

Abstract
ALDH3A1 (aldehyde dehydrogenase 3A1) is abundant in the mouse cornea but undetectable in the lens, and ALDH1A1 is present at lower (catalytic) levels in the cornea and lens. To test the hypothesis that ALDH3A1 and ALDH1A1 protect the anterior segment of the eye against environmentally induced oxidative damage, Aldh1a1(−/−)/Aldh3a1(−/−) double knock-out and Aldh1a1(−/−) and Aldh3a1(−/−) single knock-out mice were evaluated for biochemical changes and cataract formation (lens opacification). The Aldh1a1/Aldh3a1- and Aldh3a1-null mice develop cataracts in the anterior and posterior subcapsular regions as well as punctate opacities in the cortex by 1 month of age. The Aldh1a1-null mice also develop cataracts later in life (6–9 months of age). One- to three-month-old Aldh-null mice exposed to UVB exhibited accelerated anterior lens subcapsular opacification, which was more pronounced in Aldh3a1(−/−) and Aldh3a1(−/−)/Aldh1a1(−/−) mice compared with Aldh1a1(−/−) and wild type animals. Cataract formation was associated with decreased proteasomal activity, increased protein oxidation, increased GSH levels, and increased levels of 4-hydroxy-2-nonenal- and malondialdehyde-protein adducts. In conclusion, these findings support the hypothesis that corneal ALDH3A1 and lens ALDH1A1 protect the eye against cataract formation via nonenzymatic (light filtering) and enzymatic (detoxification) functions.
The cornea serves as a physical and biochemical protective barrier between the external milieu and internal ocular tissues. The anterior segment of the eye absorbs the major fraction of the UV light entering the eye and acts as a filter, protecting the lens and retina from UV-induced damage (1). UV radiation-induced pathologic changes in the eye are associated with the generation of reactive oxygen species (ROS)2 that cause lipid peroxidation (LPO), protein modification and denaturation, and DNA damage (2, 3). These effects may cause reduction of corneal epithelial cell viability and cell death by apoptotic pathways (4) as well as protein denaturation with the loss of tertiary structural relationships in the lens fibers, essential for transparency (3). Exposure to solar radiation, particularly within the UV wavelengths, is a risk factor for the development of age-related cataracts and macular degeneration in the retina as well as some corneal diseases (5, 6).
The cornea expresses several proteins that minimize the damaging effects of UV radiation to the more internal structures. The corneas of all mammals are rich in antioxidant enzymes, such as superoxide dismutase (SOD), glutathione peroxidase, glutathione reductase, and catalase, that protect other proteins against the oxidative effects of ROS generated by UV light (7). Nuclear ferritin, detected in chicken corneal epithelium, protects against UV-induced DNA oxidative damage (8). The corneal epithelium of most mammalian species expresses low molecular weight scavengers of ROS, such as glutathione, α-tocopherol, ascorbate, and NADPH (9, 10). NADPH is essential for the regeneration of GSH from its oxidized form (GSSG) through the glutathione reductase/peroxidase system, serving as an indirect antioxidant by maintaining the reducing power of GSH (11). NADPH may also function as a direct antioxidant by reducing glutathiyl and tyrosyl radicals generated during oxidative stress (12). Additionally, this molecule directly absorbs UV light (11) and protects biomolecules through direct binding. For example, bound NADPH is known to offset H2O2-induced inactivation of catalase (13). Corneal crystallins, which are proteins expressed in high levels similar to lens crystallins, are thought to also play a protective role against UV-induced damage (14). The NADP+-dependent isocitrate dehydrogenase, a bovine corneal crystallin, plays a protective role against UV-induced oxidative injury by preventing LPO, protein oxidation, oxidative DNA damage, and intracellular peroxide generation (15). Albumin, the most prevalent protein in the corneal stroma, may serve as an antioxidant scavenger of hydrogen peroxide and protects the cornea against oxidative damage (16). Two aldehyde dehydrogenases, ALDH3A1 and ALDH1A1, are of interest (17, 18). ALDH3A1 represents 5–50% of the corneal water-soluble proteins in mammalian species (19), although it appears to be absent from the lens, whereas ALDH1A1 is found mainly in the lens and to a lesser extent (<5%) in the cornea (17). Early studies have suggested a protective role of ALDH3A1 against UV-induced damage in the cornea. SWR/J mice are susceptible to UV-induced cornea clouding (20), apparently from a lack of stable ALDH3A1 expression due to several mutations in the Aldh3a1 gene (21, 22). Although the biological basis is not well characterized, one established mechanism by which both ALDH3A1 and ALDH1A1 protect the corneal epithelium from UV-induced oxidative damage is the detoxification of highly reactive products of LPO, such as 4-hydroxy-2-nonenal (4-HNE) and malondialdehyde (MDA). ALDH3A1 metabolizes 4-HNE (23), whereas ALDH1A1 metabolizes both 4-HNE and MDA (24). Thus, the presence of ALDH1A1 may complement one or more of the functions ascribed to ALDH3A1 and may provide a means of oxidizing MDA, which is a poor substrate of ALDH3A1 (23). Possible additional functions of ocular ALDH3A1 include direct absorption of UV radiation, chaperone-like activity, and scavenging of UV-generated ROS via the -SH groups of the cysteine and methionine residues (reviewed in Ref. 25).
Herein, we establish that the Aldh1a1(−/−), and Aldh3a1(−/−) single knock-outs as well as the Aldh1a1(−/−)/Aldh3a1(−/−) double knock-out mice develop premature cataracts (lens opacification), documenting the importance of these proteins and their combined roles in preventing ocular toxicity. These studies have important implications for cataract formation in humans.
MATERIALS AND METHODS
Animals
Transgenic Aldh1a1(−/−), Aldh3a1(−/−), and Aldh1a1(−/−)/Aldh3a1(−/−) mice and their wild type counterparts (C57BL/6JX129/sv) were bred. Generation of Aldh1a1(−/−) and Aldh3a1(−/−) single knock-out animals has been previously described (26, 27). Aldh1a1(−/−)/Aldh3a1(−/−) double knock-out mice were generated by breeding F3 homozygous hybrids of Aldh1a1(−/−) and Aldh3a1(−/−) mice. C57BL/6JX129/sv mice are F3 hybrids of control C57BL/6J and 129/sv mice, the genetic background of all knock-out lines used in this study. Animals were maintained in a temperature-controlled room (21–22 °C) on a 12-h light/dark cycle and supplied with food and water ad libitum. Four to twenty animals of each genetic strain and gender, at various ages, were used for each experiment. All animal use was conducted in compliance with Institutional Animal Care and Use Committee of the University of Colorado and was performed according to the published National Institutes of Health guidelines.
Clinical Assessment of Cornea and Lens
The clinical transparency of the cornea and lens in the wild type, Aldh1a1(−/−) and Aldh3a1(−/−) single knock-out, and double knock-out mice of 1–3 and 6–9 months of age was assessed using slit lamp biomicroscopy (FS-2; Nikon, Melville, NY). Mydriasis was induced by a topical application of 1:1 solution of 1% tropicamide (Alcon, Forth Worth, TX) and 10% phenylephrine (Akorn, Buffalo Grove, IL) in conscious animals. Observations lasted 3–5 min per eye and were recorded by a digital camera. To avoid bias, the observer was “blinded” to the genetic background of the mice being examined. To assess the extent of lens opacification, a cataract scoring system ranging from 0 to 3 (where 0 = no opacity, 1 = mild, 2 = moderate, and 3 = severe) based on slit lamp biomicroscopy was utilized. The assessment was made during the examination to minimize misinterpretation of photographic light and positional artifacts. Animals were randomly reexamined for consistency during the assessment. The anterior, middle, and posterior regions of the lens were examined. Quantitative analyses of light scattering from cornea and lens were performed by selecting a line of pixels through the center of the eye with cornea peaks normalized to an arbitrary value of 100, assuming that all images had the same corneal clarity (28).
Protein Studies
To determine any changes in protein expression due to the absence of ALDH1A1 and ALDH3A1 proteins, corneal and lenticular expression of alcohol dehydrogenase and aldose reductase, which are two major 4-HNE-metabolizing enzymes, as well as antioxidant enzymes (catalase, Cu,Zn-SOD, and glutathione peroxidase) and corneal crystallins (transketolase) were evaluated in all Aldh genetic stocks by Western blot and SDS-PAGE analyses.
Preparation of Tissue Extracts
Wild type, Aldh1a1(−/−), Aldh3a1(−/−), and Aldh1a1(−/−)/Aldh3a1(−/−) double knock-out mice were sacrificed by CO2 inhalation followed by cervical dislocation. Corneas and lenses were then immediately removed from enucleated eyes by circumferential sclerotomy posterior to the limbus. Lenses (from one animal), and corneas (pooled from at least three animals) were homogenized in lysis buffer containing 25 mM Tris, 0.25 M sucrose (pH 7.4), 0.5 μg/ml leupeptin, 0.5 μg/ml aprotinin, 1 μg/ml pepstatin, and 100 μg/ml phenylmethanesulfonyl fluoride (Sigma). Lysates were sonicated for 5 s on ice using a Branson sonifier 250 (VWR Scientific, Willard, OH) and then cooled on ice for 15 s for a total of three cycles. Lysates were centrifuged at 10,000 × g for 30 min at 4 °C, and supernatants were collected for analysis by Western blotting. Protein concentrations were measured using the BCA method (Pierce).
SDS-PAGE and Western Immunoblotting
For silver staining, 10 μg of protein was loaded and subjected to electrophoresis using a 10% SDS-polyacrylamide gel. For Western blot analysis, 10–30 μg of protein from lens and cornea were subjected to electrophoresis and immunoblotted according to previously described methods (29). The primary antibodies included affinity-purified rabbit anti-ALDH1A1 (26), monoclonal and polyclonal anti-ALDH3A1 (23), monoclonal anti-β-actin (Sigma), antibodies to Cu,Zn-SOD (Calbiochem), Mn-SOD, aldose reductase (a gift from Dr. M. Petrash, University of Washington, St. Louis, MO), catalase (Research Diagnostic, Concord, MA), glutathione peroxidase (Cortex Biochem, San Leandro, CA), transketolase (30), alcohol dehydrogenase (31), and γ-glutamylcysteine synthase (GCS) (a gift from Dr. T. Kavanagh, University of Washington, Seattle, WA). Anti-rabbit, anti-sheep, and anti-mouse immunoglobulin G-conjugated horse-radish peroxidase antibodies were obtained from Jackson Laboratories (West Grove, PA). Labeled proteins were detected by enhanced chemiluminescence (PerkinElmer Life Sciences).
ALDH3A1 Enzymatic Assay
ALDH3A1 activity was determined using either 5 mM benzaldehyde (substrate) and 2.5 mM NADP+ (coenzyme) or 5 mM propionaldehyde (substrate) and 1 mM NAD+ (coenzyme) as described previously (29). ALDH3A1 activity was normalized to total protein concentration measured by the BCA method (Pierce).
Determination of Reduced GSH Levels
Eyes from wild type, Aldh1a1(−/−), Aldh3a1(−/−), and Aldh1a1(−/−)/Aldh3a1(−/−) double knock-out mice 6 months of age were excised, and the lenses and corneas were separated and combined for each animal. The tissues were weighed using an analytical scale (model CAHN C-33; Waltham, MA), sonicated on ice in KPBS buffer (50 mM KH2PO4, 50 mM K2HPO4, pH 7.4, 17.5 mM EDTA, 50 mM serine, and 50 mM boric acid), and incubated in the dark at room temperature for 30 min in the presence of monobromobimane (for the development of the GSH-monobromobimane derivative). The reaction was stopped by the addition of 70% perchloric acid, the samples were centrifuged at 10,000 × g for 10 min at 4 °C, and the supernatant solution was removed and analyzed for GSH on a Hitachi high pressure liquid chromatograph (model L-2420; San Jose, CA) equipped with a fluorometric detector (model L-2480) as previously described with minor modifications (32).
Determination of Protein-Carbonyls and 4-HNE- and MDA-adducted Proteins
Lenses were dissected from 6-month-old wild type and Aldh1a1(−/−)/Aldh3a1(−/−) double knock-out mice, and the soluble and insoluble fractions were processed as previously described (33). Equal aliquots of samples from each animal strain were processed for detection of carbonyl groups using the Oxyblot® detection kit (Chemicon International, Temecula, CA). Briefly, the soluble and insoluble fractions were treated with 2,4-dinitrophenylhydrazine to derivatize the carbonyl groups to 2,4-dinitrophenylhydrazone. The 2,4-dinitrophenylhydrazone-derivatized protein samples were separated on 10% polyacrylamide gels, transferred to polyvinylidene difluoride membranes, and probed using specific antibody for the 2,4-dinitrophenylhydrazone-derivatized residues of oxidatively damaged proteins. The same samples were processed for detection of 4-HNE and MDA adducts by immunoblotting using polyclonal antibodies developed against 4-HNE- and MDA-adducted proteins (a gift from Dr. D. Petersen, University of Colorado Health Sciences Center, Denver, CO). Labeled proteins were detected by enhanced chemiluminescence (PerkinElmer Life Sciences).
Proteasome Activity Assay
Lens homogenates from wild type, Aldh1a1(−/−), and Aldh3a1(−/−) single and double knock-out mice were diluted to 0.5 mg/ml in PBS and assayed for chymotrypsin-like proteasome activity as described previously (34). The chymotrypsin-like activity was measured using a fluorogenic substrate, succinyl-Leu-Leu-Val-Tyr-amido-methylcoumarin (Sigma). Samples were assayed at 37 °C for 1 h in a black 96-well plate (Costar; Corning Inc.) using a temperature-controlled fluorescence plate reader (Spectra MAX Gemini EM, Sunnyvale, CA). The measured excitation and the emission wavelengths were 360 and 430 nm, respectively. Identical samples were prepared with the proteasome inhibitor, N-benzyloxycarbonyl-Leu-Leu-leucinal (MG132; Sigma), to determine specific activity to proteasome degradation.
UVB Exposure
One- to three-month-old wild type, Aldh1a1(−/−), and Aldh3a1(−/−) single and double knock-out mice were exposed to UVB light for 2.5 min (0.05 J/cm2) and 10 min (0.2 J/cm2), and lens opacification was evaluated 12 and 48 h after UV exposure by slit lamp biomicroscopy. Animals were first anesthetized by subcutaneous injection of xylazine (10 mg/kg) and ketamine (150 mg/kg) and were placed directly under the UV lamp with the head propped on its side so that each eye faced the lamp one at a time. Both eyes were exposed to equal amounts of energy. The UV source (a 115-V, 60-Hz, 0.16-A non-ozone-producing lamp with a peak wavelength of 302 nm; UVP Inc., San Gabriel, CA) was mounted horizontally in an enclosed box. The distance from the lamp to the eye was 27 cm. The energy output of the lamp at this distance was measured to be 350 milliwatts/cm2 using a UVX digital radiometer attached to a UVX-31 300-nm sensor (UVP Inc.). Heat transmission from the lamp to the bottom of the box was negligible. In addition, the chymotrypsin-like activity of the lens 20 S proteasome was determined.
Statistical Analysis
Data are expressed as mean values ± S.E. Statistical analyses were done using Sigmastat®, version 3.10 (Systat Software, Point Richmond, CA), or SAS®, version 9.1 (SAS Institute Inc., Cary, NC). All comparisons of mean (or median, if not normally distributed) values between groups were performed using one-way analysis of variance (or Kruskal-Wallis test based on ranks), with post hoc pairwise comparisons based on Bonferroni’s t test. A comparison of the proportion of mice with lens opacification between single or double knock-out mice and control mice was performed using Fisher’s exact test. A level of p < 0.05 was considered statistically significant. Each experiment was repeated three times.
RESULTS
Aldh1a1(−/−)/Aldh3a1(−/−) Double Knock-out Mice
The tail DNA analyses of the mice (Fig. 1) confirmed the genotype of the double knock-out and wild type mice. The Aldh1a1 genotype was determined by Southern blot analysis (26), whereas the Aldh3a1 genotype was determined by PCR (27) (supplemental Fig. 1). The homozygous mutant mice were born alive in the expected Mendelian gender ratio with no malformations or reduced survival or growth, indicating that Aldh1a1(−/−)/Aldh3a1(−/−) double knock-out mice are healthy and fertile. The absence of ALDH1A1 and ALDH3A1 proteins in the double knock-out mice in lens and cornea was verified using silver staining (Fig. 1A) and Western blot analysis (Fig. 1B). ALDH1A1 protein (55 kDa) was missing from the lens of Aldh1a1(−/−) and Aldh1a1(−/−)/Aldh3a1(−/−) mice; ALDH3A1 (51 kDa) was missing from the cornea of the Aldh1a1(−/−)/Aldh3a1(−/−) mice but not from the cornea of Aldh1a1(−/−) mice (Fig. 1A). It is also obvious that no other new proteins are present in the area of ~50 kDa in either cornea or lens of the Aldh1a1(−/−)/Aldh3a1(−/−) mice as a compensatory mechanism. Immunoblot analyses using specific antibodies confirmed that the Aldh1a1/Aldh3a1-null mutant was generated (Fig. 1B). ALDH1A1 was detected in the lens and cornea of the wild type and Aldh3a1(−/−) mice but not in Aldh1a1(−/−) and Aldh1a1(−/−)/Aldh3a1(−/−) mice. ALDH3A1 was detected in the cornea but not in the lens of wild type and in Aldh1a1(−/−) mice. As expected, ALDH3A1 was absent from the cornea and lens of Aldh3a1(−/−) and double knock-out mice. No traces of ALDH enzymatic activity were detected in the cornea or lens of Aldh1a1(−/−)/Aldh3a1(−/−) mice using benzaldehyde and NADP+ or propionaldehyde and NAD+ as substrates and coenzymes, respectively (Table 1), which suggests that no compensation by any other ALDH has occurred in the Aldh1a1(−/−)/Aldh3a1(−/−) mice.
FIGURE 1. Detection of ALDH1A1 and ALDH3A1 proteins in lens and cornea.
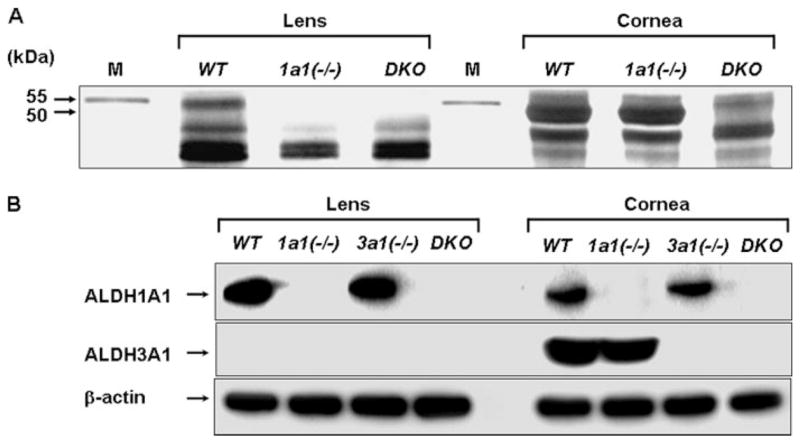
A, SDS-polyacrylamide gel with silver staining of proteins (10 μg) extracted from lens and cornea from wild type (WT), Aldh1a1(−/−) (1a1(−/−)), and double knock-out mice (DKO). The positions of the protein bands corresponding to ALDH1A1 and ALDH3A1 are indicated by the arrows (55 and 50 kDa, respectively). M, molecular weight markers. B, Western blot analyses of ALDH1A1 and ALDH3A1 proteins (20 μg) in wild type, Aldh1a1(−/−), and Aldh3a1(−/−) (3a1(−/−)) single and double knock-out mice. β-Actin expression was used to show equal loading.
TABLE 1.
ALDH activity in cornea and lens in wild type (WT) and Aldh1a1(−/−)/Aldh3a1(−/−) double knock-out mice (DKO)
| Substrate | Activitya |
|---|---|
| nmol NAD(P)H/min/mg protein | |
| NADP+/benzaldehyde | |
| WT cornea | 3430.5 ± 3.1 |
| WT lens | 9.1 ± 3.6 |
| DKO cornea | NDb |
| DKO lens | ND |
|
| |
| NAD+/propionaldehyde | |
| WT cornea | 853.7 ± 9.9 |
| WT lens | 2.9 ± 8.6 |
| DKO cornea | ND |
| DKO lens | ND |
Specific activities are expressed as nmol of NAD(P)H/min/mg protein as mean ± S.E. from triplicates in three separate experiments and were assayed using benzaldehyde and NADP+ or propionaldehyde and NAD+ as substrates and coenzymes, respectively.
ND, no enzymatic activity detected.
Lenticular Opacification
Two age groups of mice (1–3 and 6–9 months old) were evaluated for cataract phenotype by slit lamp biomicroscopy. We quantified the cataract phenotype using a grading scale ranging from 0 to 3 (where 0 = no opacity, 1 = mild, 2 = moderate, and 3 = advanced). The lenses of wild type mice were clear in most animals, although a 17 and 26% incidence of cataract was detected in the young and older animals, respectively (Table 2 and Fig. 2A). The Aldh1a1(−/−) mice had a consistent lenticular opacification detected in ~63% of the older mice only (Table 2). Cataract was almost undetectable in young Aldh1a1(−/−) mice, whereas older animals developed mild anterior subcapsular cataract as compared with the age-matched wild type counterpart (supplemental Table 1B and Fig. 2B). Cataract was observed in Aldh3a1(−/−) mice as early as 1 month of age as compared with wild type mice; this opacification was severe in older mice as compared with both younger Aldh3a1(−/−) and wild type mice (Table 2). Approximately 60 and 80% of the young and old Aldh3a1-null mice, respectively, had mild to moderate lens opacification in the anterior subcapsular region (supplemental Table 1, A and B, and Fig. 2C). The Aldh1a1(−/−)/Aldh3a1(−/−) mice exhibited the highest proportion of cataracts at all ages with the location being anterior subcapsular cataract, as compared with wild type mice (Fig. 2D, Table 2, and supplemental Table 1, A and B). The cataract incidence was higher in older (94%) than in younger mice (79%) in the double knock-out as compared with wild type mice (Table 2). By quantitative analyses, opacification in the anterior portion of the lens was denser in the double knock-out Aldh1a1(−/−)/Aldh3a1(−/−) as compared with single knock-out Aldh1a1(−/−) or Aldh3a1(−/−) and wild type mice (Fig. 2, A–D). Cataract morphology was documented in the anterior subcapsular and posterior capsular regions in a double knock-out mouse of more than 12 months of age (supplemental Fig. 2). Anterior subcapsular lenticular opacities (supplemental Fig. 2B) and punctate, refractile opacities were visible in the posterior cortex (supplemental Fig. 2, C and D) in contrast with the embryonal nucleus, which was relatively clear (supplemental Fig. 2, B and C). Age-matched wild type mice had clear lenses, although a small proportion developed mild cataracts with age (data not shown).
TABLE 2. Cataract detection in wild type, Aldh1a1(−/−), and Aldh3a1(−/−) single and Aldh1a1(−/−)/Aldh3a1(−/−) double knock-out (DKO) mice by in vivo slit lamp biomicroscopy.
Cataract was documented in wild type, Aldh1a1(−/−), and Aldh3a1(−/−) single and Aldh1a1(−/−)/Aldh3a1(−/−) double knock-out mice 1–3 and 6–9 months of age.
| Age | Genotype | Total mice | Mice with cataract | Percentage | p valuea |
|---|---|---|---|---|---|
| months | % | ||||
| 1–3 | Wild type | 18 | 3 | 17 | |
| Aldh1a1(−/−) | 15 | 3 | 20 | 0.99 | |
| Aldh3a1(−/−) | 16 | 10 | 62 | 0.012 | |
| DKO | 14 | 11 | 79 | <0.001 | |
| 6–9 | Wild type | 23 | 6 | 26 | |
| Aldh1a1(−/−) | 16 | 10 | 63 | 0.046 | |
| Aldh3a1(−/−) | 12 | 10 | 83 | 0.003 | |
| DKO | 17 | 16 | 94 | <0.001 |
p values comparing proportion of mice with cataracts among Aldh1a1(−/−) and Aldh3a1(−/−) single and double knock-out mice versus wild type mice (Fisher’s exact test). p < 0.05 was considered statistically significant. When comparing single and double knock-out mice, for the 1–3-month-old group, both Aldh3a1(−/−) and double knock-out mice were significantly different from the Aldh1a1(−/−) mice (Fisher’s exact test, p < 0.05). However, there was no significant difference between the double knock-out and the Aldh3a1(−/−) mice (p = 0.44). For the 6–9-month-old group, the DKO mice were significantly different from the Aldh1a1(−/−) mice only (Fisher’s exact test, p < 0.05). No other significant differences between knock-out mice groups were identified.
FIGURE 2. Slit lamp biomicroscopy and corresponding densitometry tracings of the anterior capsule and cortex of wild type, Aldh1a1(−/−), and Aldh3a1(−/−) single and double knock-out mice, 6 months of age.
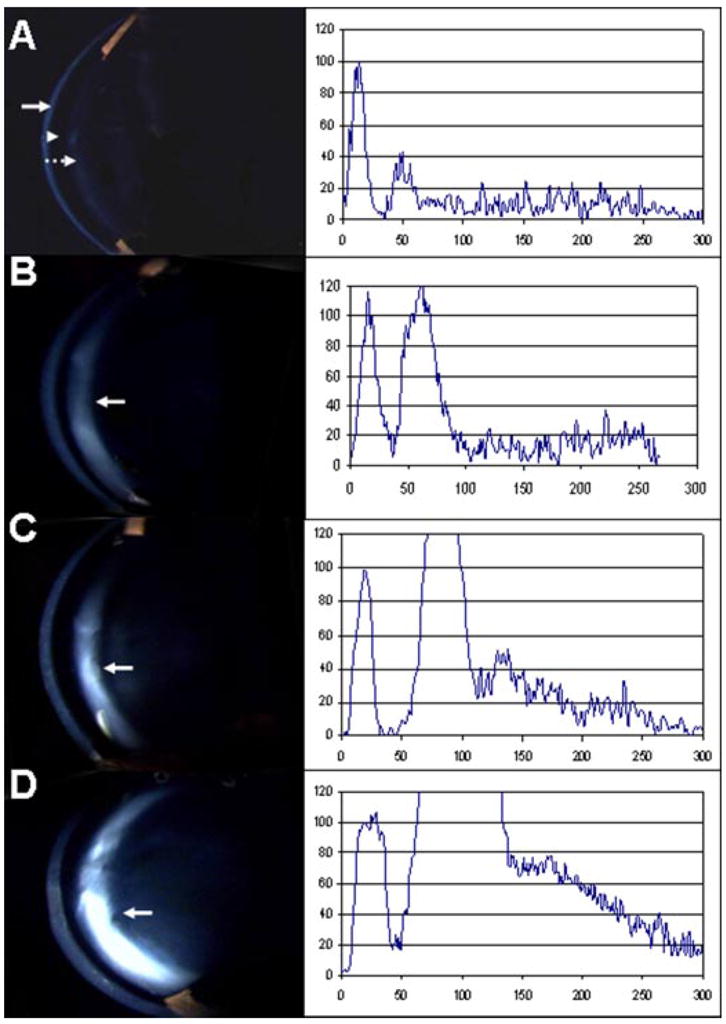
The left light band is the cornea (arrow). The adjacent dark region is the water-filled anterior chamber (arrowhead). The next light band is the anterior capsule and lens epithelium (dashed arrow), and the remainder of the image is the central lens. Densitometry scans of pixel intensity through the center of each image are shown on the right side of the slit lamp photographs with cornea peaks normalized to an arbitrary value of 100, assuming that all images had the same corneal clarity. A, wild type. The lens is clear with a relatively flat graph after the peak due to the capsule and epithelium. B, Aldh1a1(−/−). There is mild scattering from the anterior subcapsular cataract (arrow). C, Aldh3a1(−/−). There is denser anterior subcapsular cataract (arrow). D, Aldh1a1(−/−)/Aldh3a1(−/−). There is definite and denser anterior subcapsular cataract (arrow).
4-HNE-metabolizing and Antioxidant Enzymes in Aldh Transgenic Mice
There were no differences in the corneal and lenticular expression of alcohol dehydrogenase and aldose reductase, two major 4-HNE-metabolizing enzymes, as well as antioxidant enzymes (catalase, Cu,Zn-SOD, glutathione peroxidase) and corneal crystallins (transketolase) between wild type and each Aldh stock (supplemental Fig. 3, A and B). Each membrane was stripped and reprobed with anti-β-actin antibody to document equal loading.
Increased Levels of Reduced GSH in the Lens and Cornea of Aldh1a1(−/−), Aldh3a1(−/−), and Double Knock-out Mice
The level of reduced GSH was measured in 6-month-old animals. Compared with wild type animals, there was a 2.3, 22.3, and 29.3% GSH increase in the lens of Aldh1a1(−/−), Aldh3a1(−/−), and double knock-out mice, respectively (Table 3). In addition, a 6.4, 25.8, and 45.2% GSH increase occurred in the cornea of Aldh1a1(−/−), Aldh3a1(−/−), and double knock-out mice, respectively, compared with the wild type animals. There was an overall significant difference in GSH for both the lens and cornea, which was primarily evident in the double knock-out mice (Table 3). In addition, increased levels of GSH were also closely correlated with an increase in the content of GCS, which is a heterodimer composed of a catalytic or heavy (GCSh) (Fig. 3A) and modulatory or light (GCSl) (Fig. 3B) subunit, respectively. However, GCSl was undetectable in lens extracts, perhaps due to the antibody sensitivity. Although GCSh(−/−) mice are lethal, GCSl(−/−) mice are viable with low levels of GSH (35, 36).
TABLE 3. Levels of GSH in lenticular and corneal tissues extracted from wild type, Aldh1a1(−/−), Aldh3a1(−/−) single, and Aldh1a1(−/−)/Aldh3a1(−/−) double knock-out (DKO) mice at 6 months of age.
The data are expressed as pmol/mg of tissue. All values represent mean ± S.E. (n = 6 per group). Statistical analyses were performed using one-way analysis of variance. There was an overall significant difference in GSH for both the lens and cornea (lens, F3,20 = 3.316, p = 0.041; cornea, F3,20 = 3.50, p = 0.034). Based on post hoc comparisons, this difference was primarily evident in the double knock-out mice.
| Genotype | Tissue | GSH (mean ± S.E.) | p valuea |
|---|---|---|---|
| pmol/mg tissue | |||
| Wild type | Lens | 3940 ± 234 | |
| Aldh1a1(−/−) | Lens | 4030 ± 398 | NSb |
| Aldh3a1(−/−) | Lens | 4819 ± 333 | NS |
| DKO | Lens | 5095 ± 269 | 0.052 |
| Wild type | Cornea | 345 ± 38 | |
| Aldh1a1(−/−) | Cornea | 367 ± 42 | NS |
| Aldh3a1(−/−) | Cornea | 434 ± 36 | NS |
| DKO | Cornea | 501 ± 34 | 0.025 |
All p values presented in the table represent the post hoc Bonferroni’s t test comparing the designated group with the wild type mice.
NS, not significant.
FIGURE 3. Increased GCS expression in the lens and cornea of the double knock-out mice (6 months of age).
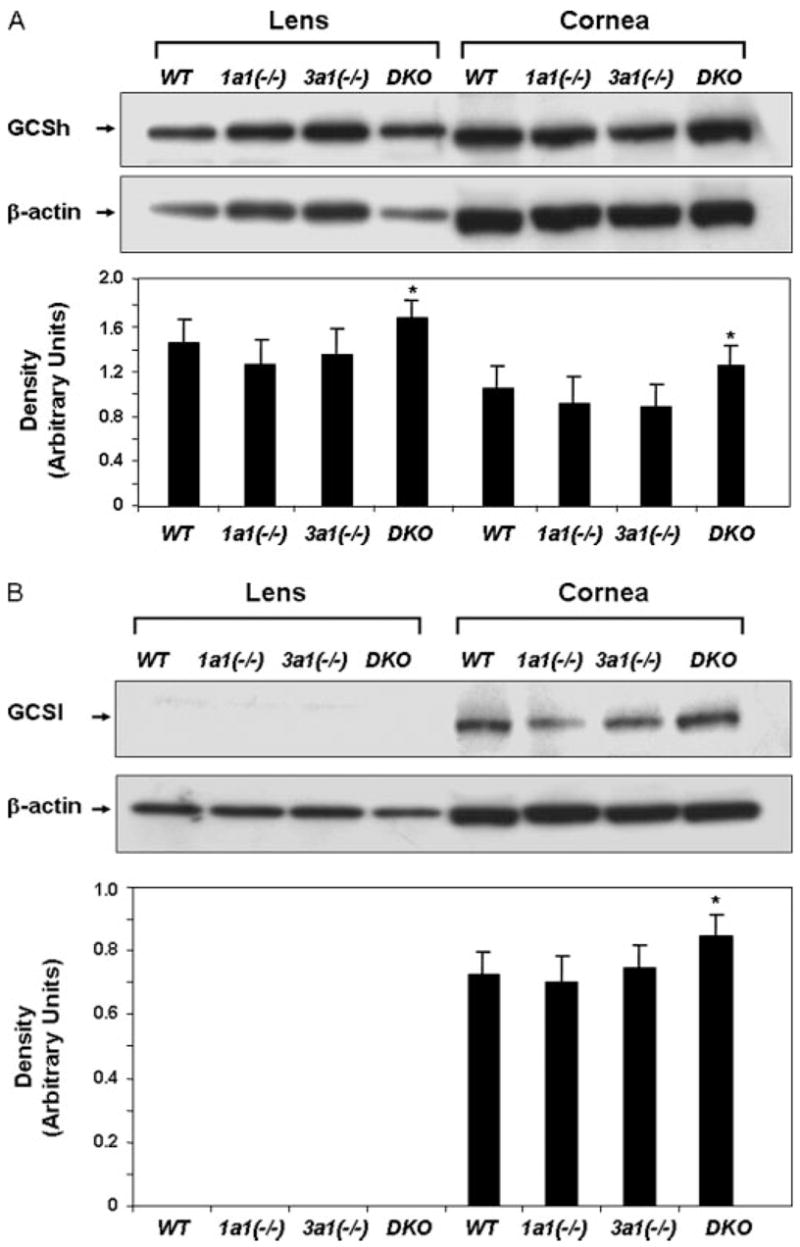
Shown is Western blot analysis of GCSh (73 kDa) (A) and GCSl (30 kDa) (B) subunits in lens and cornea lysates. Thirty μg of cellular protein was loaded per lane, and equal loading was confirmed by probing the membranes with β-actin antibody. S.E. was less than 10% in all cases. *, p < 0.05, Student’s unpaired t test, compared with respective wild type control mice.
Decreased Proteasomal Function in Double Knock-out Mice
Because lens opacification results from aggregation and denaturation of proteins, we assessed the lens proteasomal activity in Aldh1a1/Aldh3a1-null mice by determining the chymotrypsin-like activity of the proteasome in the soluble fraction of lenses of all groups of mice at various ages (1–3 and 6–9 months of age). A greater difference in chymotrypsin-like proteasome activity was observed in the 6–9-month-old group, with the double knock-out mice having the lowest mean value of proteasome activity as compared with age-matched wild type animals (Fig. 4).
FIGURE 4. Decrease of chymotrypsin-like proteasome activity in Aldh1a1(−/−)/Aldh3a1(−/−) double knock-out mice.
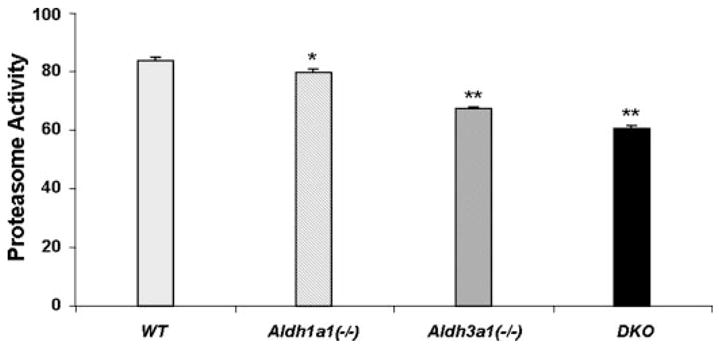
Lenses from wild type, Aldh1a1(−/−), and Aldh3a1(−/−) single and double knock-out mice, 6 months of age, were extracted, and fresh lysates were processed for chymotrypsin-like proteasome activity. The activities are expressed as the rate of substrate conversion/min × mg of protein. Values represent mean ± S.E. (n = 3/group). An overall significant difference in proteasome activity was observed among the different genotypes (F3,8 = 264.2, p < 0.001). Post hoc Bonferroni’s t test indicated that all groups were significantly different from each other. *, p < 0.05 comparing Aldh1a1(−/−) to wild type and p < 0.001 comparing Aldh1a1(−/−) to either Aldh3a1(−/−) or double knock-out mice. **, p < 0.001 comparing the designated group to any other group.
Increased Carbonylation and 4-HNE- and MDA-adducted Proteins in the Lens of Double Knock-out Mice
Carbonylated proteins (Fig. 5A) as well as 4-HNE- and MDA-adducted proteins (Fig. 5B) were detected in the lens extracts from wild type and double knock-out mice. These data illustrate the increase in oxidized proteins as well as the increase of 4-HNE- and MDA-adducted proteins in the insoluble fractions of lenses in 6-month-old double knock-out mice as compared with the age-matched wild type animals.
FIGURE 5. Detection of oxidized proteins (A) and 4-HNE- and MDA-adducted proteins (B) in lens extracts from wild type and Aldh1a1(−/−)/Aldh3a1(−/−) double knock-out mice.
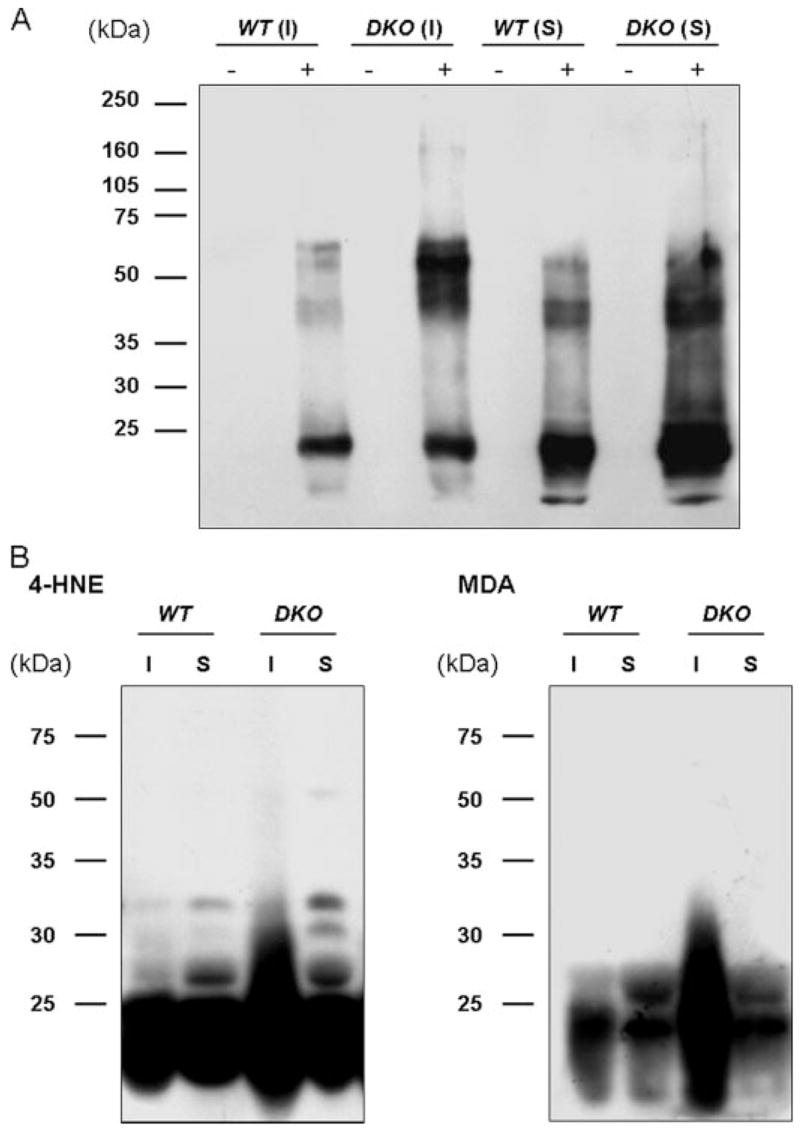
Lenses from wild type (WT) and double knock-out (DKO) mice, 6 months of age, were separated into soluble (S) and insoluble (I) fractions. A, 2,4-dinitrophenylhydrazine-derivatized (+) and nonderivatized (−) proteins were separated by SDS-PAGE (10%) and transferred to polyvinylidene difluoride membranes. Oxidized proteins were recognized by an antibody specific to the 2,4-dinitrophenylhydrazone-derivatized residues and detected by enhanced chemiluminescent reagents. B, equal amounts of lens homogenates were subjected to SDS-PAGE (10%) and transferred to polyvinylidene difluoride membranes. 4-HNE- and MDA-adducted proteins were detected by Western blotting. Protein molecular mass (kDa) markers are given on the left.
UVB-induced Corneal Edema and Cataract Formation
All Aldh genetic stocks exhibited anterior subcapsular lens opacification and corneal edema at both UVB doses tested, which were more evident in Aldh3a1(−/−) and Aldh1a1(−/−)/Aldh3a1(−/−) double knock-out mice (supplemental Fig. 4A and supplemental Table 2, A and B). Exposure to a high UV dose (0.2 J/cm2) led to the development of severe corneal edema in all Aldh-deficient mice that was much more pronounced in Aldh3a1(−/−) and Aldh1a1(−/−)/Aldh3a1(−/−) double knock-out mice compared with Aldh1a1(−/−) and wild type animals (supplemental Fig. 4B and supplemental Table 2B). In addition, samples from the same animals were used to determine the chymotrypsin-like activity of the proteasome. UVB caused significant inhibition of the proteasome activity in all Aldh genetic stocks, especially in Aldh3a1(−/−) and Aldh1a1(−/−)/Aldh3a1(−/−) double knock-out mice (Fig. 6).
FIGURE 6. Decrease of chymotrypsin-like proteasome activity in Aldh genetic stocks after UVB exposure.

Wild type, Aldh1a1(−/−), and Aldh3a1(−/−) single and Aldh1a1(−/−)/Aldh3a1(−/−) double knock-out mice, 1–3 months of age, were exposed to two different doses of UVB light (0.05 and 0.2 J/cm2). The lenses were subsequently extracted and processed for chymotrypsin-like proteasome activity. The activities were normalized to percentage of control. Values represent mean ± S.E. (n = 3/group).
DISCUSSION
We report accelerated cataract formation and document the phenotype in mice with single and double knock-out of the genes encoding ALDH1A1 and ALDH3A1, establishing the importance of these two enzymes in the mechanism of cataract formation. A combined absence of ALDH3A1 and ALDH1A1 activity in these mice significantly increased lens opacification, decreased proteasomal activity, and increased oxidative damage of proteins, based on increased levels of carbonylated as well as 4-HNE- and MDA-adducted proteins in the lens of the double knock-out mice as compared with wild type animals.
UV radiation leads to the production of ROS in the lens and increases the risk of cataract formation in mammals. ROS initiate LPO, which generates aldehydes, such as 4-HNE and MDA, that are potent electrophiles. Both MDA and 4-HNE adduct to nucleophilic groups of proteins, resulting in protein cross-linking, denaturation, and aggregation (37). Levels of protein carbonyls, as well as 4-HNE- and MDA-adducted proteins, therefore provide a sensitive indicator of cellular protein oxidative damage (38, 39). Increased levels of LPO are associated with the accumulation of 4-HNE and/or MDA in human corneal pathologies (7), lenses with cataracts (40, 41), and retinas with macular degeneration (42).
We described herein the lenticular phenotype of the Aldh genetic stocks, by slit lamp biomicroscopy, in vivo. Although lens opacities were documented in all Aldh-deficient groups examined, we found cataracts in most Aldh3a1(−/−) single knock-out mice as early as 1 month of age, whereas the wild type control mice had typically clear lenses. In Aldh1a1(−/−) single knock-out mice, lens opacification was less frequent than in both the Aldh3a1(−/−) single knock-out and Aldh1a1(−/−)/Aldh3a1(−/−) double knock-out mice and was also observed to occur later in life. The double knock-out mice showed denser lens opacification in the anterior subcapsular region at all ages when compared with single knock-outs (Aldh1a1(−/−) and Aldh3a1(−/−)) and wild type mice. For this reason, we documented the double knock-out cataract phenotype in detail. Moreover, the percentage of mice with cataracts was significantly higher in the double knock-out mice and was evident as early as 1 month of age; this trend increased over time as compared with age-matched control wild type animals. Over 12 months of age, the double knock-out mice developed anterior and posterior subcapsular cataracts with punctate and refractile lenticular opacities with a relatively clear embryonal nucleus. Based on these results, we postulate that both ALDH1A1 and ALDH3A1 act additively in protecting against cataract formation. However, Aldh1a1/Aldh3a1 null mice do not have altered amounts of other antioxidant enzymes; hence, the potential protective effect of ALDH1A1 and ALDH3A1 against UV-induced cataract formation should be considered a direct sequel. Specifically, the absence of ALDH3A1 from the corneal epithelium and stroma in the knock-out mice (23), where it constitutes ~50% of the total water-soluble protein content in wild type mice (27), increased lens opacification. This effect is most likely due to the ability of ALDH3A1 to act as a filter in the cornea by absorbing light (43, 44). ALDH1A1 may protect the lens against cataract formation by detoxifying aldehydic products of LPO in both the cornea and lens. Further support for this mechanism is provided by the fact that the antimalarial drug chloroquine induces prominent anterior cataracts in rats (45) and that this drug binds and inhibits ALDH1A1 (46). The role of ALDH1A1 as a light filter is rather unlikely, because this enzyme is not expressed in the cornea at levels comparable with ALDH3A1. Therefore, the combined absence of these two enzymes results in increased oxidative damage and aldehyde-adducted proteins in the lens, and this is probably the basis for cataract formation.
In this study, we found that all of the Aldh genetic stocks had increased levels of GSH. The double knock-out mice have the highest mean values of GSH as compared with age-matched control animals in both lens and cornea (Table 3), indicating a possible adaptive response to oxidative stress in these animals. Response to oxidative stress often involves changes in GSH content, which is replaced through either enzymatic reduction of its disulfide (GSSG) or through de novo synthesis. GSH is a major regulator of the tissue redox environment, in both lens and cornea (47, 48). GSH is synthesized in the cell by the sequential actions of GCS and glutathione synthase in a series of six-enzyme-catalyzed reactions, which have been termed as the γ-glutamyl cycle (49). GCS activity appears to be rate-limiting, being subject to feedback inhibition by physiological concentrations of GSH. GCS is a heterodimer comprising a catalytic (heavy subunit, GCSh; 73 kDa) and regulatory (light subunit, GCSl; 30 kDa) polypeptide. GCSh is responsible for the catalytic activity, whereas the smaller GCSl modulates the Km of the enzyme for glutamate and its sensitivity to feedback inhibition by GSH (35). Both subunits are expressed in ocular tissues (47), and both are inducible by electrophilic compounds like 4-HNE (50). The increased levels of GSH correlated with an increase in the content of GCSh and GCSl subunits (Fig. 6, A and B). ALDH3A1 oxidizes 4-HNE with high specificity (23), whereas ALDH1A1 metabolizes both 4-HNE and MDA (24). Therefore, we attribute the increased levels of GSH in the double knock-out mice to accumulation of 4-HNE and MDA derived from UV-induced lipid peroxidation as a consequence of the lack of ALDH1A1 and ALDH3A1.
We also demonstrated a decrease in chymotrypsin-like proteasome activity (Fig. 4) in the double knock-out mice and in Aldh-null mice exposed to UV (Fig. 6). The proteasome is the major proteolytic system that removes oxidatively damaged proteins (51). As oxidized proteins aggregate, they tend to inhibit the multicatalytic protease (52). The significant decrease in proteolytic activity in the double knock-out mice is probably due to increased levels of protein carbonylation (Fig. 5A). The age-related accumulation of oxidized proteins is caused, in part, by loss of the chymotrypsin-like activity of the proteasome in the double knock-out mice, and the resultant carbonylated proteins are less susceptible to proteolysis. The absence of ALDH3A1 and ALDH1A1 proteins in the double knock-out mice results in the accumulation of highly oxidized proteins as compared with age-matched wild type controls. These findings suggest that both ALDH3A1 and ALDH1A1 protect the proteasome against oxidative stress and further substantiate the protective roles of both enzymes in ocular tissues.
The 4-HNE- and MDA-modified proteins increase in the double knock-out mice. The lens insoluble protein fractions from the double knock-out mice had higher levels of protein carbonylation (Fig. 5A) as well as proteins adducted to 4-HNE and MDA (Fig. 5B) compared with age-matched wild type controls. We have also observed increased 4-HNE protein adducts in both lens and cornea of Aldh3a1(−/−) knock-out mice (53). Cataract formation in the double knock-out mice correlates with the increased levels of 4HNE- and MDA-adducted proteins. The combined oxidative effects of increased carbonylated and 4-HNE- and MDA-adducted proteins, in concert with decreased proteasome activity are likely to be the basis of the cataracts we observed in the animals. The decrease in proteasome activity and the formation of carbonylated as well as 4-HNE- and MDA-adducted proteins almost certainly contribute significantly to the mechanisms causing protein aggregation and cataract formation in the double knock-out mice. Therefore, without ALDH activity inhibiting the formation of protein aggregates and, consequently, lens opacification by detoxification of LPO-derived aldehydes, the denaturing effects of UV-induced oxidative stress are not protected effectively.
In conclusion, our data suggest that ALDH1A1 in the lens and cornea together with ALDH3A1 in the cornea protect the mouse lens against opacification through both catalytic and noncatalytic properties. Detoxification of lipid peroxide aldehydes and scavenging of free oxygen radicals in the lens by ALDH1A1 prevent protein cross-linking and aggregation. Less expected was our finding that the absence of ALDH3A1 alone leads to lens opacification, considering that this enzyme is essentially undetectable in wild type mouse lenses by our assays. Indeed, Aldh3a1-null mice do not even show corneal opacification (27). A nonenzymatic role for corneal ALDH3A1 has been suggested based on the high concentration (50% of water-soluble protein), which is above what is needed for a solely catalytic role. Thus, the simplest explanation for the opaque lenses in Aldh3a1 null mice is that ALDH3A1 absorbs UV light in the wild type cornea, thus minimizing the amount of UV radiation that reaches the lens. The concept that ALDH3A1 acts as a corneal filter for UV light was proposed by Abedinia et al. (43) on the basis of its abundance in the mammalian cornea and enrichment of UV-absorbing tryptophans among its amino acids. Subsequently, Mitchell and Cenedella (44) called the abundant intracellular corneal proteins, which include ALDH3A1, “absorbins.” The use of ALDHs for nonenzymatic, optical roles finds precedence in the lens, where they occur in elephant shrews (η-crystallin) (54) and mollusks (Ω-crystallin) (55–58). Although η-crystallin of the elephant shrew lens has retinaldehyde dehydrogenase activity, Ω-crystallins in mollusk lenses lack enzymatic activity, consistent with an entirely structural role. Although our data support the idea that ALDH3A1 protects the lens by filtering UV light in the mouse cornea, we cannot exclude the possibility that the absence of corneal ALDH3A1 results in the accumulation of peroxidic aldehydes or other ROS in the cornea that leak into the lens and contribute to lens opacification. Unlike ROS, aldehydes are generally long lived compounds that can diffuse through the cell and react with biological targets distant from the site of origin (59). ALDH3A1 is an active enzyme in the mouse cornea (21), and its absence in SWR/J mice, in which ALDH3A1 is unstable (21, 22), results in corneal opacification when the mice are exposed to UV irradiation (20). Our present UV treatments of Aldh3a1-null mice are consistent with the results for the SWR/J mice regarding the corneal opacification and show further that the absence of this enzyme contributes to UV-induced lens opacification. In addition, we have recently shown that ALDH3A1 may act as an antioxidant in cultured stromal fibroblasts (60). Taken together, our data are consistent with ALDH1A1 and ALDH3A1 in the lens and cornea providing additive safeguards to protect lens clarity using a combination of enzymatic and nonenzymatic properties (Scheme 1). The distribution of lens protective mechanisms between lens and cornea is consistent with the utilization of a gene sharing strategy in both the lens and cornea, as expressed in the refracton hypothesis (61), and with the co-evolution of these two transparent tissues for their optical function.
SCHEME 1. Possible mechanisms by which ALDH3A1 and ALDH1A1 protect the eye from UV-induced cataract formation.
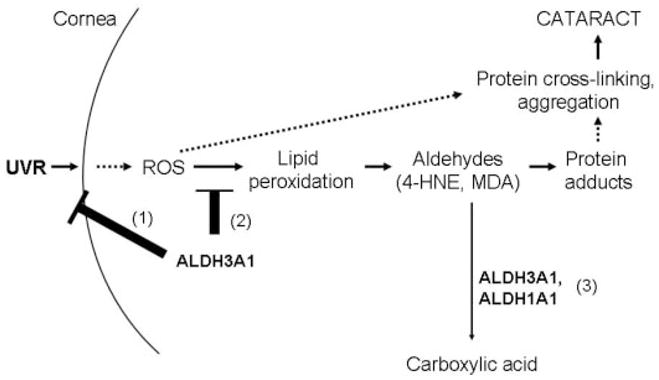
1, ALDH3A1 in the cornea directly absorbs UV light, acting as a UV filter. 2, ALDH3A1 potentially acts as a ROS scavenger, thereby preventing protein cross-linking and aggregation. 3, combined role of ALDH1A1 and ALDH3A1 in detoxification of 4-HNE and MDA derived from lipid peroxidation of cellular membranes, protecting ocular tissues from protein cross-linking and aggregation, which would lead to proteasome inhibition and eventually cataract formation.
Acknowledgments
We thank Dr. David Thompson for valuable discussions and critical review of this manuscript and Tina Fay for technical assistance.
Footnotes
This work was supported by National Institutes of Health Grants EY11490 (to V. V.), EY08282 (to J. B. B.), and EY13969 (to G. D.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables 1 and 2 and Figs. 1–4.
The abbreviations used are: ROS, reactive oxygen species; LPO, lipid peroxidation; SOD, superoxide dismutase; 4-HNE, 4-hydroxy-2-nonenal; MDA, malondialdehyde; GCS, γ-glutamylcysteine synthase; WT, wild type.
References
- 1.Ringvold A. Acta Ophthalmol Scand. 1998;76:149–153. doi: 10.1034/j.1600-0420.1998.760205.x. [DOI] [PubMed] [Google Scholar]
- 2.Spector A. FASEB J. 1995;9:1173–1182. [PubMed] [Google Scholar]
- 3.Spector A. J Ocul Pharmacol Ther. 2000;16:193–201. doi: 10.1089/jop.2000.16.193. [DOI] [PubMed] [Google Scholar]
- 4.Shinohara T, Singh DP, Chylack LT., Jr J Ocul Pharmacol Ther. 2000;16:181–191. doi: 10.1089/jop.2000.16.181. [DOI] [PubMed] [Google Scholar]
- 5.Cruickshanks KJ, Klein BE, Klein R. Am J Public Health. 1992;82:1658–1662. doi: 10.2105/ajph.82.12.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tomany SC, Cruickshanks KJ, Klein R, Klein BE, Knudtson MD. Arch Ophthalmol. 2004;122:750–757. doi: 10.1001/archopht.122.5.750. [DOI] [PubMed] [Google Scholar]
- 7.Buddi R, Lin B, Atilano SR, Zorapapel NC, Kenney MC, Brown DJ. J Histochem Cytochem. 2002;50:341–351. doi: 10.1177/002215540205000306. [DOI] [PubMed] [Google Scholar]
- 8.Cai CX, Birk DE, Linsenmayer TF. Mol Biol Cell. 1998;9:1037–1051. doi: 10.1091/mbc.9.5.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Haaften RI, Haenen GR, Evelo CT, Bast A. Drug Metab Rev. 2003;35:215–253. doi: 10.1081/dmr-120024086. [DOI] [PubMed] [Google Scholar]
- 10.Ringvold A, Anderssen E, Kjonniksen I. Invest Ophthalmol Vis Sci. 1998;39:2774–2777. [PubMed] [Google Scholar]
- 11.Atherton SJ, Lambert C, Schultz J, Williams N, Zigman S. Photochem Photobiol. 1999;70:823–828. [PubMed] [Google Scholar]
- 12.Kirsch M, De GH. FASEB J. 2001;15:1569–1574. doi: 10.1096/fj.00-0823hyp. [DOI] [PubMed] [Google Scholar]
- 13.Kirkman HN, Rolfo M, Ferraris AM, Gaetani GF. J Biol Chem. 1999;274:13908–13914. doi: 10.1074/jbc.274.20.13908. [DOI] [PubMed] [Google Scholar]
- 14.Piatigorsky J. Prog Retin Eye Res. 1998;17:145–174. doi: 10.1016/s1350-9462(97)00004-9. [DOI] [PubMed] [Google Scholar]
- 15.Lee SM, Koh HJ, Park DC, Song BJ, Huh TL, Park JW. Free Radic Biol Med. 2002;32:1185–1196. doi: 10.1016/s0891-5849(02)00815-8. [DOI] [PubMed] [Google Scholar]
- 16.Nees DW, Fariss RN, Piatigorsky J. Invest Ophthalmol Vis Sci. 2003;44:3339–3345. doi: 10.1167/iovs.02-1161. [DOI] [PubMed] [Google Scholar]
- 17.King G, Holmes R. J Exp Zool. 1998;282:12–17. [PubMed] [Google Scholar]
- 18.Piatigorsky J. J Ocul Pharmacol Ther. 2000;16:173–180. doi: 10.1089/jop.2000.16.173. [DOI] [PubMed] [Google Scholar]
- 19.Piatigorsky J. Ann N Y Acad Sci. 1998;842:7–15. doi: 10.1111/j.1749-6632.1998.tb09626.x. [DOI] [PubMed] [Google Scholar]
- 20.Downes JE, Swann PG, Holmes RS. Cornea. 1994;13:67–72. doi: 10.1097/00003226-199401000-00011. [DOI] [PubMed] [Google Scholar]
- 21.Shiao T, Tran P, Siegel D, Lee J, Vasiliou V. Pharmacogenetics. 1999;9:145–153. [PubMed] [Google Scholar]
- 22.Pappa A, Sophos NA, Vasiliou V. Chem Biol Interact. 2001;130:181–191. doi: 10.1016/s0009-2797(00)00233-7. [DOI] [PubMed] [Google Scholar]
- 23.Pappa A, Estey T, Manzer R, Brown D, Vasiliou V. Biochem J. 2003;376:615–623. doi: 10.1042/BJ20030810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manzer R, Qamar L, Estey T, Pappa A, Petersen DR, Vasiliou V. DNA Cell Biol. 2003;22:329–338. doi: 10.1089/104454903322216671. [DOI] [PubMed] [Google Scholar]
- 25.Estey T, Piatigorsky J, Lassen N, Vasiliou V. Exp Eye Res. 2006;84:3–12. doi: 10.1016/j.exer.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 26.Fan X, Molotkov A, Manabe S, Donmoyer CM, Deltour L, Foglio MH, Cuenca AE, Blaner WS, Lipton SA, Duester G. Mol Cell Biol. 2003;23:4637–4648. doi: 10.1128/MCB.23.13.4637-4648.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nees DW, Wawrousek EF, Robison WG, Jr, Piatigorsky J. Mol Cell Biol. 2002;22:849–855. doi: 10.1128/MCB.22.3.849-855.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alizadeh A, Clark J, Seeberger T, Hess J, Blankenship T, Fitzgerald PG. Invest Ophthalmol Vis Sci. 2004;45:884–891. doi: 10.1167/iovs.03-0677. [DOI] [PubMed] [Google Scholar]
- 29.Pappa A, Chen C, Koutalos Y, Townsend AJ, Vasiliou V. Free Radic Biol Med. 2003;34:1178–1189. doi: 10.1016/s0891-5849(03)00070-4. [DOI] [PubMed] [Google Scholar]
- 30.Xu ZP, Wawrousek EF, Piatigorsky J. Mol Cell Biol. 2002;22:6142–6147. doi: 10.1128/MCB.22.17.6142-6147.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deltour L, Foglio MH, Duester G. J Biol Chem. 1999;274:16796–16801. doi: 10.1074/jbc.274.24.16796. [DOI] [PubMed] [Google Scholar]
- 32.Day BJ, van Heeckeren AM, Min E, Velsor LW. Infect Immun. 2004;72:2045–2051. doi: 10.1128/IAI.72.4.2045-2051.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gerido DA, Sellitto C, Li L, White TW. Invest Ophthalmol Vis Sci. 2003;44:2669–2674. doi: 10.1167/iovs.02-1311. [DOI] [PubMed] [Google Scholar]
- 34.Bulteau AL, Moreau M, Nizard C, Friguet B. Free Radic Biol Med. 2002;32:1157–1170. doi: 10.1016/s0891-5849(02)00816-x. [DOI] [PubMed] [Google Scholar]
- 35.Dalton TP, Chen Y, Schneider SN, Nebert DW, Shertzer HG. Free Radic Biol Med. 2004;37:1511–1526. doi: 10.1016/j.freeradbiomed.2004.06.040. [DOI] [PubMed] [Google Scholar]
- 36.Yang Y, Dieter MZ, Chen Y, Shertzer HG, Nebert DW, Dalton TP. J Biol Chem. 2002;277:49446–49452. doi: 10.1074/jbc.M209372200. [DOI] [PubMed] [Google Scholar]
- 37.Truscott RJ. Ophthalmic Res. 2000;32:185–194. doi: 10.1159/000055612. [DOI] [PubMed] [Google Scholar]
- 38.Grune T, Reinheckel T, Davies KJ. FASEB J. 1997;11:526–534. [PubMed] [Google Scholar]
- 39.Stadtman ER, Levine RL. Ann N Y Acad Sci. 2000;899:191–208. doi: 10.1111/j.1749-6632.2000.tb06187.x. [DOI] [PubMed] [Google Scholar]
- 40.Ansari NH, Wang L, Srivastava SK. Biochem Mol Med. 1996;58:25–30. doi: 10.1006/bmme.1996.0028. [DOI] [PubMed] [Google Scholar]
- 41.Ates NA, Yildirim O, Tamer L, Unlu A, Ercan B, Muslu N, Kanik A, Hatungil R, Atik U. Eye. 2004;18:785–788. doi: 10.1038/sj.eye.6700718. [DOI] [PubMed] [Google Scholar]
- 42.Verdejo C, Marco P, Renau-Piqueras J, Pinazo-Duran MD. Eye. 1999;13:183–188. doi: 10.1038/eye.1999.48. [DOI] [PubMed] [Google Scholar]
- 43.Abedinia M, Pain T, Algar EM, Holmes RS. Exp Eye Res. 1990;51:419–426. doi: 10.1016/0014-4835(90)90154-m. [DOI] [PubMed] [Google Scholar]
- 44.Mitchell J, Cenedella RJ. Cornea. 1995;14:266–272. doi: 10.1097/00003226-199505000-00007. [DOI] [PubMed] [Google Scholar]
- 45.Drenckhahn D, Lullmann-Rauch R. Exp Eye Res. 1977;24:621–632. doi: 10.1016/0014-4835(77)90120-8. [DOI] [PubMed] [Google Scholar]
- 46.Graves PR, Kwiek JJ, Fadden P, Ray R, Hardeman K, Coley AM, Foley M, Haystead TAJ. Mol Pharmacol. 2002;62:1364–1372. doi: 10.1124/mol.62.6.1364. [DOI] [PubMed] [Google Scholar]
- 47.Ganea E, Harding JJ. Curr Eye Res. 2006;31:1–11. doi: 10.1080/02713680500477347. [DOI] [PubMed] [Google Scholar]
- 48.Lou MF. Prog Retinal Eye Res. 2003;22:657–682. doi: 10.1016/s1350-9462(03)00050-8. [DOI] [PubMed] [Google Scholar]
- 49.Meister A, Anderson ME. Annu Rev Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 50.Iles KE, Liu RM. Free Radic Biol Med. 2005;38:547–556. doi: 10.1016/j.freeradbiomed.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 51.Davies KJ. Biochimie (Paris) 2001;83:301–310. doi: 10.1016/s0300-9084(01)01250-0. [DOI] [PubMed] [Google Scholar]
- 52.Friguet B, Stadtman ER, Szweda LI. J Biol Chem. 1994;269:21639–21643. [PubMed] [Google Scholar]
- 53.Vasiliou V, Merkouris M, Nees DW, Piatigorsky J, Petersen DR, Pappa A. Invest Ophthalmol Vis Sci. 2004;45:3807. abstr. [Google Scholar]
- 54.Graham C, Hodin J, Wistow G. J Biol Chem. 1996;271:15623–15628. doi: 10.1074/jbc.271.26.15623. [DOI] [PubMed] [Google Scholar]
- 55.Montgomery MK, Fall-Ngai MJ. J Biol Chem. 1992;267:20999–21003. [PubMed] [Google Scholar]
- 56.Zinovieva RD, Tomarev SI, Piatigorsky J. J Biol Chem. 1993;268:11449–11455. [PubMed] [Google Scholar]
- 57.Tomarev SI, Piatigorsky J. Eur J Biochem. 1996;235:449–465. doi: 10.1111/j.1432-1033.1996.00449.x. [DOI] [PubMed] [Google Scholar]
- 58.Piatigorsky J, Kozmik Z, Horwitz J, Ding L, Carosa E, Robison WG, Jr, Steinbach PJ, Tamm ER. J Biol Chem. 2000;275:41064–41073. doi: 10.1074/jbc.M005625200. [DOI] [PubMed] [Google Scholar]
- 59.Esterbauer H, Schaur RJ, Zollner H. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 60.Lassen N, Pappa A, Black WJ, Jester JV, Day BJ, Min E, Vasiliou V. Free Radic Biol Med. 2006;41:1459–1469. doi: 10.1016/j.freeradbiomed.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 61.Piatigorsky J. Cornea. 2001;20:853–858. doi: 10.1097/00003226-200111000-00015. [DOI] [PubMed] [Google Scholar]