

Abstract
The type 1 human immunodeficiency virus presents a conical capsid formed by several hundred units of the capsid protein, CA. Homodimerization of CA occurs via its C-terminal domain, CA-C. This self-association process, which is thought to be pH-dependent, seems to constitute a key step in virus assembly. CA-C isolated in solution is able to dimerize. An extensive thermodynamic characterization of the dimeric and monomeric species of CA-C at different pHs has been carried out by using fluorescence, circular dichroism (CD), absorbance, nuclear magnetic resonance (NMR), Fourier transform infrared (FTIR), and size-exclusion chromatography (SEC). Thermal and chemical denaturation allowed the determination of the thermodynamic parameters describing the unfolding of both CA-C species. Three reversible thermal transitions were observed, depending on the technique employed. The first one was protein concentration-dependent; it was observed by FTIR and NMR, and consisted of a broad transition occurring between 290 and 315 K; this transition involves dimer dissociation. The second transition (Tm ~ 325 K) was observed by ANS-binding experiments, fluorescence anisotropy, and near-UV CD; it involves partial unfolding of the monomeric species. Finally, absorbance, far-UV CD, and NMR revealed a third transition occurring at Tm ~ 333 K, which involves global unfolding of the monomeric species. Thus, dimer dissociation and monomer unfolding were not coupled. At low pH, CA-C underwent a conformational transition, leading to a species displaying ANS binding, a low CD signal, a red-shifted fluorescence spectrum, and a change in compactness. These features are characteristic of molten globule-like conformations, and they resemble the properties of the second species observed in thermal unfolding.
Keywords: circular dichroism, chemical denaturation, fluorescence, NMR, protein stability, thermal denaturation
The viral genome and several proteins of HIV are encased in a conical shell that is assembled from a single protein subunit (CA, p24) (Gelderblom 1991; Welker et al. 2000). Initially, this protein is synthesized as part of the polyprotein Gag precursor that assembles into an immature capsid at the cell membrane (Wills and Craven 1991). After proteolytic cleavage of Gag, CA forms the mature capsid that encloses the viral RNA, the nucleocapsid protein, and the reverse transcriptase (Fuller et al. 1997). In vitro, CA spontaneously assembles into helical structures and cones resembling the viral capsids (Groß et al. 1997, 1998; Ganser et al. 1999; Li et al. 2000). Several findings suggest that proper and correct capsid assembly is critical for viral infectivity (von Schwedler et al. 1998; Tang et al. 2001; Forshey et al. 2002). For instance, inhibition of CA assembly by mutations has a lethal effect (Groß et al. 1997; von Schwedler et al. 1998; Tang et al. 2001); furthermore, the alteration of capsid stability reduces virus replication (Forshey et al. 2002). CA is capable of different modes of association (Rosé et al. 1992; Dorfman et al. 1994; Misselwitz et al. 1995), and there is a wide body of evidence that dimerization through its C-terminal domain is an important driving force in virus assembly (Wang and Barklis 1993; Borsetti et al. 1998). Understanding the energetics, stability, and folding governing the CA–CA recognition interface could shed light into the poorly understood area of protein self-association and, in turn, could allow the use of such interface as a pharmacological target. Although some insight has been gained in the kinetics of protein self-association, very little is known about the structural aspects and the precise nature of interactions involved during protein self-association (Neet and Timm 1994; Honig and Yang 1995; Jaenicke and Lillie 2000). In fact, it is usually assumed that self-association proceeds via a partially exposed hydrophobic core in a molten globule-like species (Jaenicke and Lillie 2000).
The CA protein of HIV-1 is formed by two independent domains separated by a flexible linker (Gamble et al. 1996, 1997; Gitti et al. 1996; Momany et al. 1996). The N-terminal domain (residues 1–146 of the intact protein), CA-N, is composed of five coiled-coil α-helices, with two additional short α-helices following an extended proline-rich loop (Gamble et al. 1996; Gitti et al. 1996; Momany et al. 1996). The C-terminal domain (residues 147–231), CA-C, is a dimer both in solution and in the crystal form (Gamble et al. 1997); furthermore, the dimerization constant of CA-C is nearly the same (10 ± 3 μM) as that of the intact CA (18 ± 1 μM) (Gamble et al. 1997). Each CA-C monomer is composed of a 310 helix followed by an extended strand and four α-helices connected by short loops (Fig. 1 ▶). The dimerization interface is largely formed by the mutual docking of α-helix 2 from each monomer (residues Ser178–Val191), which buries Trp184 in dimer interface (Fig. 1 ▶). The two additional aromatic residues in each monomer, Tyr164 and Tyr169, are located in the hydrophobic core of each monomer, well away from the dimer interface. The folding and association of CA-C involves a monomeric intermediate that rearranges and dimerizes to yield the native dimer (Mateu 2002). The energetic role of the side chains at the dimerization interface has been determined by thermodynamic analysis using alanine mutants (del Álamo et al. 2003). These studies have shown that the side chain of Trp184 (Gamble et al. 1997; del Álamo et al. 2003), and those of Ile150, Leu151, Arg154, Leu172, Glu175, Val181, Met185, and Leu189 are key for CA-C dimerization (del Álamo et al. 2003).
Figure 1.

Structure of CA-C dimer. X-ray structure of CA-C showing the dimeric structure of the domain. The monomers are depicted in different colors (gray and green). The side chains of Tyr164, Tyr169, and Trp184 are indicated as sticks in each monomer. The figure was produced with PyMOL (DeLano Scientific) and using the PDB file for CA-C (accession no. 1A43).
In this work our aim has been to thoroughly characterize the thermodynamics of unfolding of a dimeric protein at different pHs and several protein concentrations, trying to get insight into the general mechanism governing the formation of the oligomer interfaces. The use of different biophysical probes, which give complementary information, is required to test how the different regions and elements of quaternary, tertiary, and secondary structure of CA-C unfold. To that end, the techniques used in this work were fluorescence, circular dichroism (CD), absorbance, nuclear magnetic resonance (NMR), Fourier transform infrared spectroscopy (FTIR), and size-exclusion chromatography (SEC). The ΔHm and ΔCp of unfolding of monomeric CA-C are similar to those found in other monomeric proteins of similar size. On the other hand, the enthalpy and the entropy of dissociation of dimeric CA-C are small when compared with those of other small oligomeric proteins of similar size that dissociate and unfold in a coupled transition. This is due to the fact that the CA-C dimer dissociates into the monomeric species that has most of the native secondary and tertiary structure. Finally, we have found that monomeric CA-C undergoes a conformational transition during thermal unfolding before being fully unfolded. This transition leads to an intermediate, which could resemble the monomeric molten globule-like species of CA-C detected at low pH, but it is different to the species whose self-association originates the dimeric form.
Results
pH-induced unfolding experiments
The pH-dependent changes in the secondary and tertiary structure of CA-C were monitored by several spectroscopic techniques, which gave complementary information about the unfolding of the protein. Since the measured dissociation constant was about 10 μM (Gamble et al. 1997; Mateu 2002; del Álamo et al. 2003), several protein concentrations were tested: 1 mM (100% dimeric CA-C, used in the FTIR and NMR experiments), 200 μM (90% dimeric CA-C), 20 μM, (~40% monomeric CA-C), and 2 μM (~75% monomeric CA-C, used in the intrinsic fluorescence, quenching, and ANS-binding experiments).
Steady-state fluorescence measurements
Fluorescence has been used to map pH-dependent transitions in the tertiary structure of the protein (Pace and Scholtz 1997) by following the changes in the maximum wavelength and the average emission intensity, 〈λ〉.
The emission fluorescence spectrum of CA-C at physiological pH and at a concentration of 200 μM showed a maximum at 340 nm, and thus, the spectrum was dominated by the emission of the sole tryptophan residue. As the pH was increased above neutral pH, the spectra became red-shifted toward 350 nm, due to basic unfolding of the protein. Although the pKa of this titration could not be determined, its apparent high value is consistent with deprotonation of Tyr, Arg, and/or Lys residues (Cantor and Schimmel 1980). As the pH was decreased, the spectra were also red-shifted toward 350 nm, due to acid unfolding (Fig. 2A ▶), yielding a titration curve with a pKa of 4.3 ± 0.2. This value was close to the ionization constant of the side chains of Glu, Asp, and/or the C-terminal carboxylate (Cantor and Schimmel 1980). The same bell-shaped profile, with one transition midpoint at low pH and another at high pH, was observed when the 〈λ〉 was plotted versus the pH (Fig. 2A ▶). The acidic transition yielded a pKa of 4.2 ± 0.2, which agrees with that determined using the maximum wavelength approach.
Figure 2.

pH-induced unfolding of CA-C followed by fluorescence, ANS binding, and FTIR. Steady-state fluorescence: The 〈λ〉 (filled squares, right axis) and the maxima wavelength (open squares, left axis) are represented vs. the pH at two different CA-C concentrations. (A) 200 μM and (B) 20 μM. Experiments were acquired at 298 K. (C) ANS binding experiments, where the maxima wavelength (open squares, left axis) and the 〈λ〉 (filled squares, right axis) are represented vs. the pH. Protein concentration was 2 μM. The lines through the data are the fittings to Equation 5. Experiments were acquired at 298 K. (D) The maximum wavelength of the tyrosine band at different pHs as followed by FTIR. The line is the fit to Equation 5. Protein concentration was 1 mM. FTIR spectra were acquired at 278 K.
The maximum wavelength or the 〈λ〉 showed the same behavior as that described at high concentrations when experiments were carried out either at a 20 μM (Fig. 2B ▶) or 2 μM (data not shown) protein concentration. At 20 μM, the maximum wavelength of the fluorescence spectrum was red-shifted (343 nm) at physiological pH (compared to the spectrum at 200 μM). This red shift was due to protein monomerization leading to increased solvent-exposure of the Trp. As the pH was either increased or decreased, the fluorescence spectra were further red-shifted toward 350 nm. The pKa determined for the acid transition from the maxima of the spectra was 4.2 ± 0.2, and that from 〈λ〉 was 4.1 ± 0.2.
ANS-binding experiments
ANS binding is used to monitor the extent of exposure of hydrophobic regions, and to detect nonnative partially folded conformations. When ANS binds to solvent-exposed hydrophobic patches, its quantum yield is enhanced and the maxima of emission is shifted from 520 nm to 480 nm (Stryer 1965; Semisotnov et al. 1991). At low pH values, the intensity of ANS in the presence of monomeric CA-C was largely enhanced, and the maximum wavelength was 510 nm (Fig. 2C ▶). As the pH was increased, the spectral intensity was reduced and the maximum wavelength shifted to 530 nm. Consistent effects were observed when the average emission intensity was examined: At low pHs, the 〈λ〉 was high, and it decreased as the pH was raised (Fig. 2C ▶). The apparent pKa determined from either set of data was 4.4 ± 0.2, which agrees, within the experimental error, with those determined by the intrinsic protein fluorescence (see above).
Examination of tryptophan and tyrosine exposure by fluorescence quenching
The solvent-accessibility of tryptophan and tyrosine residues was examined by acrylamide quenching at several pHs. Three pHs were chosen based on the steady-state fluorescence results: pH 3; at neutral pH; and pH 11, in the basic region (Fig. 2A,B ▶).
Acrylamide-quenching experiments, measured by excitation at 295 nm, yielded exponential Stern-Volmer plots in all experiments, except in that carried out at neutral pH. The Ksv parameter remained constant, within the error, at acidic and basic pHs, but it was smaller at neutral pH (Table 1). This suggests that at the extreme pHs, Trp184 was more solvent-exposed. Quenching control experiments in the presence of 5 M GdmHCl (pH 7), where the protein was unfolded (see below), yielded values of Ksv similar to those observed at acidic or basic pHs. The behavior of the Ksv, upon excitation at 280 nm, followed the same pattern as that observed at 295 nm: higher at the extreme pHs and in 5 M GdmHCl than at neutral pH. The values of υ were the same, within error, at the extreme pHs and in 5 M GdmHCl. In general, Ksv and υ were larger when excited at 280 nm than at 295 nm (Table 1), probably because tyrosine and tryptophan residues were being excited at 280 nm.
Table 1.
Quenching constants for CA-C in acrylamide at different excitation wavelengths
| 280 | 295 | |||
| Conditions | KSV(M−1) | υ(M−1) | KSV(M−1) | υ(M−1) |
| pH 3 | 5.5 ± 0.6 | 1.2 ± 0.2 | 5.4 ± 0.2 | 0.67 ± 0.06 |
| pH 7 | 4.4 ± 0.3 | 0.6 ± 0.1 | 3.2 ± 0.5 | 0.0 ± 0.1 |
| pH 11 | 6.1 ± 0.2 | 1.2 ± 0.1 | 5.0 ± 0.8 | 0.7 ± 0.2 |
| 5 M GdmHCl | 5.9 ± 0.6 | 1.0 ± 0.2 | 4.9 ± 0.6 | 0.4 ± 0.2 |
Errors are data fit errors to Equation 1. The constants were obtained by the fitting to Equation 1 of fluorescence intensity at 338 nm vs. the concentration of acrylamide (similar constants were obtained by fitting the intensities at 335, 336, and 337 nm; data not shown). Experiments were carried out at 298 K. The experiments at 5 M GdmHCl were carried out at pH 7.0.
Far-UV CD experiments
The far-UV CD spectrum of CA-C has the features of an α-helical protein, either at 20 μM or 200 μM, with minima at 222 nm and 208 nm (Mateu 2002), as expected from its three-dimensional structure (Gamble et al. 1997). However, interference from the aromatic residues at 222 nm cannot be ruled out (Woody 1995; Kelly and Price 2000). The shape of the spectrum did not change in the pH range explored, but the value of [Θ] at 222 nm was pH-dependent. At 200 μM CA-C, the mean ellipticity remained constant from pH 7 to 10, with a value of approximately −13,000 deg cm2 dmol−1 (Fig. 3A ▶, inset). As the pH was increased, the absolute value of the ellipticity became progressively smaller, although a pKa of this basic transition could not be determined because of the absence of baseline at high pHs. On the other hand, the absolute value of the ellipticity at acidic pHs showed a transition with a midpoint of 4.5 ± 0.2. This value agrees well with those obtained by other techniques (see above).
Figure 3.

pH-induced unfolding of CA-C followed by CD and SEC. (A) Mean residue ellipticity at 222 nm at 20 μM of CA-C. (Inset) Mean residue ellipticity at 222 nm at 200 μM of CA-C. (B) Plots of the elution volume at 20 μM (open circles) and 200 μM (open squares) vs. the pH. The lines (continuous for 20 μM of CA-C; dashed for 200 μM) are the fits to Equation 5 (see text). All experiments were acquired at 298 K.
A similar behavior was observed at 20 μM protein (Fig. 3A ▶). The apparent pKa of the acid transition was 4.4 ± 0.3, which is identical, within the experimental uncertainty, to that obtained at 200 μM.
SEC experiments
Size-exclusion chromatography yielded a single peak at any pH irrespective of the concentration of CA-C used (20 μM or 200 μM). This single peak indicates that the exchange time between the monomeric and dimeric forms is faster than the rate of the chromatographic process; then, the differences in the elution volumes for both concentrations (Fig. 3B ▶) must reflect the average of both species (monomer and dimer) at the concentrations used. In general, either at high pHs (pH >11) or low pHs (pH <2.5), the curves reflecting the behavior of the Ve tend to coalesce (Fig. 3B ▶). There were titrations at low pHs with pKas of 4.3 ± 0.1 (at 20 μM CA-C) and 4.1 ± 0.1 (at 200 μM CA-C). They agree, within the error, with those measured by other techniques (see above). In both species, the Ve at low pHs is well away from the void volume of the column (8.00 mL), indicating that species observed are not oligomers. Between pH 10 and 11, a tendency to increase the Ve was observed at both concentrations, which was more evident at 200 μM (Fig. 3B ▶).
FTIR experiments
Compared to CD, the main advantage of FTIR is its higher sensitivity to the presence or β-structure, random coil, or some side chains, such as those of tyrosine. For instance, the maximum wave number of the tyrosine band appears, at low and neutral pHs, at 1514 cm−1, and it should move toward smaller wave numbers (1505 cm−1) when titration of its phenol proton occurs at basic pHs. However, the tyrosine band in CA-C did show a transition with an apparent pKa of 4.3 ± 0.2 (Fig. 2D ▶), similar to those previously described. This suggests that the transition may be associated with the protonation of an acidic residue close to at least one of the two tyrosine residues (Tyr164 or Tyr169).
In conclusion, the biophysical techniques used indicate that CA-C undergoes a protein concentration-independent conformational transition at low pH with a pKa ~ 4.3 ± 0.2.
Thermal-denaturation experiments
The thermal dissociation and unfolding of dimeric and monomeric CA-C have been characterized by using several biophysical techniques.
Far-UV CD experiments
Thermal denaturation of CA-C was probed at different pHs by following the change in ellipticity at 222 nm, using either a 20 μM (data not shown) or a 200 μM (Fig. 4A ▶) protein concentration. The thermal scans at pHs 4, 5, 6, 7, 8, and 9 (Fig. 4A ▶) revealed a sigmoidal, nonconcentration-dependent, fully reversible process (Table 2). Only at pH 10, the thermal transition was not reversible (data not shown), probably due to protein modifications occurring at the high alkalinity and temperatures. The fact that the transitions were not concentration-dependent suggests that the unfolding of a monomeric species was being observed. As the pH was increased, the Tm decreased slightly (Table 2). The plot of the ΔHm, obtained from the van’t Hoff analysis of the denaturation (Equations 6, 7), versus Tm yielded a ΔCp=1.8 ± 0.5 kcal mol−1K−1 (Fig. 4A ▶, inset).
Figure 4.

Thermal denaturation followed by far- and near-UV-CD, and absorbance at different pHs. (A) Selected far-UV thermal unfolding experiments at pH 4 (open squares), 6 (filled squares), and 9 (open circles) at 200 μM of CA-C. (Inset) Temperature dependence of the enthalpy change upon unfolding at 200 μM. In this temperature range, the unfolding of CA-C was characterized by a temperature-independent heat capacity change upon unfolding of 1.8 ± 0.5 kcal mol−1 K−1. The errors in the enthalpy are fitting errors to Equation 7. (B) Selected near-UV thermal unfolding experiments at pH 4 (open circles), 7 (filled squares), and 8 (blank squares) at 200 μM of CA-C. (C) The same as (B) but at 20 μM protein concentration. (D) Selected absorbance thermal unfolding traces at pH 5 (open circles), 8 (filled squares), and 9 (open squares). The scale on the Y- axis is arbitrary and thermal unfolding traces have been shifted for the sake of clarity. Protein concentrations were 200 μM. The lines are the fittings to Equation 6, taking into account that the free energy is given by Equation 7.
Table 2.
Thermal midpoints (Tm) (in K) of CA-C at several pH values determined using different biophysical techniques
| Conditions | |||||||||
| pH | |||||||||
| Technique | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | Protein concentration |
| Fluorescence | ND | 324.0 ± 0.2 | 325.7 ± 0.3 | 325.5 ± 0.2 | 324.5 ± 0.3 | 324.8 ± 0.5 | NR | 200 | |
| ANS-binding | ND | ND | 318 ± 1 | 325 ± 1 | 325 ± 6 | 322 ± 2 | ND | 20 | |
| 325.2 ± 0.5 | 80 | ||||||||
| Anisotropy | 325 ± 3b | 20 | |||||||
| 326 ± 2 | 200 | ||||||||
| Near-UV CD | ND | 326 ± 5b | 326 ± 5b | 20 | |||||
| ND | 327 ± 0.9 | 327.5 ± 0.9 | 200 | ||||||
| Far-UV CD | 328 ± 3 | 333.0 ± 0.3 | 332.5 ± 0.5 | 332.1 ± 0.3 | 331.7 ± 0.3 | 331.5 ± 0.3 | NR | 20 | |
| 330 ± 1 | 331.0 ± 0.5 | 334.6 ± 0.3 | 332.6 ± 0.6 | 332.6 ± 0.5 | 331.8 ± 0.5 | NR | 200 | ||
| Absorbance | 331.8 ± 0.8 | 330.5 ± 0.5 | 334 ± 0.7 | 328.6 ± 0.7 | 200 | ||||
| FTIR | NR | 2000 | |||||||
| NR | NR | 304.7 ± 0.3 | 307.2 ± 0.8 | 301.2 ± 0.6 | NR | 1000d | |||
| 304.5 ± 0.6 | 530d | ||||||||
| 307.1 ± 0.8 | 1100d | ||||||||
| 307.9 ± 0.7 | 2000d | ||||||||
| 309.8 ± 0.8 | 4500d | ||||||||
| NMR | 328 ± 2a | 334 ± 5c | 200 | ||||||
Errors are data fit errors to Equation 6, using the free energy expression given by Equations 7 and 8. Thermal denaturations followed by fluorescence were repeated three times at any pH and the standard deviation of the three measurements was ±0.6°C. Thermal denaturations followed by ANS-binding were repeated three times at any pH, and the standard deviation was ±3°C in all cases. Each anisotropy measurement at any temperature was repeated four times, being the standard deviation ±0.4; the measurements were repeated twice and the standard deviation was ±4°C. .Experiments in the far and near UV CD were repeated at least twice at any pH, the standard deviations were ±0.5°C (200 μM) and ±3°C (20 μM) for the near-UV CD, and ±0.4°C for the far-UV CD experiments. Absorbance experiments were repeated twice and the standard deviation was ±0.5°C. FTIR and NMR experiments were only acquired once at any pH, due to the large amounts of protein used. ND, not determined (see text for each particular technique); NR, the thermal transition was not reversible (see text).
a Determined from the indole signal (see text).
b The transitions at 20 μM of CA-C have large associated fitting errors because of the poorer signal-to-noise ratio.
c Determined from a methyl signal, with few experimental points at the unfolding baseline (see text).
dThese transitions describe the low-temperature transition observed in FTIR experiments (see text for details). The high-temperature transition was only characterized at pH 7.0.
Near-UV CD and absorbance experiments
Changes in the ellipticity at 280 nm were followed at three selected pHs (pH 4, 7, and 8) and at a protein concentration of 200 μM (Table 2). At pH 7 and 8, a reversible sigmoidal transition was observed, but at pH 4, the steep slope of the native baseline (Fig. 4B ▶), precluded the determination of reliable Tm and ΔHm values. The thermal midpoints were lower than those obtained from far-UV CD measurements and similar to those observed in the fluorescence experiments (see below).
Experiments were also carried out at 20 μM CA-C to check for a concentration dependence (Fig. 4C ▶). At this concentration, the slope of the native baseline was very steep, probably due to the substantial amount of the monomeric form present. This makes the determination of ΔHm and Tm somewhat uncertain.
Changes in the absorbance at 280 nm were followed at pHs 4, 5, 8, and 9 (Table 2; Fig. 4D ▶) by using 200 μM protein concentration. The thermal stability of CA-C decreased at both extremes of pH (Table 2). Sigmoidal reversible curves were obtained at the four pHs; the measured Tm were close to those observed by far-UV, and, thus, higher than those observed by near-UV CD and fluorescence (see above and next paragraph). The finding that the thermal midpoint observed by absorbance agrees better with that observed by far-UV CD than with that obtained by near-UV is not new, and it has been reported in other proteins (Maldonado et al. 1998; Irún et al. 2001; Campos et al. 2004). This can be rationalized by considering that the absorbance spectrum mainly reports changes in solvent exposure (Mach et al. 1995), and the exposition to solvent is dramatically affected by changes in secondary structure. On the other hand, the near-UV reports changes in the asymmetric environment of the aromatic residues which could not be substantially altered when changes in the secondary structure occur.
Fluorescence experiments
A sigmoidal reversible transition was observed for pHs 5, 6, 7, 8, and 9 at 200 μM protein concentration (Fig. 5A ▶; Table 2). Nonreversibility was observed at pH 10, and a linear decrease in the intensity was observed at pH 4 as the temperature was raised (Fig. 5A ▶). At 20 μM of CA-C for most of the pHs, there were not native baselines; this finding did not allow a reliable determination of the thermodynamical parameters of the transition, and most importantly, it precluded any reliable conclusion about whether the process probed by fluorescence is protein concentration-dependent or independent. Then, the calculation of Tm was carried out at a 200 μM protein concentration, where large baselines of the native and unfolded species could be observed. However, at this concentration, the steepness of the native and unfolded baselines precluded a reliable determination of ΔCp.
Figure 5.
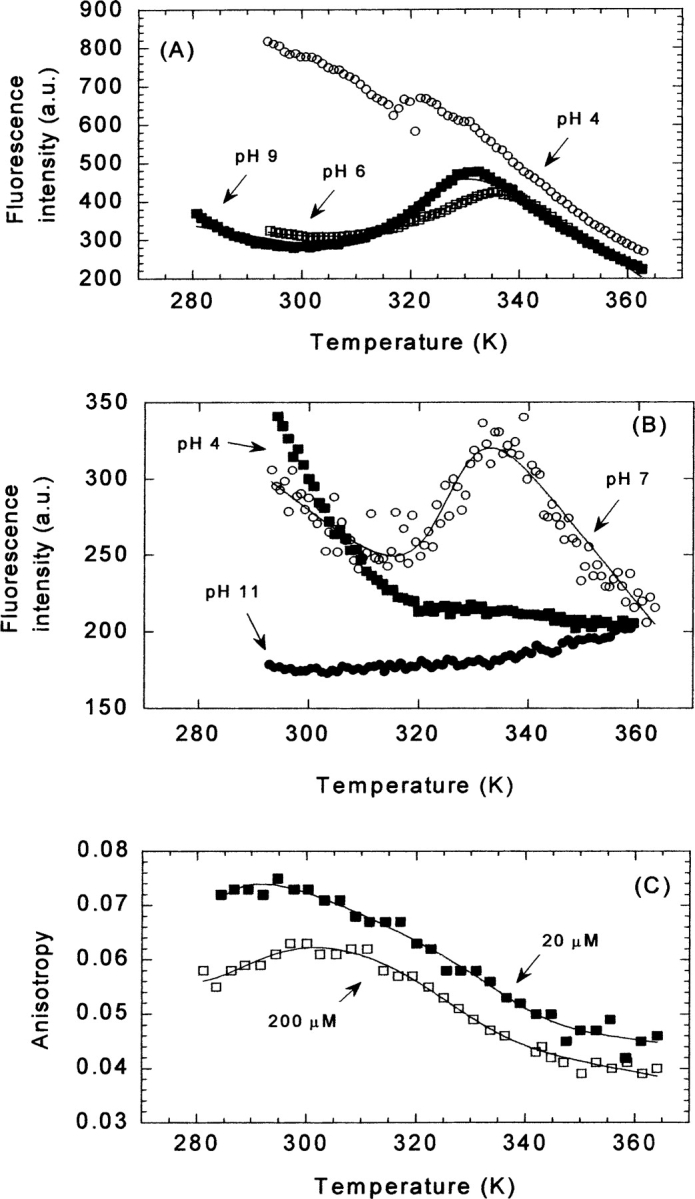
Thermal denaturation followed by protein fluorescence, ANS fluorescence, and anisotropy at different pHs. (A) Thermal unfolding traces followed by fluorescence at selected pHs for 200 μM of CA-C: pH 4 (open circles), pH 6 (white squares), and pH 9 (filled squares). (B) The thermal denaturation experiments followed by the emission intensity at 520 nm of 20 μM CA-C and 100 μM of ANS at several pHs: pH 4 (filled squares), pH 7 (open circles), and pH 11 (filled circles). The scale on the Y-axis for both fluorescence measurements is arbitrary. (C) The change in anisotropy at 20 μM (filled squares) and 200 μM of CA-C (blank squares) at pH 7. The lines are the fittings to Equation 6, taking into account that the free energy is given by Equation 7.
The thermal midpoints at 200 μM of CA-C were lower than those determined by far-UVCD experiments (Table 2).
Thermal ANS binding experiments
A sigmoidal reversible transition was observed at pHs 6, 7, 8, and 9 when thermal denaturation was followed by excitation at 380 nm of samples containing ANS (Table 2). The Tm were close to those obtained by fluorescence and near-UV and smaller than those observed by far-UV and absorbance, suggesting that those different techniques were reporting separated processes. In agreement with the previous spectroscopic techniques, the thermal stability decreased at the extremes of pH. The thermal midpoint was not protein concentration dependent between 20 μM and 80 μM of CA-C (Table 2). The plot of the ΔHm versus Tm yielded a poor value of ΔCp, due to the large fitting errors associated to ΔHm.
Experiments at pH 4, 5, 10, and 11 did not show sigmoidal transitions (Fig. 5B ▶). Conversely, at pH 10 and 11, where CA-C does not bind ANS (Fig. 2 ▶), increasing the temperature caused a linear increase of the emission fluorescence intensity (Fig. 5B ▶).
Thermal anisotropy measurements
The anisotropy gives a measurement of the mobility (global or local) of the tryptophan (Lakowicz 1999), as the temperature is raised. Experiments were carried out at 20 μM and 200 μM CA-C at pH 7 (Fig. 5C ▶). The Tm were not concentration-dependent, and they agree with those obtained by near-UV CD, ANS binding, and fluorescence (Table 2).
FTIR measurements
Conversely to that observed by other techniques, thermal FTIR experiments carried out at pH 7 showed the presence of two transitions (Fig. 6A ▶): (1) a transition with a Tm of 305 K, which had not been observed by any of the previous techniques, and occurring with only small changes in the width of the amide I band, and (2) a transition accounting for most of the change in the width of the band, and with the same Tm (335 K) (Table 2) than those probed by far-UV CD or absorbance. The first thermal transition was reversible at physiological pH, as long as the temperature was not raised above 330 K. At pH 7, the transition observed at high temperatures was not reversible, probably due to the high protein concentrations used. Since it seems that the high-temperature transition was the same as that observed by the above techniques, a pH-dependent study of the low-temperature transition was carried out.
Figure 6.
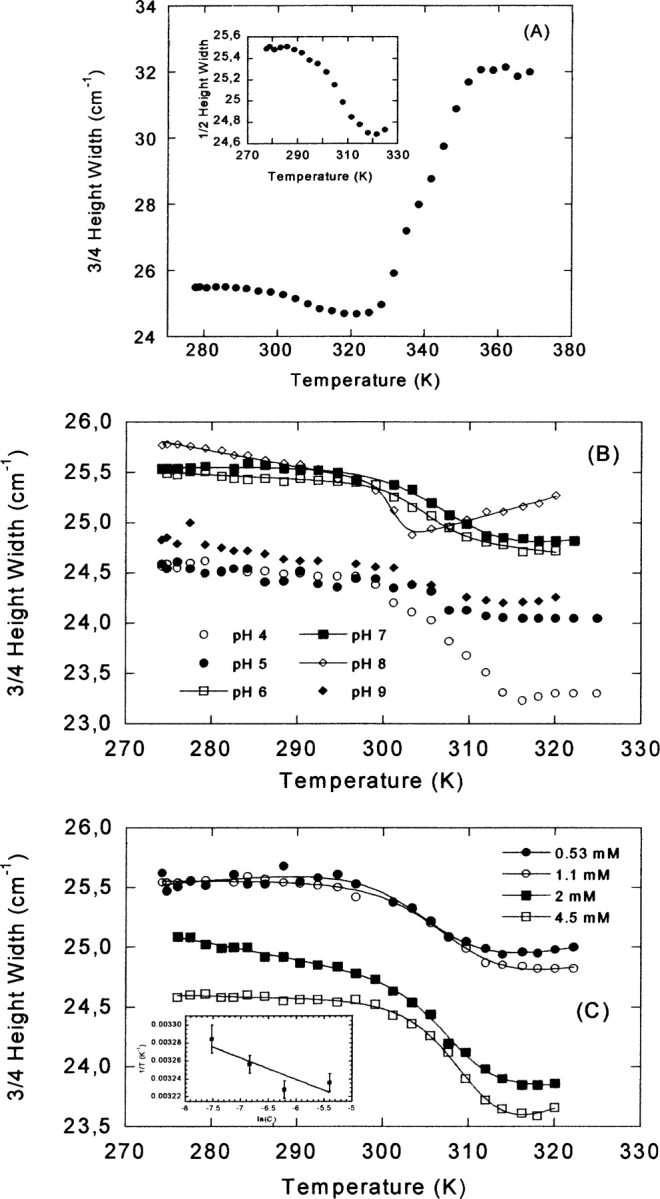
Thermal denaturation followed by FTIR. (A) The complete temperature range (280–370 K) for the FTIR experiments at pH 7, by measuring the 3/4 height width of the amide I band. Protein concentration was 1 mM. (Inset) Expansion showing the 3/4 of the height width of the band for the low-temperature transition. (B) The low-temperature transition observed by FTIR at different pHs (see label). Protein oncentration was 1 mM. (C) The low-temperature transition at different protein concentrations (see label in the figure) at pH 7. (Inset) The protein–concentration dependence of the thermal midpoint for the low-temperature transition. The errors in the temperature are fitting errors to Equation 8. The lines are the fittings to Equation 6, taking into account that the free energy is given by Equation 8.
The low-temperature transition was explored at pHs 4, 5, 6, 7, 8, and 9 (Fig. 6B ▶), but it was only fully reversible at pH 6, 7, and 8. This low-temperature transition was protein concentration-dependent at pH 7 (Fig. 6C ▶; Table 2), and it showed an increase in the Tm as the CA-C concentration was raised. This variation can be used to determine the thermodynamic parameters governing the concentration-dependent reaction. In an oligomeric system, the dependence of Tm on Ct (where here Ct is the total molar protein concentration referred to the molar mass of the monomer) is given by 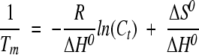 , (Marky and Breslauer 1987; Steif et al. 1993), where ΔH0 and ΔS0 refer to the temperature- independent changes in enthalpy and entropy, respectively, under standard conditions at Tm. In CA-C, the thermodynamic parameters governing the dimer dissociation were (with
, (Marky and Breslauer 1987; Steif et al. 1993), where ΔH0 and ΔS0 refer to the temperature- independent changes in enthalpy and entropy, respectively, under standard conditions at Tm. In CA-C, the thermodynamic parameters governing the dimer dissociation were (with 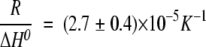 , and
, and 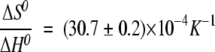 ,) (Fig. 6C ▶, inset) ΔH0=74 ± 8 kcal (mol of cooperative unit) −1 and ΔS0=230 ± 30 cal K−1 (mol of cooperative unit) −1 at 1 M (standard state) protein concentration.
,) (Fig. 6C ▶, inset) ΔH0=74 ± 8 kcal (mol of cooperative unit) −1 and ΔS0=230 ± 30 cal K−1 (mol of cooperative unit) −1 at 1 M (standard state) protein concentration.
NMR measurements
NMR spectroscopy provides a wealth of information concerning the tertiary and secondary structure of a protein at residue level. The 1D-1H-NMR spectra were acquired at pH 7 and 3 at 200 μM CA-C (and 900 μM at pH 7) (Fig. 7 ▶), where, as it has been observed by other techniques (see above), the protein is folded (pH 7), or has lost some of its tertiary structure, but it retains secondary structure (pH 3).
Figure 7.

The up-field shifted methyl region of the NMR spectra at pH 7 and pH 3 at different temperatures. (From top to bottom) NMR spectrum at pH 3 and 320 K; pH 7 and 320 K; pH 3 and 298 K; and pH 7 and 298 K.
The 1D-1H-NMR spectra at 298 K and pH 3 showed a larger number of signals clustered between 8.0 and 8.5 ppm compared to that at pH 7; furthermore, there were only two methyl signals up-field shifted (Fig. 7 ▶). This result suggests that CA-C at pH 3 is less structured than at pH 7 (Wüthrich 1986).
We focused on the methyl and indol signals to follow the temperature changes, since those signals are well-separated from the rest of the resonances of the spectrum. In the spectrum at pH 7 and 330 K, all the amide protons were clustered between 8.0 and 8.5 ppm and the majority of the up-field shifted protons of the methyl region had disappeared, suggesting that most of the tertiary structure was lost. Conversely, at pH 3 and 320 K, the dispersion of the amide region and one of the two up-field shifted methyl protons observed at 298 K had disappeared (Fig. 7 ▶). Then, the NMR experiments confirmed that CA-C is less stable at low pH.
At pH 7, the protons can be classified in three groups, according to their temperature behavior: (1) Class I, protons appearing at −0.26, −0.20, and −0.037 ppm (Fig. 8A ▶) were visible at low temperatures (between 278 and 288 K), but in the interval 290–310 K, the signals became broad and disappeared; (2) Class II, proton appearing at 0.2 ppm (Fig. 8B ▶), whose chemical shift slightly increased with temperature between 280K and 320K, and then it did show a sigmoidal behavior. Although there were few experimental data in the unfolded region, fitting to Equations 6 and 7 was assayed and the thermal midpoint obtained was 334 ± 5 K, close to those obtained by absorbance and far-UV CD; and (3) Class III, consisting of the indole proton of Trp184 (Fig. 8C ▶), whose chemical shift decreased with temperature. The signal became too broad between 290 K and 310 K, and it could not be observed again until temperatures higher than 310 K.
Figure 8.

Temperature dependence of the chemical shifts of protons in the NMR spectra. At pH 7 (see text): (A) Class I, (B) Class II, (C) Class III. At pH 3 (see text): (D) Class I, (E) Class II (methyl proton), (F) Class II (indole proton). Conditions were 200 μM of protein in phosphate buffer (pH 7) (25 mM) or deuterated acetic acid buffer (pH 3) (25 mM) with 0.1 M NaCl.
On the other hand, at pH 3, only two different classes of protons could be observed: (1) Class I, proton appearing at −0.20 ppm at low temperatures, whose chemical shift increased with temperature until 310 K, where it became very broad and disappeared (Fig. 8D ▶); and (2) Class II, composed of the proton of the indole proton of Trp184 and a methyl proton at 0.2 ppm. Both protons could be observed in the whole range of temperatures explored. The methyl proton is probably the proton at the same chemical shift at pH7 (Fig. 8E ▶). Conversely to that observed at pH7, the indole proton of the tryptophan at pH3 was visible over all the explored temperature range (Fig. 8F ▶).
Conformational stability and thermodynamic parameters of monomeric CA-C at pH 7
The inherently large fitting errors in the ΔHm (obtained by some of the described techniques) yielded a ΔCp value, for unfolding of monomeric CA-C, with a large uncertainty (1.8 ± 0.5 kcal mol−1 K−1). Then, it was necessary to obtain a better estimate of the value of ΔCp by using other approaches. We have used an approach first developed by Pace and Laurents (1989), where the ΔG at a selected pH is measured at different temperatures by means of the linear extrapolation model (LEM) (Clarke and Fersht 1993; Pace and Scholtz 1997), and these data are combined with those obtained from thermal denaturations at identical pH. The GdmHCl chemical denaturation of CA-C measured by CD (where unfolding of the monomeric species is being probed) (Mateu 2002) follows the LEM. The isothermal chemical denaturation experiments by CD were carried out at several temperatures in the range from 278 to 313 K at pH 7, and their free energies were obtained (Fig. 9 ▶). From the shape of the free-energy stability curve, the ΔHm, ΔCp, and Tm of the unfolding of the monomeric species could be obtained.
Figure 9.

The GdmHCl-denaturation thermodynamical parameters at pH 7 as monitored by the change in ellipticity at 222 nm in the far-UV CD spectra. (A) Temperature dependence of the m-value from CD measurements. Errors bars are fitting errors to the LEM equation. (B) The temperature dependence of the [GdmHCl]1/2 (left axis, open squares) and ΔG (right axis, filled squares) values. The error bars are fitting errors to the LEM equation. The errors are larger at the higher temperatures, because the native baselines were shorter. The solid line represents the nonlinear least square fit of the data to 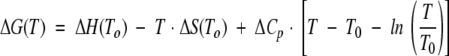 , which is similar to Equation 7 except that here T0, the reference temperature, was taken as 298 K. The use of this equation avoids a bias in the fitting due to the larger number of experimental data around the thermal midpoint. From this equation, the ΔHm, Tm, and ΔCp can be easily obtained. The experimental data at the Tm (right side of figure) were obtained from CD thermal denaturation experiments. ΔG values were obtained by using the mean m-value over all the temperatures. The temperature-dependence of ΔG was consistent with a temperature-independent heat capacity change, ΔCp, of 1.14 ± 0.06 kcal mol−1 K−1.
, which is similar to Equation 7 except that here T0, the reference temperature, was taken as 298 K. The use of this equation avoids a bias in the fitting due to the larger number of experimental data around the thermal midpoint. From this equation, the ΔHm, Tm, and ΔCp can be easily obtained. The experimental data at the Tm (right side of figure) were obtained from CD thermal denaturation experiments. ΔG values were obtained by using the mean m-value over all the temperatures. The temperature-dependence of ΔG was consistent with a temperature-independent heat capacity change, ΔCp, of 1.14 ± 0.06 kcal mol−1 K−1.
The m-values (Fig. 9A ▶) were constant, within the error, in the temperature range explored. Although this tendency does not agree with the LEM model, a similar behavior has been observed in other proteins when chemical denaturations have been followed by CD (Pace and Scholtz 1997; Zweifel and Barrick 2002), and it can be rationalized by considering the presence of large slopes in the native or unfolded baselines. There was a good agreement between the data obtained from the isothermal chemical denaturation measurements and those from thermal denaturation experiments (Fig. 9B ▶); this finding validates the use of the LEM, and most importantly, indicates that the same denatured state of CA-C is being probed by thermal and chemical denaturation measurements. A bell-shaped curve was observed when the [GdmHCl]1/2 and the free-energy values were represented against the temperature (Fig. 9B ▶). The temperature dependence of ΔG at pH 7 was consistent with a temperature- independent ΔCp of 1.14 ± 0.06 kcal mol−1 K−1 (similar, within the error, to that obtained by far-UV measurements; see above), a Tm of 332.7 ± 0.1 K (which agrees with that determined previously by far-UV CD and NMR), and an ΔHm of 54 ± 2 kcal mol−1.
Discussion
The equilibrium thermal denaturation of CA-C follows a four-state mechanism
Dissociation of dimeric CA-C
The low-temperature transition probed by FTIR in thermal denaturation experiments was clearly concentration- dependent (Fig. 6 ▶), and it must reflect the thermal dissociation of the dimer. It is important to keep in mind that dimer dissociation was not only observed by FTIR at the very high concentrations used (~1 mM), but also by NMR at lower concentrations (200 and 900 μM). Furthermore, there are two pieces of evidence, from the NMR experiments at pH 7, which suggest that a transition occurs: (1) The indole signal disappeared in the temperature range from 290 to 310 K (Fig. 8C ▶); the tryptophan is a key residue in the dimerization interface (Gamble et al. 1997; Mateu 2002), and it must be involved in a slow conformational equilibrium, within the NMR time scale as the temperature was increased; and (2) there were methyl signals which started broadening and then disappeared at ~290 K (Fig. 8A,B ▶). Since the rest of the signals of the spectrum did not broaden and disappear (data not shown), this finding must indicate than only some particular regions were involved in the transition. Recently, the changes in the breadth and the shape of the signal of an indole proton have been explained as due to the presence of intermediates species in the unfolding of the monomeric B1 domain of protein G (Ding et al. 2004).
The conformational states of monomeric CA-C
As concluded from the variation in Tm (Table 2), the stability of monomeric CA-C increased from pH 4 to a plateau around pH 6, and then decreased toward the alkaline region. Interestingly, there are two groups of techniques, which show two different midpoints. Far-UV CD, NMR and absorbance showed a Tm which was higher (~6 K) than that obtained by near-UV CD, ANS binding, and fluorescence anisotropy. The noncoincidence of protein unfolding curves, following different spectroscopic properties, is classical evidence of the accumulation of an equilibrium folding intermediate in the unfolding process (Luo et al. 1995, 1997).
There are at least three conformational states for the monomeric species (Fig. 10 ▶): (1) the monomeric form observed above 308 K, as a result of dimer dissociation (see above); (2) the species obtained from a conformational rearrangement, whose Tm was probed by ANS binding, fluorescence anisotropy, and near-UV CD; and (3) the final denatured state, obtained in a transition whose Tm was mapped by far-UV CD, absorbance, and NMR. In all cases, since the transitions were sharp and well separated by ~6 K, we fitted each transition to a two-state curve (Equations 6, 7; Table 2). Since different techniques show the same thermal midpoints, we can rule out that the different Tms are the result of the fitting procedure. Moreover, to further rule out any skew in the calculations, we carried out a global fitting analysis of the far and near-UV CD data at 20 μM and 200 μM of protein concentration; the analysis shows that there are three states with Tm1=329 ± 3 and Tm2=335 ± 3, which are similar to those obtained from the individual two-state fitting of each curve. The dominant monomeric species between both temperatures binds ANS, whereas the monomer obtained as a direct product of dimer dissociation does not. This finding has important implications for the mechanism of oligomer formation, since it has been suggested that assembly of oligomers occurs via a molten globule-like species (Ptitsyn 1995; Jaenicke and Lillie 2000). On the other hand, given the structural features of the rearranged monomer, it is tempting to suggest that this species could be similar to that observed at low pH at 298 K.
Figure 10.

The equilibrium species observed in unfolding of CA-C at pH 7. U, denatured monomer; N′ partially rearranged monomer; N, folded monomer; N2, native dimer. The figures at the bottom of the scheme are for illustrative purposes only, and the rearranged reaction ongoing from N to N′ might not involve fraying of a particular α-helix. The techniques used in this work are indicated below the arrows; the temperature where the transition was observed is at the top of the arrow. The red letters indicate the transitions mapped by thermal denaturation (this work). The blue letters indicate evidence for the detection of the different transitions by chemical denaturation experiments in previous work (Mateu 2002; del Álamo et al. 2003).
At low pH CA-C, in either the monomeric or dimeric form, undergoes a conformational transition, with a pKa of 4.3 (Figs. 2 ▶, 3 ▶). The low pKa suggests that the titration corresponds to one acidic group. Since the titration was also followed by the change in the amide I band corresponding to tyrosine residues in FTIR, it must be an acidic residue close to at least one of the two tyrosine residues in CA-C (Tyr164 or Tyr169), probably Asp166. It is important to stress here that this pKa is an apparent value since it is the average of the titration midpoints of the corresponding residues in the folded and the unfolded CA-C species, appearing as the pH was lowered (Anderson et al. 1990). We could not determine from that value how many kilocalories CA-C was destabilized upon acid unfolding, but rather, we could estimate at what pH the protein was unfolded, as it has been done in other electrostatic studies (Chen et al. 2000; Forsyth and Robertson 2000). In the acidic transition some of the CA-C tertiary structure is lost (as shown by fluorescence), but some does remain, as indicated by the up-field shifted methyl protons in the NMR spectra (Fig. 7 ▶). Furthermore, the compactness of the molecule was changed as shown by the SEC experiments (Fig. 3B ▶). Concomitantly, hydrophobic regions of CA-C became accessible to the solvent, as indicated by ANS experiments at 2 μM of protein concentration (Fig. 2C ▶). Then, the acidic transition must involve exposure of hydrophobic patches belonging to each monomer, and thus the species could have similar structural features to those observed in thermal unfolding experiments. The interface region of this monomeric species is not native-like, as concluded by the quenching experiments and NMR spectra at acidic pH. The quenching experiments showed that the tryptophan is as exposed at low pHs (pH 3) as it is when the protein is fully unfolded (Table 1). This solvent exposure was further confirmed by the NMR experiments at pH 3 where at the lower temperature explored (278 K), the chemical shift of the indole proton was close to that observed for random-coil models: 10.27 ppm in CA-C at pH 3 (Fig. 8F ▶) compared to 10.22 ppm in random-coil models (Wüthrich 1986), and 10.62 ppm observed in CA-C at pH 7 (Fig. 8C ▶).
To sum up, our thermal denaturation data suggest that unfolding of the dimeric CA-C occurs via a four-state mechanism. The dissociation step occurs at temperature midpoints about 307 K, to yield a monomeric folded intermediate, which undergoes a conformational transition at temperatures ~325 K, and it finally unfolds at ~333 K. These series of thermal intermediates agree with the findings of previous chemical denaturation experiments followed by fluorescence and far- UV CD, which monitored exclusively dimer dissociation/monomer rearrangement and monomer unfolding, respectively (Mateu 2002). It is important to stress here that the thermal transition observed by fluorescence cannot be assigned to any particular dissociation and/or conformational rearrangement, since it did not show a clear concentration-independent behavior. Probably, the thermal denaturation fluorescence is reporting several chemical events, as it has been discussed in the chemical denaturation experiments followed by fluorescence (Mateu 2002). Based on the results from FTIR, far-UV CD, anisotropy, and ANS-binding experiments, it can be suggested that thermal fluorescence is measuring dimer dissociation and monomer rearrangement occurring as the temperature changed. Finally, it must be borne in mind that the results obtained here are observed only in equilibrium, and that the sequence of species suggested might not be observed when the kinetic folding pathway of CA-C is described.
Thermodynamic parameters governing the unfolding and dissociation of the monomeric and dimeric forms of CA-C: Comparison with theoretical methods
The ΔCp observed upon protein unfolding is largely the result of changes in hydration of groups, which are buried in the native folded state, and become exposed to solvent upon unfolding (Robertson and Murphy 1997). Taking into account the changes in the accessible surface area, ASA, of a monomer of CA-C upon unfolding calculated fromits X-ray structure (Gamble et al. 1997; Worthylake et al. 1999) (ΔASAnonpolar=4298 Å2 and ΔASApolar=1385 Å2), we obtain ΔCp=(0.45 ± 0.02) (ΔASAnonpolar)+(−0.26 ± 0.03) (ΔASApolar)=1.6 ± 0.2 kcal mol−1 K−1, which is similar to that determined by far-UVCD (1.8 ± 0.5 kcal mol−1K−1), and by the Pace and Laurents (1989) approach (1.14 ± 0.06 kcal mol−1 K−1). Also, the measured value is close to that determined in other proteins of similar size (Robertson and Murphy 1997).
The use of the above theoretical expression also allows us to estimate the ΔCp for the CA-C dissociation reaction, assuming that the monomers retain, upon dissociation, most of the native structure they had when they were forming the dimer species. We think this is a reasonable assumption because (1) the far-UV CD spectra of the monomeric and dimeric species are similar (Mateu 2002), (2) there are protons at similar chemical shifts in the NMR spectra of monomeric and dimeric species (J.L. Neira, M. del Álamo, and M.G. Mateu, unpubl.), and (3) there are only small changes in the width of the FTIR amide I band during the low-temperature transition (Fig. 6A ▶). Based on the X-ray structure, the estimated changes in accessible surface area upon dimer formation per monomer are (Worthylake et al. 1999): ΔASAnonpolar = 611 Å2 and ΔASApolar = 245 Å2. These values yield a ΔCp = 211 ± 10 cal mol−1K−1 per monomer. This low value explains why a dissociation transition was not observed by DSC measurements in the protein concentration range from 8 μM to 70 μM (data not shown), and why it was not observed by most of the techniques described here. The CA-C dissociation reaction is governed by a ΔH0 = 74 ± 8 kcal (mol of cooperative unit) −1 and ΔS 0 =230 ± 30 cal K−1 (mol of cooperative unit) −1 at 1 M standard state, which are also small when compared to the corresponding values of other oligomeric proteins (Backmann et al. 1998 and references therein). However, it must be borne in mind that in many small proteins or domains, dissociation and unfolding of dimers occur concomitantly, although it does not happen in many large oligomeric proteins (Neet and Timm 1994; Jaenicke and Lillie 2000). In CA-C, the values measured (ΔH0 and ΔS0) and calculated (ΔCp) reflect exclusively the dissociation of the dimer.
The free energy of the dissociation of the dimer/conformational rearrangement of the CA-C monomer at 298 K has been obtained from chemical denaturation measurements followed by fluorescence, and it was calculated to be 12.1 kcal mol−1 (at 1 M standard state) (Mateu 2002). Also, the free energy of the specific dissociation step at 298 K, obtained by dilution of the protein in gel filtration analysis was 6.9 kcal mol−1 (at 1 M standard state) (del Álamo et al. 2003). If it is assumed, based on the above calculations, that the ΔCp for the CA-C dissociation reaction is small, by using the calculated ΔH0 and ΔS0, the free energy of dissociation would be ΔG=5 ± 8 kcal mol−1 at 298 K. Although the large error in this latter value may prevent a quantitative comparison, these calculations indicate that both the dissociation and a conformational reorganization of the monomer produced may significantly contribute to the combined dissociation–rearrangement transition measured by fluorescence in chemical denaturation experiments (Mateu 2002; del Álamo et al. 2003).
Possible biological implications
Although the role of the pH in HIV maturation in vivo is controversial, the capsids of many viruses are pH-sensitive; for instance, foot-and-mouth disease virus and mengovirus are dissociated at low pH, and the capsids of rhinovirus and poliovirus are conformationally altered at low pH; furthermore, morphological changes around neutral pH have been observed in several viral systems (Johnson 1996). Moreover, the shape of the assemblies formed by CA (Groß et al. 2000; Erlich et al. 2001) and the association rates to form such assemblies are pH-dependent (Lanman et al. 2002). In such studies (Groß et al. 2000), a conformational change is suggested to occur between pH 6 and 7, which leads to a more extended conformation of CA at high pHs. The SEC experiments in this work indicate that there were no significant changes in the CA-C compactness at neutral and basic pHs (Fig. 3B ▶). Only the fluorescence and CD measurements indicated a new conformational transition (Figs. 2 ▶, 3 ▶). This transition was not characterized by a solvent exposure of hydrophobic patches, but there were changes in (1) the secondary structure (Fig. 3A ▶) and (2) the environment of tryptophan and tyrosine residues, which became solvent exposed (Table 1; Fig. 2A,B ▶). Since HIV capsid assembly depends on both CA-C and CA-N, it is suggested, as a working hypothesis, that most of the observed differences at neutral and basic pHs in CA must be due to the CA-N domain.
On the other hand, based on our thermal denaturation experiments, it could be argued that at physiological temperatures CA-C should be in the monomeric form. However, it must be taken into account that in vivo, CA-C is a domain of CA, whose associative properties are highly influenced by the CA-N domain (Lanman et al. 2002). The low dimerization affinity/thermal stability of the isolated CA-C dimer might be essential to allow disassembly (del Álamo and Mateu 2005) and/or to mediate a wide range of macromolecular interactions during capsid assembly, as it has been suggested for other protein domains (Tang et al. 1999).
Conclusions
The conformational propensities of the dimeric and monomeric forms of the dimerization domain of HIV-1 have been studied. The results described stress the importance of using several biophysical techniques, which give complementary information, to describe protein unfolding. The thermodynamic parameters of the thermal dissociation and unfolding of the dimeric and monomeric species of CA-C have been determined. The enthalpy and the entropy of dissociation of CA-C are small when compared with those of other small oligomeric proteins, due to the fact that the direct product of dissociation is essentially folded. The monomeric species undergoes, before being fully unfolded, at least one conformational transition, to give a species which could resemble the molten globule-like form of CA-C detected at low pH. Then, the monomer species, whose assembly causes dimer formation, does not show the molten globule-like properties, as it has been observed in other oligomers.
Materials and methods
Materials
Trizma base and acid, phosphate salts, ANS, and NaCl were from Sigma. GdmHCl ultrapure was from ICN Biochemicals. Exact concentrations of GdmHCl were calculated from the refractive index of the solution (Pace and Scholtz 1997). Standard suppliers were used for all the other chemicals. Dialysis tubing was from Spectrapore, with a molecular weight cutoff of 3500 Da. Water was deionized and purified by a Millipore system.
Protein expression and purification
The wild-type CA-C protein was expressed in Escherichia coli BL21(DE3) and purified as described (Mateu 2002; del Álamo et al. 2003). Protein was stored in 25 mM sodium phosphate buffer (pH 7.3). Protein stocks were run in SDS-PAGE gels and found to be >97% pure. Purity was also confirmed by MALDI mass spectrometry analysis (data not shown). Protein concentration was calculated by using the extinction coefficients of model compounds (Pace and Scholtz 1997).
Fluorescence measurements
Fluorescence spectra were collected in a Cary Eclipse spectrofluorometer (Varian) interfaced with a Peltier cell. A 1-cm pathlength quartz cell (Hellma) was used. Experiments were acquired at 298 K, unless indicated otherwise. Protein concentrations used were either 2 μM, 20 μM, or 200 μM, unless otherwise stated.
Steady-state fluorescence measurements
Protein samples were excited at 280 nm and 295 nm between pH 2 and 12 to characterize a possible different behavior of tryptophan and tyrosine residues. The slit widths were 5 nm for the excitation and emission wavelengths. Experiments were recorded between 300 nm and 400 nm. The signal was acquired for 1 sec and the increment of wavelength was set to 1 nm. Blank corrections were made in all spectra.
In the pH-induced unfolding experiments, the pH was measured before and after completion of experiments with an ultra-thin Aldrich electrode in a Radiometer (Copenhagen) pH meter. Three-point calibration of the pH meter was performed by using standards from Radiometer. The salts and acids used in buffer preparation were pH 2.0–3.0, phosphoric acid; pH 3.0– 4.0, formic acid; pH 4.0–5.5, acetic acid; pH 6.0–7.0, monosodium dihydrogen phosphate; pH 7.5–9.0, Tris acid; pH 9.5– 11.0, sodium carbonate; and pH 11.5–13.0, sodium phosphate.
Steady-state ANS binding
ANS binding was detected by collecting fluorescence spectra at different pHs in the presence of 100 μM dye and at a protein concentration of 2 μM. Excitation wavelength was 370 nm, and emission was measured from 430 to 700 nm. Slit widths were 5 nm for excitation and emission wavelengths. Stock solutions of ANS were prepared in water, using a molar extinction coefficient of 6.8 × 103M−1 cm−1 at 370 nm (Mann and Matthews 1993).
Fluorescence quenching experiments
Quenching of intrinsic tryptophan and tyrosine fluorescence by acrylamide (Lakowicz 1999) was examined at different pHs. Excitation was carried out at 280 nm (for quenching of tyrosine and tryptophan residues) and 295 nm (for quenching of the tryptophan residue); emission was measured from 300 nm to 400 nm. The slit widths were set to 5 nm for both excitation and emission wavelengths. The dynamic and static quenching constants were obtained by fitting the fluorescence intensity at different wavelengths (in the range from 330 to 340 nm) to the Stern-Volmer equation (Lakowicz 1999):
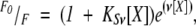 |
(1) |
where Ksv is the Stern-Volmer constant for collisional quenching and υ is the static quenching constant. The range of quencher concentrations was 0–0.7 M. Protein concentrations were 2 μM in all cases. Control experiments carried out at pH 7 with 20 and 200 μM of CA-C, after correction of inner filter effects, yielded the same quenching constants (data not shown).
Thermal denaturation measurements
Thermal unfolding curves were determined using three different techniques:
1. Thermal denaturation following the emission fluorescence of tryptophan and tyrosine residues.
Changes were followed by excitation at 280 nm, and acquisition of the emission intensity either at 335 nm or 350 nm (both wavelengths yielded the same unfolding curves; data not shown). The excitation and emission slit widths were set to 5 nm. The scan rate was either 30 K/h or 60 K/h. Both scan rates yielded the same curve (data not shown). Experiments were repeated three times, with new samples.
2. Thermal denaturation following the emission fluorescence of ANS.
Two different protein concentrations were assayed: 20 μM and 80 μM, with 100 μM and 160 μM of ANS, respectively. No differences were observed in the Tms obtained at both concentrations. We used two different procedures to ascertain that no scan-rate dependence was observed. First, the temperature was raised manually in the temperature range from 290 K to 360 K; a spectrum was acquired every 3 K after 5 min of equilibration in the Peltier cell. Excitation wavelength was 380 nm, and the emission spectrum was collected in the interval range from 430 nm to 600 nm. Spectra were acquired every 1 nm. The slit widths were set at 5 nm for excitation and emission wavelengths. Second, we used the automatic thermal scan rate of the fluorimeter. Excitation wavelengths were 360 nm, 370 nm, and 380 nm, and emission wavelengths were 480 nm and 520 nm. The same unfolding curves were obtained by collecting the data at both emission wavelengths (data not shown), and similar curves were obtained at any excitation wavelength (data not shown). The excitation and emission slit widths were set to 5 nm; points were acquired every 0.2 K. The scan rate was 60 K/h. No differences were observed between the experiments acquired using the first procedure and the latter procedure; thus, the thermal-unfolding ANS experiments were not scan-rate dependent. In both procedures, experiments were repeated three times, with new samples.
3. Thermal denaturation following the change in anisotropy.
The steady-state anisotropy, 〈r〉, is defined by (Lakowicz 1999):
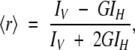 |
(2) |
where IV and IH are, respectively, the vertical and the horizontal components of the polarized fluorescence light, and G is a correction factor for the different fluorescence response to the two polarized components. The G factor was typically 0.43 for the emission wavelength of 330, and 0.56 for the emission wavelength of 350 nm. Fluorescence anisotropy was recorded in the L arrangement. Excitation wavelength was set at 280 nm and emission wavelengths were either 350 nm or 330 nm. There were no differences between both emission wavelengths, but those acquired at 350 nm and at 20 μM of CA-C had a lower signal-to-noise ratio. The excitation and emission slit widths were set to 10 nm, and the average time was 5 sec. The temperature of the Peltier cell was increased manually every 3 K and the anisotropy measurements were taken after averaging four measurements for each temperature. Each denaturation was repeated twice.
Circular dichroism and absorbance measurements
Protein concentrations were either 20 μM or 200 μM, unless otherwise stated.
Steady-state experiments
Circular dichroism spectra were collected on a Jasco J810 spectropolarimeter fitted with a thermostated cell holder and interfaced with a Neslab RTE-111 water bath. The instrument was periodically calibrated with (+) 10-camphorsulphonic acid. Isothermal wavelength spectra at different pHs were acquired at a scan speed of 50 nm/min with a response time of 2 sec and averaged over four scans.
The chemical denaturation measurements in the far-UV were performed using 20 μM of CA-C in 25 mM phosphate buffer (pH 7.3), using 0.1-cm pathlength cells (Hellma). The denaturation reaction was fully reversible, as shown by the sigmoidal curves obtained by starting from dilution of protein samples at 7 M GdmHCl (data not shown). Every chemical denaturation experiment was repeated at least three times with new samples. All spectra were corrected by subtracting the proper baseline.
Near-UV spectra at different pHs were acquired in a 0.5-cm pathlength cell (Hellma). The mean residue ellipticity, [Θ], was calculated as:
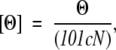 |
(3) |
where Θ is the measured ellipticity, l is the pathlength cell (in cm), c is the protein concentration (in M), and N is the number of amino acids.
Thermal denaturation experiments
Thermal denaturation experiments were performed at constant heating rates of 60 K/h and 30 K/h and a response time of 8 sec at different pHs. Thermal scans were collected in the far-UV region at 222 nm from 298 K (or 278 K) to 363 K (or 368 K) in 0.1-cm pathlength cells. The reversibility of thermal transitions was tested by recording a new scan after the sample was cooled down to 278 K or 298 K, and comparing to the spectra obtained before heating. The possibility of drifting of the CD spectropolarimeter was tested by running two samples containing buffer, before and after the thermal experiments. No difference was observed between the scans. Every thermal denaturation experiment was repeated at least twice with new samples. In all cases, the samples were transparent and no precipitation was observed, after the heating.
Circular dichroism and absorbance thermal denaturation experiments in the near-UV were simultaneously measured at 280 nm in a Jasco J-710 spectropolarimeter using a protein concentration of 200 μM in a 0.5-cm pathlength cell (Hellma). Every thermal denaturation experiment was repeated at least twice with new samples.
Analysis of the pH, thermal, and chemical denaturation curves, and free energy determination
The average emission intensity in fluorescence spectra, 〈λ〉, was calculated as (Royer 1995):
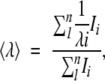 |
(4) |
where Ii is the fluorescence intensity measured at a wavelength λi. This parameter was used to map the changes in the solvent exposure of aromatic residues.
The pH-denaturation experiments were analyzed assuming that both species, protonated and deprotonated, contributed equally to the physical property being observed:
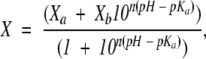 |
(5) |
where n is the Hill coefficient, which gives a measurement of the cooperativity of the transition; X is the physical property being observed (ellipticity, 〈λ〉, the maximum wavelength, Ve or the maximum wavenumber of the FTIR band); Xa is the physical property being observed at low pHs; Xb is the physical property observed at high pHs; and pKa is the apparent pK of the titrating group. The apparent pKa reported was obtained from three measurements for each technique. In all experimental data in this work, the n value was ~1.0.
Chemical denaturation data were obtained by following the change in the ellipticity at 222 nm in CD experiments. The denaturation data were fitted to the two-state equation:
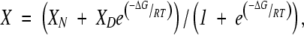 |
(6) |
where XN = αN + βN[D] and XD = αD + βD[D] are the corresponding fractions of the folded and the unfolded states, respectively, for which a linear relationship with denaturant is usually assumed, and X is the physical property (ellipticity) being observed, R is the gas constant, and T is the temperature in K. The chemical denaturation curves were analyzed by using the LEM: ΔG = m([D] 50% − [D]) (Pace and Laurents 1989; Pace and Scholtz 1997), where ΔG is the free energy of denaturation, [D] is the denaturant concentration, and [D]50% is that at the midpoint of the transition.
The change in free energy upon temperature in Equation 6 is given by the Gibbs-Helmholtz expression:
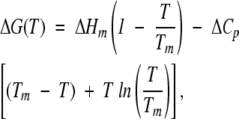 |
(7) |
where ΔHm is the van’t Hoff enthalpy change. Substitution of this expression in Equation 6, leads to the calculation of ΔHm, Tm, and ΔCp of the thermal unfolding experiments. In the FTIR experiments, where a concentration-dependent behavior was observed (see Results), the free energy is given by (Backmann et al. 1998):
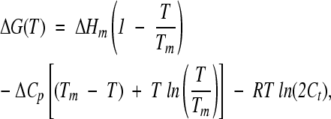 |
(8) |
where Ct is the molar concentration of the protein expressed in dimer equivalents.
Fitting by nonlinear least-squares analysis to the above equations was carried out by using the general curve fit option of Kaleidagraph (Abelbeck software) on a PC computer. A global fitting analysis of the far and the near-UV CD data, at 20 μM and 200 μM of protein concentration, was also carried out as described elsewhere (Irún et al. 2001). These experiments were chosen because of their better signal-to-noise ratio. The results obtained by both fitting procedures agree within the experimental uncertainty (see Discussion).
Size-exclusion chromatography (SEC)
Analytical SEC experiments were carried out by using an analytical gel filtration Superdex 75 HR 16/60 column (Amersham Biosciences) running on an AKTA FPLC system at 298 K. Flow rates of 1 mL/min were used and aliquots of 100 μL were loaded into the column after equilibration. The column was equilibrated with four column volumes of elution buffer (25 mM potassium phosphate [pH 7.3], containing 150 mM NaCl to avoid nonspecific interactions with the column). The column was calibrated using the gel filtration low molecular weight calibration kit (Amersham Biosciences). The standards used and their corresponding Stokes radii were ribonuclease A (16.4 Å), chymotrypsinogen (20.9 Å), ovoalbumin (30.5 Å), and bovine serum albumin (35.5 Å) (Hinkle et al. 1999). Protein elution volumes, Ve, were monitored by following absorbance at 280 nm. The CA-C concentration was in all cases either 20 μM or 200 μM. The reported Ve is the result of four independent measurements with fresh new samples.
Nuclear magnetic resonance spectroscopy
1D-1H NMR experiments were carried out in a Bruker DRX-500 spectrometer at 298 K with 32 K data points and using the WATERGATE sequence (Piotto et al. 1992) to eliminate the water signal. Typically, 512 scans were acquired, and the spectral width was 6000 Hz in all cases. CA-C concentration was 200 μM at pH 7 in 25 mM phosphate buffer in 90% H2O/10% 2H2O or at pH 3 in deuterated acetate buffer 25 mM in 90% H2O/10% 2H2O. No corrections were done for isotope effects. Thermal denaturation experiments at both pHs were also carried out at 0.9 mM of protein concentration. No differences were observed either in the chemical shifts or in their temperature behavior at 0.9 mM when compared with those at 0.2 mM (data not shown).
The spectra were processed using the BRUKER-XWINNMR software working on a PC workstation. An exponential window function and polynomial base line corrections were applied. The final 1D-1H NMR data contained 64,000 data points. 1H chemical shifts were quoted relative to internal TSP.
Thermal denaturation FTIR experiments
Protein was dried in a Speed Vac concentrator (Savant) and dissolved in deuterated buffer at the desired pH. The buffer was composed of 0.1 M NaCl, 0.1 mM ethylenediaminetetracetate, 0.02% NaN3, 10 mM sodium acetate, 10 mM N-(1-morpholino)- propane-sulfonic acid, and 10 mM 3-(cyclohexylamino)-1-propane- sulfonic acid. No corrections were done for the isotope effects in the measured pH. Samples of CA-C, at a final concentration between 0.53 mM and 4 mM, were placed amid a pair of CaF2 windows separated by a 50 μm-thick spacer in a Harrick demountable cell. Spectra were acquired on a Bruker FTIR-66S instrument equipped with a deuterated triglycine sulphate detector and fitted with a water bath. The cell container was continuously filled with dry air. Usually 50 scans per sample were taken, averaged, apodized with a Happ-Genzel function, and Fourier transformed to give a final resolution of 2 cm−1. The contributions of buffer spectra were subtracted, and the resulting spectra were used for analysis. The scanning rate was 50K/h, and spectra were acquired every 2.5K. At pH 7, the range of explored temperatures was 280–370 K, and the sigmoidal curve was fully reversible only for the low-temperature transition (see Results). The thermal experiments, carried out to detect the low-temperature transition, were acquired up to temperatures not higher than 325 K. Hence, the first thermal denaturation was fully reversible at most of the pHs (see Results). Protein concentration was 1 mM. The thermal transitions were analysed by taking the 3/4 of the height width of the band; similar results were obtained by measuring the half of the width of the height band.
Acknowledgments
This work was supported by grants from the Ministerio de Sanidad y Consumo (MSC) (FIS 01/0004-02) and Generalitat Valenciana (GV04B-402) and by an institutional grant from the Urbasa Group to the Instituto de Biología Molecular y Celular to J.L.N.; from the Spanish Ministerio de Educación y Ciencia (MEC) (BFU2004-01411) to J.S.; and from the MSC (FIS 01/0004–01) and Ministerio de Ciencia y Tecnología (BIO2003–04445) to M.G.M.; and by an institutional grant from the Fundación Ramón Areces to the Centro de Biología Molecular “Severo Ochoa.” M.C.L.-M. was the recipient of a predoctoral fellowship from MSC. F.N.B. and M.B. were recipients of two predoctoral fellowships from the Ministerio de Educación, Cultura y Deporte. We thank Dr. Claudio Cavasotto for the calculations of the solvent-accessible surface area of folded CA-C. We deeply thank May García, María del Carmen Fuster, Javier Casanova, María T. Garzón, Helena López, and Eva Martínez for excellent technical assistance. We thank the two anonymous reviewers for their suggestions, critical thoughts, and improvements to the manuscript.
Abbreviations
ASA, accessible surface area
ΔASAtotal, the total accessible surface area exposed upon unfolding
ANS, 8-anilino-1-naphtalenesulfonate
CA, capsid protein of HIV (p24)
CA-C, C-terminal domain of CA
CA-N, N-terminal domain of CA
CD, circular dichroism
DSC, differential scanning calorimetry
FTIR, Fourier transform infrared spectroscopy
GdmHCl, guanidinium chloride
[GdmHCl]1/2, the GdmHCl concentration at the midpoint of the chemical denaturation
ΔCp, the heat capacity change of unfolding
ΔHm, the thermal enthalpy change at the denaturation midpoint
ΔS, the entropy change
HIV, human immunodeficiency virus
LEM, linear extrapolation model
NMR, nuclear magnetic resonance
SEC, size-exclusion chromatography
TSP, 3-(trimethylsilyl) propionic acid-2,2,3,3-2H4-sodium salt
Tm, the thermal denaturation midpoint
UV, ultraviolet
Ve, elution volume in SEC experiments.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041324305.
References
- Anderson, D.E., Becktel, W.J., and Dahlquist, F.W. 1990. pH-induced denaturation of proteins: A single salt bridge contributes 3–5 kcal/mol to the free energy of folding of T4 lysozyme. Biochemistry 29 2403–2408. [DOI] [PubMed] [Google Scholar]
- Backmann, J., Schäfer, G., Wyns, L., and Bönisch, H. 1998. Thermodynamics and kinetics of unfolding of the thermostable trimeric adenylate kinase from the archeon Sulfolobus acidocaldarius. J. Mol. Biol. 284 817–833. [DOI] [PubMed] [Google Scholar]
- Borsetti, A., Öhagen, Å., and Göttlinger, H.G. 1998. The C-terminal half of the human immunodeficiency virus type 1 Gag precursor is sufficient for efficient particle assembly. J. Virol. 72 9313–9317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos, L.A., Bueno, M., López-Llano, J., Jiménez, M.A., and Sancho, J. 2004. Structure of stable protein folding intermediates by equilibrium φ-analysis: The apoflavodoxin thermal intermediate. J. Mol. Biol. 344 239–255. [DOI] [PubMed] [Google Scholar]
- Cantor, C.R. and Schimmel, P.R. 1980. Biophysical chemistry. W.H. Freeman, New York.
- Chen, H.A., Pfuhl, M., McAlister, M.S.B., and Driscoll, P.C. 2000. Determination of pKa values of carboxyl groups in the N-terminal domain of rat CD2: Anomalous pKa of a glutamate on the ligand binding surface. Biochemistry 39 6814–6824. [DOI] [PubMed] [Google Scholar]
- Clarke, J. and Fersht, A.R. 1993. Engineered disulfide bonds as probes of the folding pathway of barnase: Increasing the stability of proteins against the rate of denaturation. Biochemistry 32 4322–4329. [DOI] [PubMed] [Google Scholar]
- del Álamo, M. and Mateu, M.G. 2005. Electrostatic repulsion, compensatory mutations, and long-range non-additive effects at the dimerization interface of the HIV capsid protein. J. Mol. Biol. 345 893–906. [DOI] [PubMed] [Google Scholar]
- del Álamo, M., Neira, J.L., and Mateu, M.G. 2003. Thermodynamic dissection of a low-affinity protein–protein interface involved inhuman immunodeficiency virus assembly. J. Biol. Chem. 278 27923–27929. [DOI] [PubMed] [Google Scholar]
- Ding, K., Louis, J.M., and Gronenborg, A.M. 2004. Insights into conformation and dynamics of protein GB1 during folding and unfolding by NMR. J. Mol. Biol. 335 1299–1307. [DOI] [PubMed] [Google Scholar]
- Dorfman, T., Bukovsky, A., Öhagen, Å., Hoglund, S., and Göttlinger, H.G. 1994. Functional domains of the capsid protein of human immunodeficiency virus. J. Virol. 68 8180–8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich, L.S., Liu, T., Scarlata, S., Chu, B., and Carter, C.A. 2001. HIV-1 capsid proteins forms spherical (immature-like) and tubular (mature-like) particles in vitro: Structure switching by pH-induced conformational changes. Biophys. J. 81 586–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forshey, B.M., von Schwedler, U.K., Sundquist, W.I., and Aiken, C. 2002. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J. Virol. 76 5667–5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsyth, W.R. and Robertson, A.D. 2000. Insensitivity of perturbed carboxyl pKa values in the ovomucoid third domain to charge replacement at a neighboring residue. Biochemistry 39 8067–8072. [DOI] [PubMed] [Google Scholar]
- Fuller, T.W., Wilk, T., Gowen, B.E., Kräuslich, H.G., and Vogt, V.M. 1997. Cryo-electron microscopy reveals ordered domains in the immature HIV-1 particle. Curr. Biol. 7 729–738. [DOI] [PubMed] [Google Scholar]
- Gamble, T.R., Vajdos, F.F., Yoo, S., Worthylake, D.K., Houseweart, M., Sundquist, W.I., and Hill, C.P. 1996. Crystal structure of human cyclophylin A bound to the amino terminal domain of HIV-1 capsid. Cell 87 1285–1294. [DOI] [PubMed] [Google Scholar]
- Gamble, T.R., Yoo, S., Vajdos, F.F., von Schwedler, U.K., Worthylake, D.K., Wang, H., McCutcheon, J.P., Sundquist, W.I., and Hill, C.P. 1997. Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science 278 849–853. [DOI] [PubMed] [Google Scholar]
- Ganser, B.K., Li, S., Kliskho, V.Y., Finch, J.T., and Sundquist, W.I. 1999. Assembly and analysis of conical models for the HIV-1 core. Science 283 80–83. [DOI] [PubMed] [Google Scholar]
- Gelderblom, H.R. 1991. Assembly and morphology of HIV: Potential effect of structure on viral function. AIDS 5 617–638. [PubMed] [Google Scholar]
- Gitti, R.K., Lee, B.M., Walker, J., Summers, M.F., Yoo, S., and Sundquist, W.I. 1996. Structure of the amino-terminal core domain of the HIV-1 capsid protein. Science 273 231–235. [DOI] [PubMed] [Google Scholar]
- Groß, I., Hohenberg, H., and Kräulisch, H.G. 1997. In vitro assembly properties of purified bacterially expressed capsid proteins of human immunodeficiency virus. Eur. J. Biochem. 249 592–600. [DOI] [PubMed] [Google Scholar]
- Groß, I., Hohenberg, H., Huckangel, C., and Kräulisch, H.G. 1998. N-terminal extension of human immunodeficiency virus capsid protein converts the in vitro assembly phenotype from tubular to spherical particles. J. Virol. 72 4798–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groß, I., Hohenberg, H., Wilk, T., Wiegers, K., Grättinger, M., Müller, B., Fuller, S., and Kräulisch, H.G. 2000. A conformational switch controlling HIV-1 morphogenesis. EMBO J. 19 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinkle, A., Goranson, A., Butters, C.A., and Tobacman, L.S. 1999. Roles for the troponin tail domain in thin filament assembly and regulation. A deletional study of cardiac troponin T. J. Biol. Chem. 274 7157–7164. [DOI] [PubMed] [Google Scholar]
- Honig, B. and Yang, A.S. 1995. Free energy balance in protein folding. Adv. Protein Chem. 46 27–58. [DOI] [PubMed] [Google Scholar]
- Irún, M.P., García-Mira, M.M., Sánchez-Ruiz, J.M., and Sancho, J. 2001. Native hydrogen bonds in a molten globule: The apoflavodoxin thermal intermediate. J. Mol. Biol. 306 877–888. [DOI] [PubMed] [Google Scholar]
- Jaenicke, R. and Lillie, H. 2000. Folding and association of oligomeric and multimeric proteins. Adv. Protein Chem. 53 329–401. [DOI] [PubMed] [Google Scholar]
- Johnson, J.E. 1996. Functional implications of protein–protein interaction in icosahedral viruses. Proc. Natl. Acad. Sci. 93 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly, S.M. and Price, N.C. 2000. The use of circular dichroism in the investigation of protein structure and function. Curr. Protein Pept. Sci. 1 349–384. [DOI] [PubMed] [Google Scholar]
- Lakowicz, J.R. 1999. Principles of fluorescence spectroscopy, 2nd ed. Plenum Press, New York.
- Lanman, J., Sexton, J., Sakalian, M., and Prevelige Jr., P.E. 2002. Kinetic analysis of the role of intersubunit interactions in human immunodeficiency virus type 1 capsid protein assembly in vitro. J. Virol. 76 6900–6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, S., Hill, C.P., Sundquist, W.I., and Finch, J.T. 2000. Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature 407 409–413. [DOI] [PubMed] [Google Scholar]
- Luo, J., Iwakura, M., and Matthews, C.R. 1995. Detection of an intermediate in the thermal unfolding of a cysteine-free form of dihydrofolate from Escherichia coli. Biochemistry 34 10669–10675. [DOI] [PubMed] [Google Scholar]
- Luo, Y., Kay, M., and Baldwin, R.L. 1997. Co-operativity of folding of apomyoglobin pH 4 intermediate studied by glycine and proline mutations. Nat. Struct. Biol. 4 925–930. [DOI] [PubMed] [Google Scholar]
- Mach, H., Volkin, D.B., Burke, C.J., and Middaugh, C.R. 1995. Ultraviolet absorption spectroscopy. In Protein stability and folding (Methods in molecular biology, Vol. 40) (ed. B.A. Shirley), pp. 91–114. Humana Press, Towota, NJ. [DOI] [PubMed]
- Maldonado, S., Jiménez, M.A., Langdon, G.M., and Sancho, J. 1998. Cooperative stabilization of a molten-globule apoflavodoxin fragment. Biochemistry 37 10589–10596. [DOI] [PubMed] [Google Scholar]
- Mann, C.J. and Matthews, C.R. 1993. Structure and stability of an early folding intermediate of Escherichia coli trp aporepressor measured by far-UV stopped-flow circular dichroism and 8-anilino-1-naphthalene sulfonate binding. Biochemistry 32 5282–5290. [DOI] [PubMed] [Google Scholar]
- Marky, L.A. and Breslauer, K.J. 1987. Calculating thermodynamic data for transitions of any molecularity from equilibrium melting curves. Biopolymers 26 1601–1620. [DOI] [PubMed] [Google Scholar]
- Mateu, M.G. 2002. Conformational stability of dimeric and monomeric forms of the C-terminal domain of human immunodeficiency virus. J. Mol. Biol. 318 519–531. [DOI] [PubMed] [Google Scholar]
- Misselwitz, R., Hausdorf, G., Welfe, K., Höhne, W.E., and Welfe, H. 1995. Conformation and stability of recombinant HIV-1 capsid protein p24 (rp24). Biochem. Biophys. Acta 1250 9–18. [DOI] [PubMed] [Google Scholar]
- Momany, C., Kovari, L.C., Prongay, A.J., Keller, W., Gitti, R.K., Lee, B.M., Gorbalenya, A.E., Tong, L., McClure, J., Ehrlich, L.S., et al. 1996. Crystal structure of the dimeric HIV-1 cpasid protein. Nat. Struct. Biol. 3 763–770. [DOI] [PubMed] [Google Scholar]
- Neet, K.E. and Timm, D.E. 1994. Conformational stability of dimeric proteins: Quantitative studies by equilibrium denaturation. Protein Sci. 3 2167–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace, C.N. and Laurents, D.V. 1989. A new method for determining the heat capacity change for protein folding. Biochemistry 28 2520–2525. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. and Scholtz, J.M. 1997. Measuring the conformational stability of a protein. In Protein structure (ed. T.E. Creighton), 2nd ed., pp. 253–259. Oxford University Press, Oxford.
- Piotto, M., Saudek, V., and Sklenar, V. 1992. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2 661–665. [DOI] [PubMed] [Google Scholar]
- Ptitsyn, O.B. 1995. Molten globule and protein folding. Adv. Protein Chem. 47 83–229. [DOI] [PubMed] [Google Scholar]
- Robertson, A.D. and Murphy, K.P. 1997. Protein structure and the energetics of protein stability. Chem. Rev. 97 1251–1267. [DOI] [PubMed] [Google Scholar]
- Rosé, S., Hensley, P., O’Shanessy, D.J., Culp, J., Debouck, C., and Chaiken, I. 1992. Characterization of HIV-1 p24 self-association using analytical chromatography. Proteins 13 112–119. [DOI] [PubMed] [Google Scholar]
- Royer, C.A. 1995. Fluorescence spectroscopy. In Protein stability and folding (Methods in molecular biology Vol. 40) (ed. B.A. Shirley), pp. 65–89. Humana Press, Towota, NJ. [DOI] [PubMed]
- Semisotnov, G.V., Rodionova, N.A., Razgulyaev, O.I., Uversky, V.N., Gripas, A.F., and Gimanshin, R.I. 1991. Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers 31 119–128. [DOI] [PubMed] [Google Scholar]
- Steif, C., Weber, P., Hinz, H.J., Flossdorf, J., Cesareni, G., and Kokkinidis, M. 1993. Subunit interactions provide a significant contribution to the stability of the dimeric four-α-helical bundle protein ROP. Biochemistry 32 3867–3876. [DOI] [PubMed] [Google Scholar]
- Stryer, L. 1965. The interaction of a naphthalene sulfonate dye with apomyoglobin and apohemoglobin: A fluorescent probe of non-polar binding site. J. Mol. Biol. 13 482–495. [DOI] [PubMed] [Google Scholar]
- Tang, K.S., Guralnick, B.J., Wong, W.K., Fersht, A.R., and Itzhaki, L.S. 1999. Stability and folding of the tumor suppressor protein p16. J. Mol. Biol. 285 1869–1886. [DOI] [PubMed] [Google Scholar]
- Tang, S., Murakami, T., Agresta, B.E., Campbell, S., Freed, E.O., and Levin, J.G. 2001. Human immunodeficiency virus type 1 N-terminal capsid mutants that exhibit aberrant core morphology are blocked in initiation of reverse transcription in infected cells. J. Virol. 75 9357–9366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schwedler, U.K., Stemmler, T.L., Kliskho, V.Y., Li, S., Albertine, K.H., Davis, D.R., and Sundquist, W.I. 1998. Proteolytic refolding of the HIV-1 capsid protein amino-terminus facilitates viral core assembly. EMBO J. 17 1555–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C.T. and Barklis, E. 1993. Assembly, processing and infectivity of human immunodeficiency virus type 1 Gag mutants. J. Virol. 67 4264–4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welker, R., Hohenberg, H., Tessmer, U., Huckhagel, C., and Kräuslich, H.G. 2000. Biochemical and structural analysis of isolated matured cores of human immunodeficiency virus type I. J. Virol. 74 1168–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills, J.W. and Craven, C. 1991. Form, function and use of retroviral gag proteins. AIDS 5 639–654. [DOI] [PubMed] [Google Scholar]
- Woody, R.W. 1995. Circular dichroism. Methods Enzymol. 246 34–71. [DOI] [PubMed] [Google Scholar]
- Worthylake, D.K., Wang, H., Yoo, S., Sundquist, W.I., and Hill, C.P. 1999. Structures of the HIV-1 capsid protein dimerization domain at 2.6 Å resolution. Acta Crystallogr. D Acta Crystallogr. 55 85–92. [DOI] [PubMed] [Google Scholar]
- Wüthrich, K. 1986. NMR of proteins and nucleic acids. John Wiley & Sons, New York.
- Zweifel, M.E. and Barrick, D. 2002. Relationships between the temperature dependence of solvent denaturation and the denaturant dependence of protein stability curves. Biophys. Chem. 101–102 221–237. [DOI] [PubMed] [Google Scholar]