

Abstract
The serine penicillin-recognizing proteins have been extensively studied. They show a wide range of substrate specificities accompanied by multidomain features. Their adaptation capacity has resulted in the emergence of pathogenic bacteria resistant to β-lactam antibiotics. The most divergent enzymatic activities in this protein family are those of the Ochrobactrum anthropi D-aminopeptidase and of the Streptomyces R61 D,D-carboxypeptidase/transpeptidase. With the help of structural data, we have attempted to identify the factors responsible for this opposite specificity. A loop deletion mutant of the Ochrobactrum anthropi D-aminopeptidase lost its original activity in favor of a new penicillin-binding activity. D-aminopeptidase activity of the deletion mutant can be restored by complementation with another deletion mutant corresponding to the noncatalytic domain of the wild-type enzyme. By a second step site-directed mutagenesis, the specificity of the Ochrobactrum anthropi D-aminopeptidase was inverted to a D,D-carboxypeptidase specificity. These results imply a core enzyme with high diversity potential surrounded by specificity modulators. It is the first example of drastic specificity change in the serine penicillin-recognizing proteins. These results open new perspectives in the conception of new enzymes with nonnatural specificities. The structure/specificity relationship in the serine penicillin-recognizing proteins are discussed.
Keywords: directed evolution; serine penicillin-recognizing proteins; penicillin binding proteins; Ochrobactrum anthropi D-aminopeptidase; Streptomyces R61 D,D-peptidase
Although less abundant than their L-counterparts, D-amino acids are encountered in a large number of compounds, exhibiting a wide range of biological activities from anti-microbial peptides to neuropeptide hormones where they can result from a post-transcriptional isomerization of an L-residue near the N terminus of the peptide (Kreil 1997; Volkmann and Heck 1998).
By contrast, the D-residues found in peptidoglycan are produced by the action of a racemase on the free amino acid. Peptidoglycan is a major constituent of the bacterial cell wall, and β-lactam antibiotics interfere with its biosynthesis (Frère and Joris 1985). The Streptomyces R61 and Actinomadura R39 (formerly Streptomyces) D,D-carboxypeptidase/transpeptidase have been widely studied as the prototype of penicillin-sensitive enzymes and of D-Ala-D-Ala carboxypeptidases/transpeptidases (Dusart et al. 1973; Perkins et al. 1973; Leyh-Bouille et al. 1977; Joris et al. 1988), but in addition to D,D-carboxypeptidases, the large family of penicillin-recognizing, active-site serine enzymes (PRPs) contains a large number of proteins exhibiting a wide variety of activities: β-lactamases, D,D-endopeptidases (Asano et al. 1996), D,L-endopeptidases (Bourne et al. 2001), D-amidases (Komeda and Asano 2000), D-esterases (Petersen et al. 2001; Wagner et al. 2002), and D-aminopeptidases (Asano et al. 1989; Fanuel et al. 1999).
The most divergent enzymatic activities in this protein family are those of the Ochrobactrum anthropi D-aminopeptidase (DAP) and of the Streptomyces R61 D,D-carboxypeptidase/transpeptidase (R61) (Fig. 1 ▶). Representative substrates of R61 and DAP are, respectively, Nα,Nα-Ac2-L-Lys-D-Ala-D-Ala and D-Ala-L-Ala-L-Ala (D-Ala-paranitroanilide is more suitable for kinetics experiments). Superposition of the substrates cleavages sites (between the two D-Alanyl residues) implies that the two enzymes stabilize the extremities of their substrates in opposite manners (Asano et al. 1992; Bompard-Gilles et al. 2000).
Figure 1.

Specificity of the D-amino and D,D-carboxipeptidases.
Although the acylation/deacylation mechanism seems to be well established (Frère et al. 1976a; Frère and Joris 1985), little is known about the factors which determine the specificity since site-directed mutagenesis modifications often result in loss of activity or protein instability rather than significant specificity modification. With the help of structural data, we have attempted to identify the factors responsible for this opposite specificity.
DAP is composed of three domains (Fig. 2A ▶). The backbone of its N-terminal domain (DAP-A) is remarkably similar to that of the R61 enzyme (Fig. 2B,C ▶), and the active-site serine residues are found in corresponding positions (Fig. 2D ▶). The two C-terminal domains, which have no equivalent in R61, are two β-barrels (DAP-B and DAP-C) each constituted of eight anti-parallel β-strands (Fig. 2A ▶). This folding motif is usually found in proteins which bind and transport hydrophobic ligands (LaLonde et al. 1994), and are classified as streptavidin-like structures. Domain C also contains a 13-residue loop (the 476–486 γ-loop), which protrudes into the active site, resulting in strong steric hindrance and contributing an aspartyl residue, which might be involved in the D-aminopeptidase specificity by interacting with the free alkylammonium group of the substrate (Fig. 2A,B,D ▶). In turn, the R61 Arg285 residue, which is expected to interact with the carboxylate of the substrate, is replaced by an Asn275 in DAP-A (Fig. 2D ▶) (Kelly and Kuzin 1995; Bompard-Gilles et al. 2000).
Figure 2.

(A) Structure of the DAP monomer. Stereo view of the DAP monomer. Domain A of DAP (the R61-like domain) is in blue and in light blue. Domains B and C of DAP are each composed of an eight-stranded β-barrel and are, respectively, in green and red. The DAP-C γ-loop is in orange. (B,C) Comparison of the folds of (C) R61 and (B) domain A of DAP. The DAP L3-loop between the α4 and α5 helices are diverging comparatively to R61 in order to accommodate the γ-loop (note that the L3-loop sometimes contains secondary structure elements; the conventional numbering of the secondary structure is that of Joris et al. [1991]). The presence of the L3-loop imposes a characteristic χ structure indicated by an arrow. The L3-loop regions are shaded. The structure of R61 is that determined in the presence of cephalosporin (McDonough et al. 2002). The Cα trace of the γ-loop is in gray and the side chain of Asp481 is shown using the standard coloring convention. (D) DAP and R61 substrate-binding sites comparison. Stereo view of the superposition of the substrate-binding sites of DAP (carbons atoms in green for domain A, red for Asp481, and gray for the γ-loop Cα trace) and R61 (carbons atoms in yellow except Arg485 in blue). Labels indicate DAP/R61 residue numbers. SwissPdb Viewer program was used to fit the Cα of the DAP/R61 residues (S62/S62, K65/K65, Y153/Y159, N155/N161, R285/N275, H287/H298, G288/T299, G289/G300, and A290/T301). The figures were produced using the MOLSCRIPT (Kraulis 1991) and Raster3D (Merritt 1994) programs.
To better understand the factors which underlie the specificities of both enzymes, we have produced the single DAP-A domain, which turned out to be inactive but could be complemented by the coexpressed B and C domains. The γ-loop of domain C was replaced by a single glycyl residue, and the Asn275Arg mutation was performed. This final modified protein exhibited a strongly decreased D-aminopeptidase and significant D,D-carboxypeptidase activities.
Results
Expression of DAP-A and DAP-BC
The expression of DAP-A (residues 1–333) was tested at 18°C, 28°C, and 37°C, with IPTG concentrations ranging from 0.1 to 1.0 mM. In all conditions, the protein was found in inclusions bodies (Fig. 3 ▶, lanes 1,2), and could not be detected in the soluble fraction, which was also devoid of D-aminopeptidase activity.
Figure 3.

Twelve percent SDS-polyacrylamide slab gel electrophoresis of variant DAP forms. MW, molecular mass standards of known sizes (10, 15, 25, 37, 50, and 75 kDa); lane 1, DAP-A insoluble fraction; lane 2, DAP-A soluble fraction; lane 3, soluble fraction of coexpressed DAP-A and DAP-BC; lane 4, insoluble fraction of coexpressed DAP-A and DAP-BC; lane 5, copurified DAP-A and DAP-BC; lane 6, purified DAP-BC.
By contrast, the DAP-BC (residues 339–520) fragment added with a His6 tail (DAP-BC-His) was recovered in the soluble fraction and could be purified with the help of the Ni-NTA column (Fig. 3 ▶, lane 6). As expected, the purified DAP-BC protein was devoid of D-aminopeptidase activity, but a low activity could be detected when the purified DAP-BC-His was added to a crude total extract of the DAP-A producing cells.
Coexpression of DAP-A and DAP-BC
Since the addition of DAP-BC to DAP-A failed to regenerate a high enzymatic activity, probably due to the accumulation of DAP-A in inclusions bodies, it was attempted to coexpress the two parts of the protein. To do so, a sequence containing a stop (TGA) codon, an RBS (AGGA), and an ATG codon was inserted into the gene coding for the wild-type enzyme, allowing the expression of the two parts of the DAP protein from a single promotor. The resulting plasmid (pDML1113) was used for coexpression of the DAP-A and DAP-BC-His proteins. Upon coexpression of the two parts of the DAP wild-type protein, the major proportion of the DAP-A fragment was again recovered in inclusion bodies, but some of it was present in the soluble fraction, indicating that the presence of DAP-BC-His allowed a better folding of the DAP-A fragment (Fig. 3 ▶, lanes 3,4). The two proteins copurified on the Ni-NTA column, which demonstrates a rather strong interaction between the two parts of the protein. According to densitography analysis of the SDS-PAGE, the molar ratio in the complex was roughly 1:1 (Fig. 3 ▶, lane 5). With D-Ala-p-nitroanilide as a substrate, the complex exhibited Km and kcat values of 5.1 mM and 214 sec−1, respectively. Interestingly, when compared to the wild-type protein, the kcat/Km value was significantly reduced, but the kcat value was not very different (Table 1). The 10-fold increase of the Km value might reflect a slight change in the active-site architecture.
Table 1.
Kinetic parameters of wild-type and variant DAP forms with D-Ala-p-nitroanilide as a substrate
| Enzymes | Km (mM) | kcat (sec−1) | kcat/Km (M−1sec−1) |
| DAP-WT | 0.46 | 580 | 1,260,000 |
| DAP-A + DAP-BC-Hisa | 5.10 | 210 | 42,000 |
| DAP-475G487 | N.D. | N.D. | 0.9 |
| DAP-475G487 + DAP-BC-Hisb | 0.51 | 33 | 65,000 |
| DAP-475G487-N275R | N.D. | N.D. | 0.6 |
D-aminopeptidase activity was measured using D-Ala-paranitroanilide as a substrate. The R39 and R61 enzymes and the purified DAP-BC-His fragment did not exhibit a measurable activity. Similarly, no hydrolysis of the substrate was detected upon incubation with a total lysate of DAP-A producing cells. The experiments were performed in 20 mM Tris-HCl buffer (pH 8.0) at 30°C. Standard deviations did not exceed 10%.
a Coexpressed.
b After overnight incubation at 4°C at a 1/8 molar ratio.
N.D., not determined.
The DAP-475G487 mutant
As suggested by Bompard-Gilles et al. (2000), it was suspected that the γ-loop (residues 476–486) is important for DAP specificity for two reasons: There is a possible interaction between residue Asp481 and the N-terminal extremity of the D-aminopeptidase peptide substrate, and the γ-loop results in steric hindrance with the D,D-carboxypeptidase substrate. The DAP-475G487 mutant was produced as a soluble protein and could be purified according to the protocol used for the wild-type enzyme. It hydrolyzed D-Ala-p-nitroanilide >1 million-fold more slowly than the wild-type DAP enzyme, but it exhibited a low but significant D,D-carboxypeptidase activity (Tables 1, 2). Deletion of the γ-loop thus represented a first step toward the inversion of specificity. When compared to the R61 enzyme, the 10,000-fold difference in the kcat/Km values was due more to kcat (550-fold lower) than to Km (18-fold higher).
Table 2.
Kinetic parameters of various enzymes and variants with the peptide substrates Nα-Ac-L-Lys-D-Ala-D-Ala and Nα,Nα-Ac2-L-Lys-D-Ala-D-Ala
| Nα-Ac-L-Lys-D-Ala-D-Ala | Nα,Nα-Ac2 -L-Lys-D-Ala-D-Ala | |||||
| Enzymes | Km (mM) | kcat (sec−1) | kcat /Km (M−1sec−1) | Km (mM) | kcat (sec−1) | kcat /Km (M−1sec−1) |
| R39a | 0.2 | 32 | 160,000 | 0.8 | 17.5 | 20,000 |
| R61a | 15 | 0.25 | 17 | 12 | 55 | 4600 |
| DAP-WT | <3 nmol/min/mgb | <3 nmol/min/mgb | ||||
| DAP- 475G487 | <3 nmol/min/mgb | 200 | 0.09 | 0.45 | ||
| DAP- 475G487-N275R | 185 | 0.2 | 1.1 | 49 | 0.62 | 13 |
D,D-carboxypeptidase activities were measured using Nα-Ac-L-Lys-D-Ala-D-Ala and Nα,Nα-Ac2-L-Lys-D-Ala-D-Ala as substrates. The activity of the DAP wild-type enzyme was not measurable, and DAP-475G487 did not exhibit a sufficient activity when using Nα-Ac-L-Lys-D-Ala-D-Ala as a substrate. The experiments were performed in 20 mM Tris-HCl buffer (pH 8.0) at 37°C. Standard deviation values did not exceed 10%.
aAs previously published (Frère and Joris 1985).
b Activity tested with 200 mM substrate. These maximum values would correspond to kcat values of 0.0015 sec−1 if Km is <<200mMand to a kcat /Km value of 0.005 M−1 sec−1 if Km is >>200 mM.
Similarly, a new penicillin-binding activity could be demonstrated with Flu-Gly-6APA (Fig. 4 ▶). Binding of the fluorescent penicillin was strongly decreased after preincubation in the presence of benzylpenicillin. The mutant also exhibited a very low β-lactamase activity with nitrocefin (Table 3).
Figure 4.

SDS-polyacrylamide slab gel electrophoresis and fluorography of wild-type DAP and DAP-475G487. Three micrograms of enzyme (wild-type DAP and DAP-475G487) were incubated with 0.5 mM Flu-Gly-6APA (a sample was preincubated for 90 min with 0.5 mM penicillin G) at 20°C in 50 mM phosphate buffer (pH 7.0). The complexes were revealed by fluorography.
Table 3.
Kinetic parameters of variant DAP forms with nitrocefin as a substrate
| Enzymes (DAP variants) | kcat (sec−1) | Km (μM) | K (μM) | k2 (sec−1) | k3 (sec−1) | k2/K (M−1 sec−1) |
| 475G487 | 0.0016a | 29a | 29 | 0.0016 | >0.0016 | 55 |
| 475G487-N275R | 0.0008a | 26a | 0.0016e | 50e | ||
| 0.00125b | 25b | 115e | 0.0058e | |||
| 0.0011c | 35c | 117f | 0.0037f | 32f | ||
| 9d |
β-Lactamase activity was measured using nitrocefin as a substrate. DAP-475G487 and DAP-475G487-N275R exhibited, respectively, classical and biphasic time courses. Wild-type DAP had no measurable activity. The experiments were performed in 50 mM sodium phosphate buffer (pH 7.0) at 30°C. Standard deviation values did not exceed 10%.
a From vss versus S0.
b,ckcat computed as k2k3 /(k2+k3) with k2=0.0058 sec−1 (b) or 0.0037 sec−1 (c)Km computed as k3K/(k2+k3) withk2=0.0058 sec−1 (b) or 0.0037 sec−1 (c).
d Measured as a Ki with Ac2-L-Lys-D-Ala-D-Ala as substrate.
e From time courses (Equations 1, 4).
f From extrapolated v0 and Equation 5.
Interestingly, after incubation of the mutant with the free DAP-BC-His fragment, the D-aminopeptidase activity was significantly recovered. This indicated that the natural fragment could displace the mutated one and partly regenerate the initial specificity of the enzyme (Table 1).
The double DAP-475G487-N275R mutant
It has been suggested that the R61 enzyme Arg285 residue which protrudes in the active site is important for the binding of the negative C-terminal extremity of the D,D-peptidase substrate (Bompard-Gilles et al. 2000; Llinas et al. 2005). The structures show that the DAP Asn275 residue lies in the same position as the R61 Arg285 residue (Bompard-Gilles et al. 2000). In order to improve the D,D-carboxypeptidase activity of the DAP-475G487 mutant, we performed the Asn275Arg mutation.
The D-aminopeptidase activity of this double DAP- 475G487-N275R mutant was also very low, but, when compared to the simple deletion mutant, its D,D-carboxypeptidase activity was increased about 30-fold, and it was, in consequence, 20-fold better as a D,D-carboxypeptidase than as D-aminopeptidase.
When the mono- and di-acetylated peptides were compared, the double mutant showed the same preference as R61, although much less marked for the diacetyl derivative, and it is thus quite different from the R39 D,D-peptidase, which prefers the mono-acetyl compound (Leyh-Bouille et al. 1972). As the simple deletion mutant, it exhibits a poor β-lactamase activity on nitrocefin. The time courses of the hydrolysis of nitrocefin by the two mutants were, however, quite different. With DAP-475G487, the time courses were linear, so that it can be concluded, that, if the three-step scheme applies, kcat=k2 and Km=K. By contrast, with DAP-475G487-N275R, the time courses were clearly biphasic. Presteady-state experiments were performed in the purpose of demonstrating a stoichiometric or quasi-stoichiometric burst and analysis of the steady-state phase allowed the derivation of the classical kcat and Km parameter. The values of v0, vss, and ka were determined at nitrocefin concentrations ranging from 20 μM to 160 μM, and the constants presented in Table 3 could be computed on the basis of Equations 1–6 and of the Henri-Michaelis equation for vss (see experimental procedures). Moreover, nitrocefin inhibited the hydrolysis of the tripeptide and, on the basis of a competitive model a Ki of 9 μM, could be calculated, not very different from the Km value. This indicated that the β-lactam and the peptides were hydrolyzed at the same active site.
Discussion
The “R61-like” domain A and domains BC of DAP
No production of the DAP domain A in a soluble form could be obtained. In contrast, the peptide containing the DAP domains BC was recovered in a soluble form and the protein was stable. To explain these different properties, the wild-type DAP enzyme might be considered as composed of three interacting protein pieces (DAP-A, DAP-B, and DAP-C). Hydrophobic interactions between domains A and C are necessary to obtain a correctly folded protein. In domain C, the hydrophobic core is buried in the cavity of the eight-stranded β-sheet. In contrast, the L3-loop hydrophobic DAP-A residues 113–119 are protruding (Bompard-Gilles et al. 2000). So the correct positioning of the DAP-A residues 113–119 is dependent on the presence of the DAP-C domain. This folding dependence is well demonstrated by the effect of DAP-BC on the solubility of DAP-A in the coproduction experiment. If the kinetic parameters of the DAP-A + DAP-BC complex are compared to those of the wild-type DAP, it appears that the continuity in the peptide chain is not absolutely essential for the activity. Moreover, the displacement of the γ-loop deleted domain C by its wild-type counterpart result in a partial restoration of the activity (DAP-475G487+DAP-BC). These data are in accordance with the hypothesis that DAP results from the fusion of genes encoding a dimer of a streptavidin-like protein (DAP-BC) and an R61-like enzyme (DAP-A). It can further be hypothesized that the PRPs core enzyme itself could result from a gene fusion. Indeed, coproduction of fragments of the TEM β-lactamase can induce the formation of an active protein (Galarneau et al. 2002).
The DAP γ-loop
The removal of 10 residues near the catalytic center and the Asn275Arg mutation did not significantly destabilize the protein. Replacement of the γ-loop (476–486) by a single glycyl residue decreased the D-aminopeptidase activity 106-fold but resulted in the appearance of a low but significant D,D-carboxypeptidase activity. Interestingly, a new penicillin binding activity was also detected. These results underline the central role of the γ-loop and of residue 275 in the determination of the catalytic properties of the enzyme. In the wild-type enzyme, steric hindrance due to the γ-loop is responsible for the absence of penicillin binding and D,D-carboxypeptidase activities. Conversely, the γ-loop, and in particular the Asp481 residue, are important for the D-aminopeptidase activity by contributing to the binding of the substrate positively charged N terminus.
The synthesis of pure peptides containing D-residues is important in industrial applications, but organic synthesis is sometimes too expensive or complex. The use of enzymes is envisaged, but the few enzymes with D-specificity are not always satisfying. The utility of the wild-type DAP enzyme in D-stereospecific aminolysis reactions is well documented (Kato et al. 1989). TheDAP γ-loop is a good candidate for future mutagenesis experiments in order to construct new D-specificity enzymes.
The R61 Arg275 residue
Addition of the Asn275Arg mutation to the deletion mutant DAP-475G487 increased the D,D-carboxypeptidase activity by a factor of 28 with practically no effect on the D-aminopeptidase activity. The arginyl residue positive charge interacts with the C-terminal negative charge of the peptide. The Asn275Arg mutation essentially reverses the specificity of the enzyme from D-aminopeptidase to D,D-carboxypeptidase. One can assume that residues Arg 198, 248, 244, and 250 in, respectively, PBP5, K15, the Staphylococcus aureus β-lactamases, and OXA-10, are equivalent to R61 Arg285. Although the Arg/active Ser distances are similar, the relative positions of the other conserved residues differ in the crystal structures. This work confirms the importance of the arginyl residue in the PRP family.
In order to try to improve the significant but low D,D-carboxypeptidase activity of the DAP-475G487- N275R mutant, it would be interesting to perform the Gly288Thr and Ala290Thr mutations in further work (Fig. 2D ▶). Interestingly, in R61, these residues (Gly288 and Ala290) are replaced by two threonines (Thr299 and Thr301), which are involved in the binding of cephalosporins (cephalothin and cefotaxime) (Kelly et al. 1989; Kuzin et al. 1995).
Conclusions
The removal of the DAP γ-loop combined with the Asn275Arg mutation has completely modified the enzyme specificity from a D-aminopeptidase to a D,D-carboxypeptidase. Interestingly, the new enzyme exhibits the same preference as R61 for the diacetyl vs the monoacetyl derivative of L-Lys-D-Ala-D-Ala and also covalently binds penicillins. Although the new D,D-carboxypeptidase kcat/Km value remains modest (13 M−1sec−1), it is worth mentioning that due to the high stability of the peptide substrate, the enzyme rate enhancement factor (Laws and Page 1989) is 2 × 107 versus 6 × 109 for R61 with the same substrate (Rhazi et al. 1999).
Materials and methods
Plasmids, bacterial strains, materials, oligonucleotide primers, and enzymes
The expression plasmid pET-28a(+) was purchased from Novagen. pDML1109 is a pUC18 plasmid containing the full-length DNA for DAP (Bompard-Gilles et al. 2000). Escherichia coli strains BL21(DE3) were supplied by Stratagene. Restriction endonucleases, Pfu DNA polymerase, and T4 DNA Ligase were purchased from Promega, D-Alanine-p-nitroanilide from Bachem, and Nitrocefin from Unipath. Nα-Ac-L-Lys-D-Ala-D-Ala and Nα,Nα-Ac2-L-Lys-D-Ala-D-Ala were those used previously (Nieto et al. 1973). Flu-Gly-6-Apa was synthesized as described by Lakaye et al.(Galleni et al. 1993). Oligonucleotides primers and dNTPs were from Eurogentech.
Site-directed mutagenesis, expression, and coexpression plasmids
The wild-type gene was amplified by PCR with oligonucleotide primers DAP-A/NcoI 5′-GGAATTCCATGGCCAAGTTTGA TACGTCTGCCC and DAP-BC/Stop/XhoI 5′-GGCTCGAG TCATGGCTGAACTCTCCTGTATTC using the pDML1109 plasmid as the template DNA. The PCR product was subcloned into pET28a yielding pDML1110. The DAP gene, deleted of the coding sequence for the two β-barrels (DAP-A or R61-like), was amplified by PCR with oligonucleotide primers DAP-A/NcoI and DAP-A/BamHI 5′-CGGGATCCTCACGATACACCCA GAGCAATATTCATCAG using the pDML1109 plasmid as the template DNA. The PCR product was cloned into the NcoI and BamHI sites of pET28a, yielding pDML1111. Two PCR fragments each containing the coding sequence for the two β-barrels, DAP-BC and RBS-DAP-BC, were obtained with oligonucleotide primers DAP-BC/NcoI 5′-CCCATGGCACG GGTGGAGGCTGATTCAGCATGG and DAP-BC/XhoI 5′- GGCTCGAGTGGCTGAACTCTCCTGTATTCAAC, DAP-BC/BamHI 5′-CGGGATCCATCATATGTCGCGGGTGGA GGCTGATTCAGCATGG and DAP-BC/XhoI 5′-GGCTCGA GTGGCTGAACTCTCCTGTATTCAAC, respectively. DAP-BC was subcloned into NcoI/XhoI sites of pET28a, yielding pDML1112. pDML1113 was constructed by cloning the RBS-DAP- BC fragment into the BamHI/XhoI site of pDML1111. In pDML1112 and pDML1113, a His6 Tag was added at the C-terminal end of DAP-BC to facilitate purification. The PCR fragment where the sequence coding for the 476–486 loop was replaced by that for a single glycyl residue was obtained with oligonucleotide primers DAP-A/NcoI and DAP-475G487 5′-CAAGCTTCCACTCCCCAGGCAGCAGCCAGACATCA GACCCGACAC using the pDML1109 plasmid as the template DNA. The fragment was subcloned into the NcoI/HindIII sites of pGem yielding pGem DAP-475G487 and pDML1110 yielding pDML1114. A single mutation was introduced into pGem DAP-475G487 with oligonucleotide primers Asn275Arg+ 5′-GGTTTTGGTCTCAGACTTCACGAAACAGGCGG and Asn275Arg- 5′-CCGCCTGTTTCGTGAAGTCTGAGACCA AAACC by the quick change method of Stratagene, yielding pGem DAP-475G487-N275R. pDML1115 was constructed by cloning the DAP-475G487-N275R fragment into the NcoI/HindIII site of pDML 1110. The resulting constructs were all verified by complete DNA sequencing.
Expression and coexpression of DAP-A and DAP-BC
E. coli BL21(DE3) cells were transformed separately with plasmids pDML 1111, pDML 1112, pDML 1113, pDML 1114, and pDML 1115, and grown at 37°C in 2XYT medium (16 g tryptone, 10 g yeast extract, 5 g NaCl for 1 L of medium) with kanamycin at a final concentration of 50 μg/mL. When the absorbance of the culture at 600 nm reached 1, expression of the protein(s) was induced by the addition of isopropyl-β-D-thiogalactoside (IPTG) to a final concentration of 1 mM. The cultures were further incubated at 28°C for 3.5 h. The cells were collected and suspended in lysis buffer (20mMsodium phosphate buffer, 5 mM MgCl2, 20 μg/mL DnaseI [pH 7.8]). The cell extracts were prepared using an LH-SGI Inceltech disruptor at 4°C. The soluble fractions were obtained by centrifugation at 30,000g for 30 min at 4°C. Proteins were analyzed by SDS-PAGE.
Purification and copurification of DAP-A and DAP-BC
The soluble fractions containing DAP-BC (pDML1111) or the two parts of the enzyme (DAP-A and DAP-BC, pDML1112) were applied to an Ni-NTA column. Washing buffers 1 (20 mM sodium phosphate buffer [pH 7.8]) and 2 (buffer 1+500 mM NaCl [pH 7.8]) were used to remove contaminants. Elution was performed with 100 mM imidazole (pH 7.8). The purified and copurified proteins behaved as homogeneous upon 12% SDS-PAGE. The quantity of purified proteins was 14 mg/L of culture (coexpression) and 4.5 mg/L of culture (DAP-BC).
Purification of DAP-475G487 and DAP-475G487-N275R
The DAP-475G487 and DAP-475G487-N275R proteins were purified as described for the wild-type enzyme (Bompard-Gilles et al. 2000).
Penicillin binding assay
Three micrograms of enzyme (DAP-WT and DAP-475G487) were incubated (0, 10, and 60 min) with 0.5 mM Fluoresceyl- Glycyl-6aminopenicillanic acid (Flu-Gly-6APA) with or without a 90-min preincubation with 0.5 mM penicillin G at 20°C in 50 mM phosphate buffer (pH 7.0). The complex was revealed by SDS-PAGE and fluorography on a Biorad Molecular Imager FX.
Enzyme assays
The D-Ala-D-Ala carboxypeptidase activity was determined using the D-amino acid oxidase method (Frère et al. 1976b).
The D-aminopeptidase activity was detected by monitoring the formation of p-nitroaniline from D-alanine-paranitroanilide at 405 nm in a 20 mM Tris buffer (pH 8.0) at 30°C.
The β-lactamase activity was tested by monitoring the hydrolysis of nitrocefin at 482 nm in 50 mM sodium phosphate buffer (pH 7.0) at 30°C.
Kinetic parameters
When detected, the hydrolysis of nitrocefin was either linear or exhibited a burst phase followed by a steady state. On the basis of the three-step scheme,
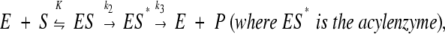 |
these two cases can be distinguished. If k3>k2, product formation is linear (and kcat=k2, Km=K), while the burst occurs if k2 is similar to or larger than k3. In this latter situation, values of the acylation (K, k2) and deacylation (k3) rate constants were computed by combining the presteady-state and steady-state data. Since the absorbance of ES* at 482nm can be expected to be close to that of P, the rate of β-lactam opening during the burst phase is
 |
(1) |
where
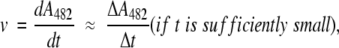 |
v0 is the extrapolated reaction rate at t=0 and kf is a pseudo-first- order rate constant.
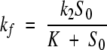 |
(2) |
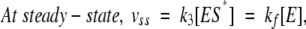 |
(3) |
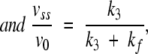 |
(4) |
From vss, the classical kcat and Km values could be derived. The values of v0 also allow the computation of k2 and K according to
 |
(5) |
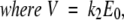 |
(6) |
Km can also be determined as a Ki using nitrocefin as an inhibitor of the tripeptide hydrolysis reaction.
Acknowledgments
M.D. and S.B. are fellows of the Fonds pour la Formation à la Recherche dans l’Industrie et l’Agriculture (F.R.I.A.) (Brussels, Belgium), B.J. is Research Associate of the Fonds National de la Recherche Scientifique (F.N.R.S.) (Brussels, Belgium). This work was supported by PAI P05/33 (Federal Government of Belgium) and grants 2.4508.01, 2.4521.01, and 2.4524.03 from the FRFC (Brussels, Belgium).
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051475305.
References
- Asano, Y., Nakazawa, A., Kato, Y., and Kondo, K. 1989. Properties of a novel D-stereospecific aminopeptidase from Ochrobactrum anthropi. J. Biol. Chem. 264 14233–14239. [PubMed] [Google Scholar]
- Asano, Y., Kato, Y., Yamada, A., and Kondo, K. 1992. Structural similarity of D-aminopeptidase to carboxypeptidase DD and β-lactamases. Biochemistry 31 2316–2328. [DOI] [PubMed] [Google Scholar]
- Asano, Y., Ito, H., Dairi, T., and Kato, Y. 1996. An alkaline D-stereo-specific endopeptidase with β-lactamase activity from Bacillus cereus. J. Biol. Chem. 271 30256–30262. [DOI] [PubMed] [Google Scholar]
- Bompard-Gilles, C., Remaut, H., Villeret, V., Prange, T., Fanuel, L., Delmarcelle, M., Joris, B., Frère, J., and Van Beeumen, J. 2000. Crystal structure of a D-aminopeptidase from Ochrobactrum anthropi, a new member of the “penicillin-recognizing enzyme” family. Struct. Fold. Des. 8 971–980. [DOI] [PubMed] [Google Scholar]
- Bourne, D.G., Riddles, P., Jones, G.J., Smith, W., and Blakeley, R.L. 2001. Characterisation of a gene cluster involved in bacterial degradation of the cyanobacterial toxin microcystin LR. Environ. Toxicol. 16 523–534. [PubMed] [Google Scholar]
- Dusart, J., Marquet, A., Ghuysen, J.M., Frère, J.M., Moreno, R., Leyh- Bouille, M., Johnson, K., Lucchi, C., Perkins, H.R., and Nieto, M. 1973. DD-carboxypeptidase-transpeptidase and killing site of β-lactam antibiotics in Streptomyces strains R39, R61, and K11. Antimicrob. Agents Chemother. 3 181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanuel, L., Thamm, I., Kostanjevecki, V., Samyn, B., Joris, B., Goffin, C., Brannigan, J., Van Beeumen, J., and Frère, J.M. 1999. Two new aminopeptidases from Ochrobactrum anthropi active on D-alanyl-p-nitroanilide. Cell Mol. Life Sci. 55 812–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frère, J.M. and Joris, B. 1985. Penicillin-sensitive enzymes in peptidoglycan biosynthesis. Crit. Rev. Microbiol. 11 299–396. [DOI] [PubMed] [Google Scholar]
- Frère, J.M., Duez, C., Ghuysen, J.M., and Vandekerkhove, J. 1976a. Occurrence of a serine residue in the penicillin-binding site of the exocellular DD-carboxy-peptidase-transpeptidase from Streptomyces R61. FEBS Lett. 70 257–260. [DOI] [PubMed] [Google Scholar]
- Frère, J.M., Leyh-Bouille, M., Ghuysen, J.M., Nieto, M., and Perkins, H.R. 1976b. Exocellular DD-carboxypeptidases-transpeptidases from Streptomyces. Methods Enzymol. 45 610–636. [DOI] [PubMed] [Google Scholar]
- Galarneau, A., Primeau, M., Trudeau, L.E., and Michnick, S.W. 2002. β-lactamase protein fragment complementation assays as in vivo and in vitro sensors of protein protein interactions. Nat. Biotechnol. 20 619–622. [DOI] [PubMed] [Google Scholar]
- Galleni, M., Lakaye, B., Lepage, S., Jamin, M., Thamm, I., Joris, B., and Frère, J.M. 1993. A new, highly sensitive method for the detection and quantification of penicillin-binding proteins. Biochem. J. 291 (Pt. 1) 19–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joris, B., Ghuysen, J.M., Dive, G., Renard, A., Dideberg, O., Charlier, P., Frère, J.M., Kelly, J.A., Boyington, J.C., Moews, P.C., et al. 1988. The active-site-serine penicillin-recognizing enzymes as members of the Streptomyces R61 DD-peptidase family. Biochem. J. 250 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joris, B., Ledent, P., Dideberg, O., Fonze, E., Lamotte-Brasseur, J., Kelly, J.A., Ghuysen, J.M., and Frère, J.M. 1991. Comparison of the sequences of class A β-lactamases and of the secondary structure elements of penicillin- recognizing proteins. Antimicrob. Agents Chemother. 35 2294–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato, Y., Asano, Y., Nakazawa, A., and Kondo, K. 1989. First stereo-selective synthesis of D-amino acid N-alkyl amide catalysed by Daminopeptidase. Tetrahedron 45 5743–5754. [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Kelly, J.A. and Kuzin, A.P. 1995. The refined crystallographic structure of a DD-peptidase penicillin-target enzyme at 1.6 Å resolution. J. Mol. Biol. 254 223–236. [DOI] [PubMed] [Google Scholar]
- Kelly, J.A., Knox, J.R., Zhao, H., Frère, J.M., and Ghaysen, J.M. 1989. Crystallographic mapping of β-lactams bound to a D-alanyl-D-alanine peptidase target enzyme. J. Mol. Biol. 209 281–295. [DOI] [PubMed] [Google Scholar]
- Komeda, H. and Asano, Y. 2000. Gene cloning, nucleotide sequencing, and purification and characterization of the D-stereospecific amino-acid amidase from Ochrobactrum anthropi SV3. Eur. J. Biochem. 267 2028–2035. [DOI] [PubMed] [Google Scholar]
- Kreil, G. 1997. D-amino acids in animal peptides. Annu. Rev. Biochem. 66 337–345. [DOI] [PubMed] [Google Scholar]
- Kuzin, A.P., Liu, H., Kelly, J.A., and Knox, J.R. 1995. Binding of cephalothin and cefotaxime to D-ala-D-ala-peptidase reveals a functional basis of a natural mutation in a low-affinity penicillin-binding protein and in extended-spectrum β-lactamases. Biochemistry 34 9532–9540. [DOI] [PubMed] [Google Scholar]
- LaLonde, J.M., Bernlohr, D.A., and Banaszak, L.J. 1994. The up-and-down β-barrel proteins. FASEB J. 8 1240–1247. [DOI] [PubMed] [Google Scholar]
- Laws, A.P. and Page, M.I. 1989. The effect of the carboxy group on the chemical and β-lactamase reactivity of β-lactam antibiotics. J. Chem. Soc. Perkins Trans. 11 1577–1581. [Google Scholar]
- Leyh-Bouille,M., Nakel,M., Frère, J.M., Johnson, K., Ghuysen, J.M., Nieto, M., and Perkins, H.R. 1972. Penicillin-sensitive DD-carboxypeptidases from Streptomyces strains R39 and K11. Biochemistry 11 1290–1298. [DOI] [PubMed] [Google Scholar]
- Leyh-Bouille, M., Dusart, J., Nguyen-Disteche, M., Ghuysen, J.M., Reynolds, P.E., and Perkins, H.R. 1977. The peptidoglycan crosslinking enzyme system in Streptomyces strains R61, K15 and rimosus. Eur. J. Biochem. 81 19–28. [DOI] [PubMed] [Google Scholar]
- Llinas, A., Ahmed, N., Cordaro, M., Laws, A.P., Frère, J.M., Delmarcelle, M., Silvaggi, N.R., Kelly, J.A., and Page, M.I. 2005. Inactivation of bacterial DD-peptidase by β-sultams. Biochemistry. 44 7738–7746. [DOI] [PubMed] [Google Scholar]
- McDonough, M.A., Anderson, J.W., Silvaggi, N.R., Pratt, R.F., Knox, J.R., and Kelly, J.A. 2002. Structures of two kinetic intermediates reveal species specificity of penicillin-binding proteins. J. Mol. Biol. 322 111–122. [DOI] [PubMed] [Google Scholar]
- Merritt, E.A. 1994. Raster3D Version 2.0. A program for photorealistic molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 50 869– 873. [DOI] [PubMed] [Google Scholar]
- Nieto, M., Perkins, H.R., Leyh-Bouille, M., Frère, J.M., and Ghuysen, J.M. 1973. Peptide inhibitors of Streptomyces DD-carboxypeptidases. Biochem. J. 131 163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins, H.R., Nieto, M., Frère, J.M., Leyh-Bouille, M., and Ghuysen, J.M. 1973. Streptomyces DD-carboxypeptidases as transpeptidases. The specificity for amino compounds acting as carboxyl acceptors. Biochem. J. 131 707–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, E.I., Valinger, G., Solkner, B., Stubenrauch, G., and Schwab, H. 2001. A novel esterase from Burkholderia gladioli which shows high deacetylation activity on cephalosporins is related to β-lactamases and DD-peptidases. J. Biotechnol. 89 11–25. [DOI] [PubMed] [Google Scholar]
- Rhazi, N., Galleni, M., Page, M.I., and Frère, J.M. 1999. Peptidase activity of β-lactamases. Biochem. J. 341 (Pt. 2) 409–413. [PMC free article] [PubMed] [Google Scholar]
- Volkmann, R.A. and Heck, S.D. 1998. Biosynthesis of D-amino acid-containing peptides: Exploring the role of peptide isomerases. EXS 85 87–105. [DOI] [PubMed] [Google Scholar]
- Wagner, U.G., Petersen, E.I., Schwab, H., and Kratky, C. 2002. EstB from Burkholderia gladioli: A novel esterase with a β-lactamase fold reveals steric factors to discriminate between esterolytic and β-lactam cleaving activity. Protein Sci. 11 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]