage were from New England Biolabs. Maxisorp immunoplates were from NUNC. Escherichia coli XL1-Blue was from Stratagene. Bovine serum albumin (BSA) and Tween-20 were from Sigma.
Oligonucleotides
Degenerate oligonucleotides for site-directed mutagenesis were designed to simultaneously randomize predefined codon positions and ensure equimolar starting frequency of wild-type and serine codons or wild-type and a codon for a homologous residue in the serine-scanning and homolog-scanning libraries, respectively. Equimolar DNA degeneracies are represented in the IUB code (K, G/T; M, A/C; R, A/G; S, G/C; Y, C/T). Degenerate codons are shown in bold and the amino acid types coded by these codons are presented in Table 1. The following mutagenic oligonucleotides were used for library constructions to randomize residues in helix-1 (H1); minihelix (MH); loop-60 (L60) and helix-4 (H4).
Serine-scanning of hGHwt
WT-Ser-H1:
CGA CTA TTT GAT AAC GCT AKS CTT CGG GCC MRC CGT CTT MRC YMG CTA GCC TYC KMC ACG TAC YMG GAG TTT GAA GAG GCC TAT
WT-Ser-MH:
ATC CCC AAG GAA CAG ARM TMC TCA TYC TYG YMG ARC YCT CAG ACC TCC CTC TGT
WT-Ser-L60:
TTC TCA GAA TCG ATT CCG WCG YCT TCC ARC MGT KMG KMG WCG YMG CAG AAA TCC AAC CTA GAG
WT-Ser-H4:
AAG AAC TAC GGG CTG CTC TMC TGC TTC MGT ARM GAC ATG KMC ARM GTC KMG WCG TYC CTG MGT AKC GTG CAG TGC MGT TCT GTG GAG GGC AGC
Homolog-scanning of hGHwt
WT-Hom-H1:
CGA CTA TTT GAT AAC GCT MTG CTT CGG GCC MAC CGT CTT MAC SAA CTA GCC TWC GAM ACG TAC SAA GAG TTT GAA GAG GCC TAT
WT-Hom-MH:
ATC CCC AAG GAA CAG ARG TWC KCC TWC MTC SAA RAC SCA CAG ACC TCC CTC TGT
WT-Hom-L60:
TTC TCA GAA TCG ATT CCG ASC SCA KCC RAC ARG GAM GAM ASC SAA CAG AAA TCC AAC CTA GAG
WT-Hom-H4:
AAG AAC TAC GGG CTG CTC TWC TGC TTC ARG ARG GAC ATG GAM ARG GTC GAM ASC TWC CTG ARG RTT GTG CAG TGC ARG TCT GTG GAG GGC AGC
Serine-scanning of hGHv
Var-Ser-H1:
CGA CTA GCT GAT AAC GCT TSG CTT CGG GCC KMC CGT CTT ARC YMG CTA GCC TYC KMC ACG TAC YMG GAG TTT GAA GAG GCC TAT
Var-Ser-MH:
ATC CCC AAG GAA CAG AKC MRC TCA TYC TSG TSG ARC YCT CAG ACC TCC CTC TGT
Var-Ser-L60:
TTC TCA GAA TCG ATT CCG WCG YCT TCC ARC ARM KMG KMG WCG YMG CAG AAA TCC AAC CTA GAG
Var-Ser-H4:
AAG AAC TAC GGG CTG CTC TMC TGC TTC ARC ARM GAC ATG TCC ARM GTC TCC WCG TMC CTG MGT WCG GTG CAG TGC MGT TCT GTG GAG GGC AGC
Homolog-scanning of hGHv
Var-Hom-H1:
CGA CTA GCT GAT AAC GCT TKG CTT CGG GCC GAM CGT CTT RAC SAA CTA GCC TWC GAM ACG TAC SAA GAG TTT GAA GAG GCC TAT
Var-Hom-MH:
ATC CCC AAG GAA CAG RTT MAC KCC TWC TKG TKG RAC SCA CAG ACC TCC CTC TGT
Var-Hom-L60:
(identical to WT-Hom-L60)
Var-Hom-H4:
AAG AAC TAC GGG CTG CTC TWC TGC TTC RAC ARG GAC ATG KCC ARG GTC KCC ASC TWC CTG ARG ASC GTG CAG TGC ARG TCT GTG GAG GGC AGC
Hormone-phage library construction
Libraries for homolog- and serine-scanning were constructed, sorted, and analyzed as described in Pal et al. (2003) and references therein. A “stop template” version of the hGH display phagemid (containing TAA stop codons at each position to be mutated) was used as the template for the Kunkel mutagenesis method (Kunkel et al. 1991). For each scan (homolog or serine) of each protein (hGHwt or hGHv), a library was constructed by simultaneously replacing the stop codons for the 35 residues to be scanned by the appropriate shotgun scanning codons listed in Table 1. Sets of four mutagenic oligonucleotides were used simultaneously for each mutagenesis reaction as listed above. The mutagenesis reaction was electroporated into Escherichia coli SS320 (Sidhu et al. 2000). Each library contained ~2 ×1010 unique members. After overnight growth at 37°C in 2YT medium supplemented with 50 μg/mL carbenicillin and M13-KO7 helper phage, library phages were concentrated by precipitation with PEG/NaCl and resuspended in PBS, 0.5% (w/v) BSA, 0.1% (v/v) Tween-20, as described previously (Sidhu et al. 2000).
Library sorting and binding assays
The phage libraries were independently sorted on two different immobilized targets: the extracellular domain of the human growth hormone receptor (hGHR-ECD) and an anti-hGH monoclonal antibody (3F6-B1.4B1) (Jin et al. 1992). Maxisorp immunoplates (96-well) were coated overnight at 4°C with capture target (either the hGHR-ECD or the anti-hGH monoclonal antibody) at 5 μg/mL and blocked for 2 h with BSA. Phage solutions from the libraries described above (1012 phage/mL) were added to the coated wells, incubated for 2 h at 25°C, and washed 12 times with PBS, 0.05% Tween-20. Bound phage particles were eluted with 0.1 M HCl, and the eluant was neutralized with 1.0 M TRIS base. Eluted phages were amplified in E. coli XL1-blue with M13-KO7 helper phage and used for further rounds of selection. Individual clones from each round of selection were grown in a 96-well format, and the culture supernatants were used directly in phage ELISAs (Sidhu et al. 2000) to detect phage that bound to either the hGHR-ECD or the anti-hGH antibody. After two or three rounds of binding selection, positive binding clones that produced phage ELISA signals at least twofold greater than signals on control plates coated with BSA were subjected to DNA sequence analysis.
DNA sequence analysis
Culture supernatants containing phage particles were used as templates for PCR reactions that amplified the hGH gene. The PCR primers were designed to add M13(-21) and M13R universal sequencing primers at either end of the amplified fragment, thus facilitating the use of these primers in sequencing reactions. Amplified DNA fragments were sequenced using Big-Dye terminator sequencing reactions with the M13(-21) universal sequencing primer, and a single sequencing reaction was sufficient to read all mutated positions. The reactions were performed in a 96-well format and analyzed on an ABI Prism 3700 96-capillary DNA analyzer (PE Biosystems). The sequences were analyzed with the program SGCOUNT as described previously (Weiss et al. 2000). SGCOUNT aligned each DNA sequence against the hGHwt or hGHv DNA sequence by using a Needleman-Wunch pairwise alignment algorithm, translated each aligned sequence of acceptable quality, and tabulated the occurrence of each natural amino acid at each position. In addition, SGCOUNT reported the occurrence of any sequences containing identical amino acid residues at all mutated positions (siblings). Approximately 100 unique sequences were analyzed for each scan, and the wild-type/ mutant ratios were used to calculate differences in binding free energy as described below.
Calculating the effects of mutations on binding energy
The wild-type/mutant ratios were used to calculate the difference in binding free energy (ΔΔGmut-wt) corresponding to each serine or homolog mutation in a manner analogous to that described for shotgun alanine-scanning (Weiss et al. 2000). Briefly, the wild-type/mutant ratios for each randomized position and each selection were substituted for the Ka,wt/Ka,mut ratio in the standard equation, as follows: ΔΔG=RTln(Ka,wt/ Ka,mut)=RTln(wild-type/mutant). This calculation provided a measure of the effect of each substitution on each selection as a change in free energy relative to that of the wild type. To obtain an estimate of the contribution to binding free energy attributable to each side chain (ΔΔGmut-wt), we used the ΔΔG values from the antibody selection (α-hGH) to correct the ΔΔG values from the hGHR-ECD selection for effects on display level or biases in the naïve library, as follows: ΔΔGmut-wt= ΔΔGhGHR-ECD − ΔΔGα-hGH.
Acknowledgments
We thank Rick Artis for helpful discussions.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051519805.
References
- Clackson, T. and Wells, J.A. 1995. A hot spot of binding energy in a hormone-receptor interface. Science 267 383–386. [DOI] [PubMed] [Google Scholar]
- Cunningham, B.C. and Wells, J.A. 1989. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science 244 1081–1085. [DOI] [PubMed] [Google Scholar]
- Cunningham, B.C. and Wells, J.A. 1993. Comparison of a structural and a functional epitope. J. Mol. Biol. 234 554–563. [DOI] [PubMed] [Google Scholar]
- Cunningham, B.C., Jhurani, P., Ng, P., and Wells, J.A. 1989. Receptor and antibody epitopes in human growth hormone identified by homolog-scanning mutagenesis. Science 243 1330–1336. [DOI] [PubMed] [Google Scholar]
- Davies, D.R. and Cohen, G.H. 1996. Interactions of protein antigens with antibodies. Proc. Natl. Acad. Sci. 93 7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayhoff, M.O., Schwartz, R.M., and Orcutt, B.C. 1978. A model of evolutionary change in proteins. pp. 345–358. Washington, D.C., National Biomedical Research Foundation.
- de Vos, A.M., Ultsch, M., and Kossiakoff, A.A. 1992. Human growth hormone and extracellular domain of its receptor: Crystal structure of the complex. Science 255 306–312. [DOI] [PubMed] [Google Scholar]
- Fellouse, F.A., Wiesmann, C., and Sidhu, S.S. 2004. Synthetic antibodies from a four-amino-acid code: A dominant role for tyrosine in antigen recognition. Proc. Natl. Acad. Sci. 101 12467–12472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellouse, F.A., Li, B., Compaan, D.M., Peden, A.A., Hymowitz, S.G., and Sidhu, S.S. 2005. Molecular recognition by a binary code. J. Mol. Biol. 348 1153–1162. [DOI] [PubMed] [Google Scholar]
- Gascoigne, N.R. and Zal, T. 2004. Molecular interactions at the T cell-antigen- presenting cell interface. Curr. Opin. Immunol. 16 114–119. [DOI] [PubMed] [Google Scholar]
- Henikoff, S. and Henikoff, J.G. 1992. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. 89 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, L., Fendly, B.M., and Wells, J.A. 1992. High resolution functional analysis of antibody-antigen interactions. J. Mol. Biol. 226 851–865. [DOI] [PubMed] [Google Scholar]
- Kunkel, T.A., Bebenek, K., and McClary, J. 1991. Efficient site-directed mutagenesis using uracil-containing DNA. Methods Enzymol. 204 125–139. [DOI] [PubMed] [Google Scholar]
- Lowman, H.B. and Wells, J.A. 1993. Affinity maturation of human growth hormone by monovalent phage display. J. Mol. Biol. 234 564–578. [DOI] [PubMed] [Google Scholar]
- Mian, I.S., Bradwell, A.R., and Olson, A.J. 1991. Structure, function and properties of antibody binding sites. J. Mol. Biol. 217 133–151. [DOI] [PubMed] [Google Scholar]
- Padlan, E.A. 1994. Anatomy of the antibody molecule. Mol. Immunol. 31 169–217. [DOI] [PubMed] [Google Scholar]
- Pal, G., Kossiakoff, A.A., and Sidhu, S.S. 2003. The functional binding epitope of a high affinity variant of human growth hormone mapped by shotgun alanine-scanning mutagenesis: Insights into the mechanisms responsible for improved affinity. J. Mol. Biol. 332 195–204. [DOI] [PubMed] [Google Scholar]
- Pal, G., Ultsch, M.H., Clark, K.P., Currell, B., Kossiakoff, A.A., and Sidhu, S.S. 2005. Intramolecular cooperativity in a protein binding site assessed by combinatorial shotgun scanning mutagenesis. J. Mol. Biol. 347 489–494. [DOI] [PubMed] [Google Scholar]
- Pawson, T. and Nash, P. 2000. Protein–protein interactions define specificity in signal transduction. Genes & Dev. 14 1027–1047. [PubMed] [Google Scholar]
- Sato, K., Simon, M.D., Levin, A.M., Shokat, K.M., and Weiss, G.A. 2004. Dissecting the Engrailed homeodomain-DNA interaction by phage-displayed shotgun scanning. Chem. Biol. 11 1017–1023. [DOI] [PubMed] [Google Scholar]
- Schiffer, C., Ultsch, M., Walsh, S., Somers, W., de Vos, A.M., and Kossiakoff, A. 2002. Structure of a phage display-derived variant of human growth hormone complexed to two copies of the extracellular domain of its receptor: Evidence for strong structural coupling between receptor binding sites. J. Mol. Biol. 316 277–289. [DOI] [PubMed] [Google Scholar]
- Sidhu, S.S., Lowman, H.B., Cunningham, B.C., and Wells, J.A. 2000. Phage display for selection of novel binding peptides. Methods Enzymol. 328 333–363. [DOI] [PubMed] [Google Scholar]
- Vajdos, F.F., Adams, C.W., Breece, T.N., Presta, L.G., de Vos, A.M., and Sidhu, S.S. 2002. Comprehensive functional maps of the antigen-binding site of an anti-ErbB2 antibody obtained with shotgun scanning mutagenesis. J. Mol. Biol. 320 415–428. [DOI] [PubMed] [Google Scholar]
- Warren, A.J. 2002. Eukaryotic transcription factors. Curr. Opin. Struct. Biol. 12 107–114. [DOI] [PubMed] [Google Scholar]
- Weiss, G.A., Watanabe, C.K., Zhong, A., Goddard, A., and Sidhu, S.S. 2000. Rapid mapping of protein functional epitopes by combinatorial alanine scanning. Proc. Natl. Acad. Sci. 97 8950–8954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida, M., Muneyuki, E., and Hisabori, T. 2001. ATP synthase—A marvelous rotary engine of the cell. Nat. Rev. Mol. Cell. Biol. 2 669– 677. [DOI] [PubMed] [Google Scholar]