

Summary
The novel E2f7 and E2f8 family members are thought to function as transcriptional repressors important for the control of cell proliferation. Here we have analyzed the consequences of inactivating E2f7 and E2f8 in mice and show that their individual loss had no significant effect on development. Their combined ablation, however, resulted in massive apoptosis and dilation of blood vessels, culminating in lethality by embryonic day E11.5. A deficiency in E2f7 and E2f8 led to an increase in E2f1 and p53, as well as in many stress-related genes. Homo- and hetero-dimers of E2F7 and E2F8 were found on target promoters, including E2f1. Importantly, loss of either E2f1 or p53 suppressed the massive apoptosis in double mutant embryos. These results identify E2F7 and E2F8 as a unique repressive arm of the E2F transcriptional network that is critical for embryonic development and control of the E2F1-p53 apoptotic axis.
Keywords: E2Fs, embryo development, apoptosis, cell cycle, transcriptional regulation
Introduction
A large body of work suggests that E2Fs function to activate and repress the transcription of many essential genes involved in cell proliferation, apoptosis and differentiation (Dimova and Dyson, 2005). The effects of deregulated E2F activity are pleiotropic and vary in different experimental settings. A decrease in E2F activity is generally associated with a reduction in the proliferation capacity of cells (Leone et al., 1998; Humbert et al., 2000b; Wu et al., 2001), whereas an increase in E2F activity is often associated with inappropriate cell proliferation and/or apoptosis (DeGregori et al., 1997). The regulation of E2F activity during the cell cycle is thought to be critical for cellular homeostasis. Thus, a significant amount of genetic currency has been invested to finely control E2F activity in cells, including by direct binding of the Rb tumor suppressor, by transcription, by post-transcriptional mechanisms involving miRNAs, and by post-translational mechanisms involving protein degradation, phosphorylation and acetylation (Muller et al., 2000; O'Donnell et al., 2005).
Further complexity in the regulation of E2F function has been afforded by the evolution of numerous family members and isoforms. The mammalian E2F proteins are encoded by eight distinct genes (E2F1-8) and specific roles for each family member in controlling cell cycle transitions and apoptosis have been reported (Bracken et al., 2004; DeGregori and Johnson, 2006). Based on structure-function studies and sequence analysis, the E2F family can be conveniently divided into two general subclasses, transcription repressors and activators. Members of the activator subclass, consisting of E2F1, E2F2 and E2F3a, accumulate late in G1 and are transiently recruited to E2F-binding elements on target promoters and participate in their acute activation. Consistent with an important function for activator E2Fs in regulating gene expression during G1-S, their overexpression in quiescent cells can potently transactivate many E2F-responsive genes and drive cells to enter S phase (DeGregori et al., 1997). Conversely, the combined loss of the three activators results in a decrease of E2F-target gene expression and a severe block in cell proliferation (Wu et al., 2001). The repressor subclass consists of E2F3b, E2F4, E2F5, E2F6, E2F7 and E2F8. A subset of this group that includes E2F3b, E2F4 and E2F5, serves to recruit Rb-related pocket proteins and associated co-repressors to target promoters during G0 and to repress their expression (Attwooll et al., 2004). As cells are stimulated to enter the cell cycle, cyclin dependent kinases (CDKs) phosphorylate pocket proteins, resulting in dissociation of E2F-pocket protein repressor complexes and derepression of E2F-target genes (Muller et al., 2000; Seville et al., 2005). The E2F6 repressor is part of a multi-subunit complex that includes polycomb group proteins as well as Mga and Max, and appears to act on a different subset of target genes than E2F4 (Ogawa et al., 2002; Giangrande et al., 2004). The mechanism of how E2F7 and E2F8 repress gene expression is less clear. While the molecular basis for how E2F repressors and activators orchestrate the acute induction of E2F targets as cells transit through G0-G1-S is fairly well understood, how E2F-target genes are subsequently downregulated as cells proceed through S-G2 phases of the cell cycle is not known.
The E2F7 and E2F8 proteins are conserved in mice and humans, and related E2F-like proteins exist in Arabidopsis (Kosugi et al., 2002; Mariconti et al., 2002; de Bruin et al., 2003; Di Stefano et al., 2003; Logan et al., 2004; Christensen et al., 2005; Logan et al., 2005; Maiti et al., 2005). These two novel factors have several unique features that distinguish them from other members in the E2F family. They lack the leucine-zipper domain required for dimerization with partner proteins DP1/2 and instead possess two DNA binding domains that are predicted to interact with each other and foster DP-independent DNA-binding activity. The expression of E2F7 and E2F8 during the cell cycle is also different from that of other E2Fs, with peak levels found later in the cell cycle during S-G2. Moreover, their overexpression in fibroblasts can, unlike that of other E2Fs, repress E2F-target gene expression and block cell proliferation (de Bruin et al., 2003; Di Stefano et al., 2003; Maiti et al., 2005), suggesting a role for these two E2Fs in facilitating cell cycle transitions. Here we utilized homologous recombination strategies to inactivate E2f7 and E2f8 in mice and rigorously investigate their functions in vivo. From these studies we conclude that E2f7 and E2f8 represent a unique repressive arm of the E2F transcriptional network that is critical for embryonic development and cell survival.
Results
E2F7 and E2F8 are essential for embryonic viability
To investigate E2F7 and E2F8 function in vivo, we utilized homologous recombination techniques and cre-loxP technology to disrupt E2f7 and E2f8 function in mice. Targeting the inactivation of E2f7 and E2f8 was achieved by flanking exon 4 of E2f7 and exons 3 and 4 of E2f8 with loxP sites (Figure 1A). Cre-mediated recombination of loxP-flanked sequences resulted in the ablation of domains required for DNA binding and in a shift of the open reading frames, leading to premature termination of translation. Pups lacking the neo cassette (conditional knockout allele: E2f7loxP or E2f8loxP; Figure 1A) or lacking both the neo cassette and the loxP-flanked regions of E2f7 or E2f8 (conventional knockout allele: E2f7- or E2f8-; Figure 1A) were identified by Southern blot and PCR genotyping analysis (Figure 1B and data not shown).
Figure 1.
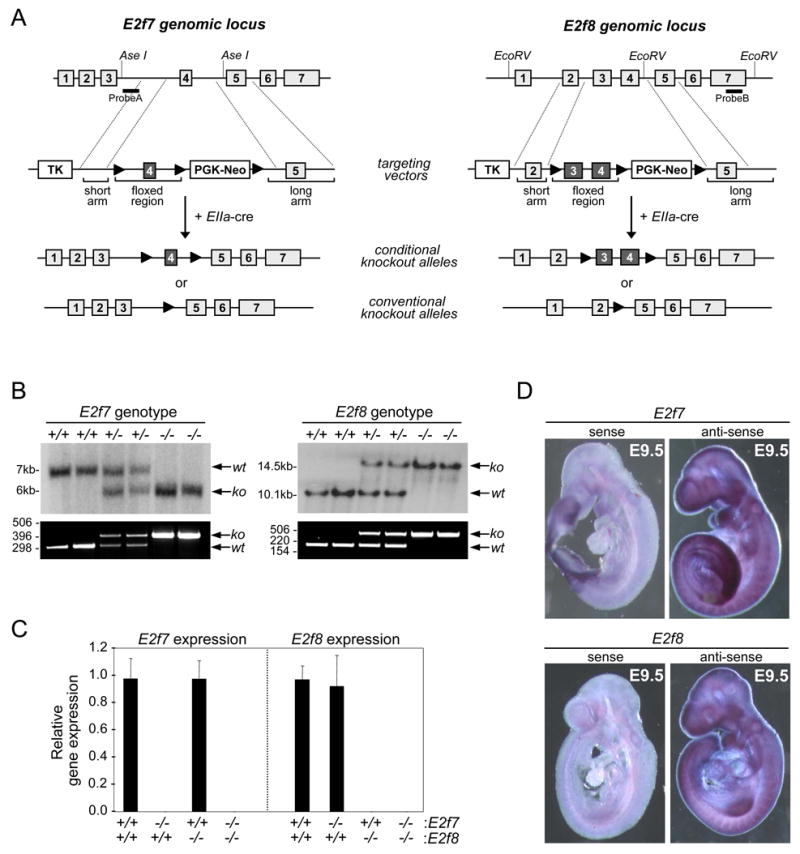
Generation of E2f7 and E2f8 knockout mice. (A) Targeting strategies used to inactivate E2f7 (left) and E2f8 (right). Top panels: partial exon/intron structures of the E2f7 and E2f8 genes. The black bars indicate the position of probes used for Southern analysis. Middle panels (targeting vectors): position of the TK and Neo cassettes, as well as loxP sites (filled triangles) are indicated. Bottom two panels (conditional knockout alleles and conventional knockout alleles): illustrate the two desired alleles resulting from possible recombination events. (B) Top panels: Southern analysis of genomic DNA isolated from conventional knockout mice using AseI for E2f7 (left) and EcoRV for E2f8 (right), and hybridized using probes A and B, respectively. Bottom panels: genotyping of tail DNA was performed using allele-specific PCR primers. (C) Real-time RT-PCR analysis of E2f7 or E2f8 expression in embryos with the indicated genotypes using primers described in Figure S7. (D) Wild type E9.5 embryos were subjected to whole mount in situ hybridization using sense and antisense probes specific to E2f7 (top panels) and E2f8 (bottom panels).
To investigate the requirement for these E2Fs in development we interbred E2f7+/- or E2f8+/- animals and found that E2f7-/- or E2f8-/- pups were born. Mutant pups developed normally through puberty and lived to old age (data not shown). Real-time RT-PCR analysis of gene expression revealed that E2f7 and E2f8 mRNA levels were unperturbed in E2f8-/- and E2f7-/- embryos, respectively (Figure 1C), indicating that the absence of abnormalities in single knockout animals is not due to simple compensation at the expression level. We then explored functional redundancy between E2F7 and E2F8 by examining the biological consequence of ablating both simultaneously. To this end, we intercrossed E2f7+/-E2f8+/- animals and analyzed the resulting offspring. Whereas E2f7-/-E2f8+/- and E2f7+/-E2f8-/- pups were born at the expected Mendelian ratios, no E2f7-/-E2f8-/- double knockout (DKO) pups were found in newborn litters (P0) (Table 1). This demonstrates that at least one allele of E2f7 or E2f8 is required for embryonic development and viability. The contribution of E2F7 and E2F8 towards postnatal development, however, does not appear to be equal. Young and adult E2f7+/-E2f8-/- mice were developmentally normal, whereas most E2f7-/-E2f8+/- animals appeared runted (Figure 2A) and died within the first three months of life (Figure 2B; P90, p<0.001).
Table 1. Genotypic analysis of embryos during development.
Genotypic analysis of embryos derived from E2f7+/-E2f8+/- or E2f7+/-E2f8-/- intercrosses at the indicated stages of development.
| E2f7+/+ | E2f7+/- | E2f7-/- | total | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| E2f8+/+ | E2f8+/- | E2f8-/- | E2f8+/+ | E2f8+/- | E2f8-/- | E2f8+/+ | E2f8+/- | E2f8-/- | ||
| E9.5
expected |
6
5 |
16
28 |
29
22 |
16
19 |
72(2)
75 |
53(1)
56 |
12
13 |
58(1)
47 |
33
34 |
299 |
| E10.5
expected |
9
7 |
21
20 |
13
13 |
7(1)
16 |
45
43 |
28(1)
28 |
4(1)
8 |
28(1)
23 |
6(7)a
14 |
172 |
| E11.5
expected |
-
- |
6
5 |
4
5 |
2
3 |
15(2)
16 |
14(2)
13 |
2(1)
3 |
8(1)
10 |
0(5)b
8 |
62 |
| E12.5
expected |
-
- |
-
- |
3
5 |
-
- |
4
4 |
17(2)
15 |
-
- |
3
4 |
0(9)b
10 |
38 |
| P0
expected |
7
8 |
22
18 |
16
10 |
24
18 |
45
39 |
17
21 |
5
10 |
18
21 |
0b
11 |
154 |
() number of dead embryos recovered; Exact binomial test:
significant (p=0.0015)
highly significant (p<0.0007).
Figure 2.
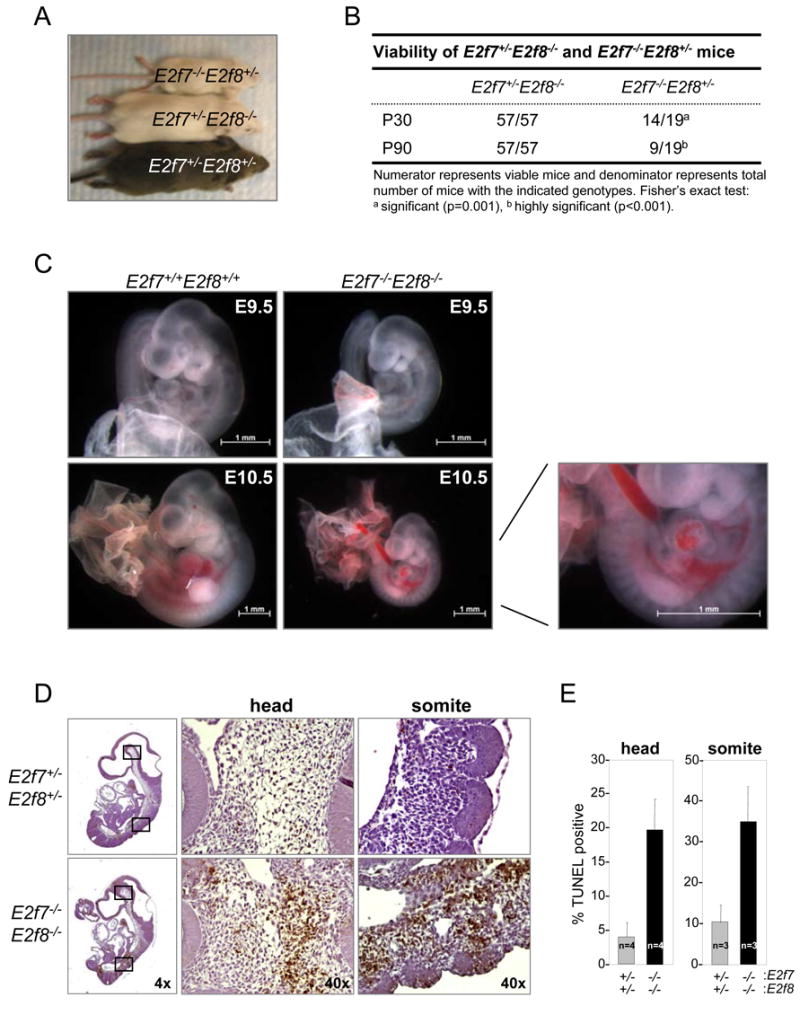
Global deletion of E2f7 and E2f8 results in developmental delay, vascular defects and widespread apoptosis in vivo. (A) Micrograph of E2f7+/-E2f8+/-, E2f7+/-E2f8-/- and E2f7-/-E2f8+/- mice at 21 days of age. (B) Tabulated number of E2f7+/-E2f8-/- and E2f7-/-E2f8+/- mice that live until 30 (P30) and 90 (P90) days of age. (C) Gross pictures of E2f7 and E2f8 double knockout embryos at E9.5 (top panels) and E10.5 (bottom panels). The right panel is a higher magnification view of the vascular defects in E10.5 E2f7-/-E2f8-/- embryos. (D) E9.5 embryos with the indicated genotypes were analyzed by TUNEL assays. Far left panels: low magnification pictures of whole embryos. Right two panels: high magnification images of representative areas demarcated by the box in the low magnification images. (E) Quantification of TUNEL-positive cells in the indicated tissue areas are presented as the average ± SD percentage of cells that are TUNEL-positive. Three sections per embryo and at least three different embryos for each genotype were analyzed.
Genetic ablation of E2f7 and E2f8 in vivo induces massive apoptosis in embryos
Analysis at earlier stages of embryonic development showed that all DKO embryos had a beating heart at embryonic day 9.5 (E9.5). These embryos were noticeably smaller than wild type littermates (Table 1 and Figure 2C, top panels) but macroscopic inspection did not reveal other obvious defects. By E10.5, only 46% of DKO embryos were alive (p=0.0015) and by E11.5 all DKO embryos were dead (p<0.0007). Live E10.5 embryos often had vascular defects in the yolk sac and in the embryo proper, which were characterized by large dilated blood vessels associated with multifocal hemorrhages (Figure 2C, bottom panels). Other phenotypes were also manifested in tissues distinct from the vasculature. Specifically, multiple areas within the head mesenchyme, branchial arch, somites and neural tube of DKO fetuses contained cells with pyknotic nuclei surrounded by bright eosinophilic cytoplasm (Figure S1A and data not shown).
These latter observations prompted us to examine proliferation and apoptosis in E2f7-/-E2f8-/- embryos more closely. Since a significant fraction of DKO embryos died by E10.5, proliferation and apoptosis were measured in E9.5 embryos. When assessed by immunohistochemistry using BrdU-specific antibodies, we observed no detectable difference in the percentage of cells incorporating BrdU between wild type and DKO embryos (Figure S2). We did observe, however, a marked increase in cells labeled by TdT-mediated dUTP nick end-labelling (TUNEL) in areas of DKO embryos previously noted to contain abundant pyknotic nuclei, confirming widespread apoptosis in these tissues (Figure 2D-E, S1B-C). In summary, global deletion of E2f7 and E2f8 resulted in a spectrum of embryonic defects impacting the vasculature and cell survival.
E2F7 and E2F8 form homo-dimers and hetero-dimers
Previous studies suggested that E2F7 and E2F8 form homo-dimers (Di Stefano et al., 2003; Logan et al., 2004; Maiti et al., 2005). Co-immunoprecipitation (Co-IP) assays using HEK 293 cells co-expressing flag-tagged and HA-tagged versions of E2F7 or E2F8 confirmed their ability to form homo-dimers (Figure 3A and data not shown). We also explored the possibility that E2F7 may physically interact with E2F8. Because immunoprecipitation-quality antibodies for E2F7 and E2F8 are not yet available, we assessed the ability of flag-tagged E2F7 to complex with endogenous E2F8 and vice versa. As shown in Figure 3B (and data not shown), these immuno-affinity purification assays revealed an association between E2F7 and E2F8. Hetero-dimerization was confirmed by Co-IP assays using HEK 293 cells co-expressing flag-E2F7 and HA-E2F8 (Figure 3C).
Figure 3.

E2F7 and E2F8 homo- and hetero-dimerize. (A) Homo-dimerization of E2F7. Lysates from cells transfected with both flag-tagged E2F7 (flag-7) and HA-tagged E2F7 (HA-7) were immunoprecipitated (IP) and immunoblotted (IB) with anti-flag and anti-HA antibodies as indicated. Immunoprecipitation with normal mouse IgG was used as a negative control. (B) Left panels: stable expression of flag-HA-E2F7 in HeLa cells. Flag-HA-E2F7 transduced HeLa cell lysates were subjected to Western blotting. The presence of a non-specific protein indicated by (**) was used as a loading control. Right panels: Endogenous E2F8 associates with flag-HA-E2F7. Nuclear extracts from flag-HA-E2F7 expressing cells were immunoprecipitated with flag antibody affinity resins as described in Experimental Procedures and eluents were immunoblotted with E2F8-specific antibodies. Mock-transduced HeLa cells (con.) were used as a negative control. (C) Hetero-dimerization of E2F7 and E2F8. Lysates derived from cells overexpressing both flag-E2F7 and HA-E2F8 were immunoprecipitated (IP) with anti-flag antibodies and immunoblotted (IB) with anti-HA antibodies (top panel) or vice versa (bottom panel). Immunoprecipitation with normal mouse IgG was used as a negative control. (D) Kinetic analysis of dimerization. HEK 293 cells overexpressing the indicated constructs were subjected to anti-flag immunoprecipitation (IP), followed by anti-HA or anti-myc immunoblotting (IB) as indicated. The amount of detected E2F7 and E2F8 was measured by densitometry and quantified relative to 1% of the input material. The relative levels indicated at the bottom of each lane are presented as the average ±SD of 3 independent experiments where Input is always equal to 1.00. (E) Bar graph presentation of the kinetic analysis of dimerization shown in (D). (F) E2F7 binds to the E2f1 promoter. Chromatin from cells overexpressing wild type flag-E2F7 (wt) or flag-E2F7DBD1,2 (mut) was immunoprecipitated (IP) with anti-flag or IgG control antibodies. Immunoprecipitated DNA was amplified using primers specific for the E2f1 promoter (E2f1), irrelevant sequences in exon 1 of E2f1 and the tubulin promoter (control and tubulin, respectively). (G) E2F8 binds to the E2f1 promoter. Cells overexpressing wild type flag-E2F8 (wt) or flag-E2F8DBD1,2 (mut) was immunoprecipitated (IP) with anti-flag or normal mouse IgG antibodies. Immunoprecipitated DNA was amplified using primers specific for the E2f1 promoter (E2f1), exon 1 of E2f1 (control) and the tubulin promoter (tubulin). (H-J) Homo-dimers and hetero-dimers of E2F7 and E2F8 occupy the E2f1 promoter. Cell extracts expressing ectopic HA-E2F7 and flag-E2F7 (H), HA-E2F8 and flag-E2F8 (I), HA-E2F7 and flag-E2F8 (K) were used for sequential ChIP assays as described in Experimental Procedures. Antibodies used for the first and second round of immunoprecipitation are indicated (1st IP and 2nd IP, respectively). Immunoprecipitated DNA collected after two rounds of ChIP was amplified using primers specific for the E2f1 promoter (E2f1) or for the tubulin promoter (tubulin). For all the ChIP experiments, real-time PCR was performed in triplicate and cycle numbers were normalized to 1% of the input DNA.
Given the redundant functions of E2F7 and E2F8 in development, we evaluated their preferred dimerization state in cells. To this end, HEK 293 cells were co-transfected with flag-E2F7, HA-E2F7 and myc-E2F8, or alternatively with flag-E2F8, HA-E2F7 and myc-E2F8. We then measured the relative amounts of homo- versus hetero-dimers by immunoprecipitation with flag-antibodies followed by immunoblotting with either HA-or myc-antibodies; the amount of E2F7 and E2F8 on blots was normalized to 1% of the input material. Quantification of three independent experiments showed that E2F7 had a greater binding affinity to itself than to E2F8 (Figure 3D-E). On the other hand, E2F8 had a greater binding affinity for E2F7 than for itself. From this analysis we conclude that, at least in cultured cells, the preferred state of dimerization is E2F7:E2F7 > E2F7:E2F8 > E2F8:E2F8.
E2f1 is a direct target of E2F7 and E2F8
We then utilized chromatin immunoprecipitation (ChIP) assays to assess the ability of E2F7 and E2F8 to bind known E2F target promoters. Quantitative real-time PCR assays showed that flagged-tagged versions of E2F7 and E2F8 were recruited to E2F binding sites present on the E2f1 and cdc6 promoters but not to irrelevant sequences in these genes (exons 1) or the tubulin promoter (Figure 3F-G, S4A-B and data not shown). This recruitment was specific since IgG failed to immunoprecipitate target promoter sequences. Moreover, anti-flag antibodies failed to immunoprecipitate target promoters from cell lysates expressing mutant forms of E2F7 or E2F8 that are incapable of binding DNA.
We then used sequential ChIP techniques to address whether E2F7 and E2F8 could be recruited to the target promoters as homo- and hetero-dimers. To this end, HEK 293 cells expressing various combinations of HA-tagged and flag-tagged versions of E2F7 and E2F8 were first immunoprecipitated with anti-flag antibodies. Immunoprecipitated DNA-protein complexes were then eluted with excess flag peptide and re-immunoprecipitated with HA-specific antibodies. The final immunoprecipitates were amplified with primers specific for the E2f1, cdc6 or tubulin promoters as described above (Figure 3H-J, S4C-E). These sequential ChIP assays showed that both homo- and hetero-dimers of E2F7 and E2F8 could occupy E2F binding sites on E2F-target promoters, including E2f1.
E2f1 expression is derepressed in E2f7-/-E2f8-/- MEFs
To determine whether the recruitment of E2F7 and E2F8 to the target promoters had any functional consequence on its expression, we initially examined E2F1 protein and mRNA levels in mouse embryo fibroblasts (MEFs) deficient for both E2f7 and E2f8. Because E2f7-/-E2f8-/- embryos died early during mouse development, we derived MEFs from E13.5 E2f7loxP/loxPE2f8loxP/loxP embryos and then ablated E2f7 and E2f8 expression in vitro using a cre recombinase-expressing retrovirus. PCR genotyping of genomic DNA and real-time RT-PCR analysis of E2f7 and E2f8 expression confirmed their efficient deletion (Figure 4A). These experiments showed that DKO cells have higher E2F1 protein and mRNA levels than control treated MEFs (Figure 4B and 4C, respectively). Interestingly, there was also an increase of p53 protein in DKO MEFs, consistent with the ability of E2F1 to induce the accumulation of p53 (Hsieh et al., 2002; Pomerantz et al., 1998; Rogoff et al., 2002; Russell et al., 2002). Introduction of an E2f1 luciferase promoter construct into wild type and DKO MEFs revealed higher luciferase reporter activity in DKO cells (Figure 4D), indicating that the higher levels of E2F1 mRNA and protein in DKO MEFs is likely due to transcriptional deregulation. Together with the ChIP assays shown in Figure 3F-J, these data suggest that the repression of E2f1 by E2F7 and E2F8 is direct.
Figure 4.

(A-F) Deregulation of E2f1 and p53 expression in MEFs deficient in E2f7 and E2f8. (A) Top panels: PCR genotyping analysis of DNA isolated from E2f7+/+E2f8+/+ and E2f7loxP/loxPE2f8loxP/loxP MEFs treated with cre-expressing retroviruses. The knockout (ko) and wild type (wt) alleles are indicated. Bottom graphs: Real-time RT-PCR analysis of E2f7 and E2f8 expression in cells. For convenience, cre-deleted loxP alleles were labeled as (-/-) at bottom of graphs. (B) Western blot analysis of lysates described in (A) using antibodies specific for E2F1 and p53 as indicated. Tubulin-specific antibodies were used as an internal loading control. (C) Expression of E2f1 in MEFs treated as in (A) was measured by real-time RT-PCR. (D) Cells treated as in (A) were co-transfected with the E2f1 firefly luciferase and thymidine kinase renilla luciferase reporter plasmids (internal control). Relative E2f1-luc reporter activity was normalized to renilla luciferase activity. A representative of at least three independent experiments is shown. (E) FACS analysis of synchronized cre-treated E2f7+/+E2f8+/+ and E2f7loxP/loxPE2f8loxP/loxP MEFs. For convenience, cre-deleted loxP alleles were labeled as (-/-). MEFs were synchronized by serum deprivation and hydroxyurea (HU) block as described in Experimental Procedures. At the indicated time points, cells were harvested and stained by propidium iodide for flow cytometry. Q: quiescent cells kept in serum-deprived medium. (F) Total RNA isolated from same synchronized MEFs samples as in (E) was analyzed by real-time RT-PCR assays specific for E2f1 expression. (G-I) MEFs deficient in E2f7 and E2f8 are hypersensitive to DNA damage induced apoptosis. (G) Cre-treated E2f7+/+E2f8+/+ and E2f7loxP/loxPE2f8loxP/loxP MEFs were mock treated with DMSO (-camptothecin) or with camptothecin (+camptothecin). At the indicated intervals cells were harvested and counted in triplicate as described in Experimental Procedures. The percentages of viable cells at the indicated time points are presented. (H) Lysates derived from MEFs treated as in (G) were analyzed at indicated time points by Western blotting using caspase-3 specific antibodies. (I) Total RNA was extracted from cells treated for 18h as in (G) and expression of the indicated p53 target genes was measured by real-time RT-PCR.
To examine whether E2F7 and E2F8 mediated repression might be cell-cycle dependent, we compared E2F-target expression in synchronized populations of wild type and DKO MEFs stimulated to progress through the cell cycle. MEFs were synchronized in G1/S by serum deprivation followed by re-stimulation with medium containing 15% serum and 1mM hydroxyurea (HU). Cells were then washed and incubated with medium lacking HU and harvested at various times following HU-release. Cell cycle progression was monitored by flow cytometry (Figure 4E). As expected, the expression of E2f1 and cdc6 in wild type cells peaked at G1/S and subsequently decreased as cells entered S phase and progressed through G2 (Figure 4F and S5A) Strikingly, expression of E2f1 and cdc6 in DKO cells continued to increase during S and G2, accumulating up to 12-fold higher levels than in wild type cells. These data suggest an important role for E2F7- and E2F8-mediated repression during S and G2, coinciding with the time of the cell cycle when these two E2Fs accumulate maximally (de Bruin et al., 2003; Maiti et al., 2005).
DKO cells are hypersensitive to DNA damage
Given the marked increase in the expression of multiple E2F-target genes in cells deficient for E2f7 and E2f8, we analyzed proliferation rates in these double mutant cells. These analyses failed to reveal any significant difference in the proliferation of DKO and wild type cells (Figure S5B and data not shown), even though DKO cells appeared to transit through S phase 2-3 hours faster than control cells (Figure 4E). Presumably, faster progression through S phase was compensated by a concomitant delay in other phases of the cell cycle as previously reported for cells lacking Rb or overexpressing dE2f1 (Resnitzky et al., 1994; Reis and Edgar, 2004). Because overexpression of E2f1 has been shown to elicit apoptosis in cells treated with DNA-damage inducing agents (Stevens and La Thangue, 2004), we tested the sensitivity of DKO cells to camptothecin and cisplatin, two DNA-damage-inducing drugs. To this end, asynchronously proliferating wild type and DKO MEFs were treated with camptothecin or cisplatin and cell viability was determined by trypan blue exclusion. These drugs induced a significant acceleration of cell death in DKO MEFs when compared to wild type MEFs (Figure 4G and data not shown). Drug-treated DKO cells detached from tissue culture plates and exhibited the characteristic blebbing morphology of apoptotic cells (Figure S5C). Consistent with this observation, drug treatment preferentially triggered cleavage of caspase-3 in DKO MEFs (Figure 4H). We also evaluated the levels of E2F1 and p53 in drug-treated DKO MEFs. As expected, E2F1 and p53 protein levels accumulated to higher levels in camptothecin-treated DKO cells than in similarly treated wild type cells (Figure S5D). The increase in p53 protein corresponded with a marked increase in the expression of p53-responsive genes, including gadd45, noxa, and pidd (Figure 5I). These results suggest that E2F7 and E2F8 may attenuate DNA-damage induced apoptosis by preventing the aberrant accumulation of E2F1 and p53.
Figure 5.
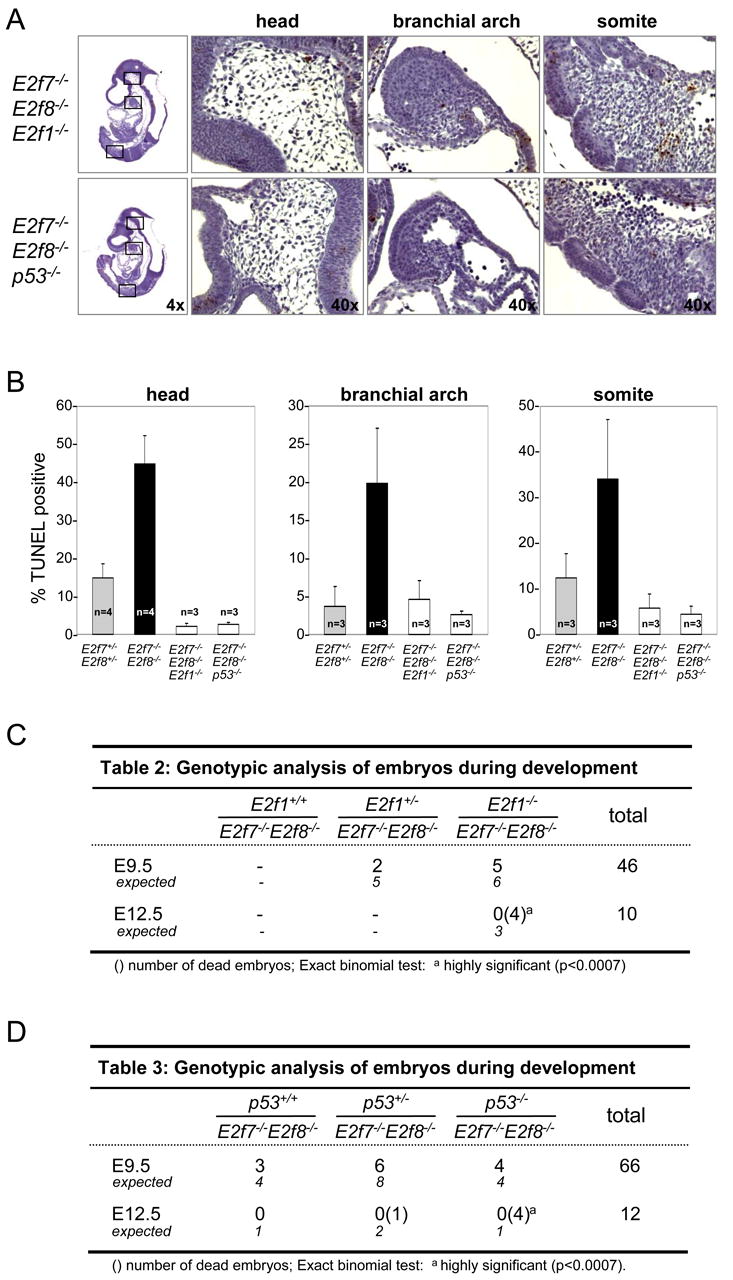
Loss of E2f1 or p53 suppresses apoptosis in E2f7-/-E2f8-/- embryos. (A) Micrographs of TUNEL stained E9.5 E2f7-/-E2f8-/-E2f1-/- (top panels) and E2f7-/-E2f8-/-p53-/- embryos (bottom panels). Far left panels: low magnification pictures of whole embryos. Right three panels: high magnification images of representative areas demarcated by the box in the low magnification images. (B) Quantification of TUNEL-positive cells in the indicated tissue areas are presented as the average ± SD percentage of cells that are TUNEL-positive. Three sections per embryo and at least three different embryos for each genotype were analyzed. For comparison purposes, data derived from E2f7+/-E2f8+/- and E2f7-/-E2f8-/- in Figure 2E, S1C was included. (C) Genotypic analysis of embryos derived from E2f1+/-E2f7+/-E2f8-/- intercrosses at the indicated stages of development. (D) Genotypic analysis of embryos derived from p53+/-E2f7+/-E2f8-/- intercrosses at the indicated stages of development.
Induction of apoptosis in E2f7-/-E2f8-/- embryos is dependent on E2F1 and p53
Given the above observations, we hypothesized that the apoptosis in E2f7-/-E2f8-/- embryos might be mediated through the induction of E2F1 and/or p53. To test this possibility, E2f7+/-E2f8-/- animals were bred with either E2f1-/- or p53-/- animals in order to generate cohorts of triple knockout embryos (TKO). TUNEL assays performed on E9.5 TKO embryos showed that the loss of either E2f1 or p53 suppressed the massive apoptosis caused by a deficiency in E2f7 and E2f8 (Figure 5A and 5B). From these results we conclude that E2F7 and E2F8 represent a critical regulatory arm of the E2F network that controls apoptosis through the E2F1-p53 axis.
Interestingly, both cohorts of TKO embryos harvested at E10.5 had dilated vessels and extensive hemorrhaging similar to DKO embryos (data not shown). Importantly, no live TKO embryos could be observed at E12.5 (Figure 5C-D). These results suggest that the widespread apoptosis observed in DKO embryos, which was suppressed in TKO embryos, is independent of vascular defects and embryonic lethality. These results also indicate that misregulated E2F7- and E2F8-target genes beyond E2f1 are likely to be involved in the mortality of DKO embryos.
Consistent with this view, global analysis of gene expression showed that among the ∼ 39,000 transcripts analyzed, 88 were upregulated and 33 were downregulated at least 3-fold in E2f7-/-E2f8-/-embryos relative to wild type embryos (Figure 6A). Real-time RT-PCR assays confirmed the altered expression of many of these genes (Figure 6B). Functional annotation of misregulated genes revealed a bias for gene products known to be activated in response to stresses, including hypoxia, nutrient deprivation and apoptosis (Figure 6C and S6). These analyses also confirmed that E2f1, cdc6 and additional E2F targets were increased in DKO versus wild type embryos, albeit the increase in their expression was only ∼2-fold (data not shown). Interestingly, a subset of the 122 transcripts (88 up- and 33 down-regulated) were partially misregulated in E2f7-/-, E2f8-/-, and/or E2f7+/-E2f8+/- embryos, underscoring the synergistic role of these two E2F factors in the control of gene expression during embryonic development.
Figure 6.

Microarray analysis of E10.5 embryos. (A) Heat maps of probe sets in Affymetrix microarrays that showed at least a 3-fold induction (left and middle) or a 3-fold reduction (right) of expression in E2f7-/-E2f8-/- relative to wild type embryos. Data are presented as the medium of 4 embryos from wild type, E2f7-/-E2f8+/+ and E2f7+/+E2f8-/- genotype groups, and the medium of 2 embryos from E2f7+/-E2f8+/- and E2f7-/-E2f8-/- genotype groups. Red and green indicate induction and reduction respectively. Probe sets representing genes of interest are indicated. (B) The expression of 8 target genes (6 up-regulated and 2 down-regulated) was evaluated by real-time RT-PCR assays. (C) Pie chart diagrams illustrate the non-random distribution of the stress-related genes among the total 88 up-regulated genes in DKO embryos.
Discussion
The E2F7 and E2F8 transcription factors likely represent the final members of the E2F family to be identified in mammals. We show here that these two novel factors are strictly required for embryonic development and are critical direct regulators of the E2F1-p53 apoptotic axis.
While disruption of E2f7 or E2f8 had little impact on mouse development, their combined ablation resulted in widespread apoptosis, vascular defects and hemorrhaging, leading to embryonic death by E11.5. Provision of even one functional allele from either locus was sufficient to carry fetuses through development all the way to birth. The contribution of E2F7 and E2F8 in postnatal development, however, does not appear to be equal. Young and adult E2f7+/-E2f8-/- mice were developmentally normal, whereas most E2f7-/-E2f8+/- animals appeared runted and died within the first three months of life. A bias in homo- versus hetero-dimerization may explain the differential requirement for E2f7 and E2f8 in postnatal development. We found that in tissue culture experiments, under conditions where expression levels can be compared (by epitope tagging) and experimentally equalized, the formation of E2F7:E2F7 homo-dimers was preferred over E2F7:E2F8 hetero-dimers, and E2F8:E2F8 homo-dimers appeared to be the least preferred state (E2F7:E2F7 > E2F7:E2F8 >E2F8:E2F8). While the molecular basis for homo- versus hetero-dimerization is not yet clear, these data suggest that inefficient homo-dimerization may compromise the ability of E2F8 to compensate for the loss of E2F7, an effect that might be aggravated in circumstances of limiting amounts of E2F8 (i.e. E2f7-/-E2f8+/-). Thus, the observed bias for E2F7 homo-dimerization may explain the stricter postnatal requirement for this subunit. While this interpretation is likely an oversimplification, these results do provide a molecular explanation for their functional redundancy in development. It will be interesting to investigate how dimerization might be impacted by the levels of E2F7 and E2F8 proteins in vivo or by tissue-specific signals.
Because of the intense interest in E2Fs as major regulators of the cell cycle and apoptosis, individual E2F family members, including E2f1 through E2f6, have been extensively studied in vivo by gene targeting approaches in mice. With the exception of E2f3 knockout mice, defects in embryos deficient for each of the known E2Fs are rather subtle (Field et al., 1996; Yamasaki et al., 1996; Lindeman et al., 1998; Humbert et al., 2000a; Murga et al., 2001; Storre et al., 2002; Rempel et al., 2004). Even the disruption of E2f3 when in a mixed genetic background yields viable mice (Humbert et al., 2000b; Wu et al., 2001). The virtual absence of cell proliferation and apoptotic defects in E2F deficient embryos has raised questions about the physiological importance of these factors, leaving the impression that E2Fs must either not be critical for the control of these processes in vivo or that there is sufficient functional redundancy among family members to accommodate for a deficiency in any single member. Previous tissue culture experiments have provided evidence for functional redundancy among E2F1-3 activators and E2F4-6 repressors (Gaubatz et al., 2000; Wu et al., 2001; Giangrande et al., 2004) in the control of proliferation. The current work underscores functional redundancy among E2F7 and E2F8 in the control of apoptosis in vivo.
Too much or unrestrained E2F activity can also compromise cell homeostasis. The importance of restraining E2F activity is highlighted by the fact that at least four independent mechanisms have evolved to achieve this. One such repressive mechanism involves the binding and inhibition of E2F proteins by the Rb tumor suppressor. Indeed, disruption of Rb leads to unregulated proliferation and ectopic apoptosis that is partly suppressed by the concomitant loss of E2F activators (Ziebold et al., 2001; Saavedra et al., 2002). A second mechanism involves binding of cyclinA/cdk2 to E2F1, E2F2 and E2F3a proteins during S phase, leading to the phosphorylation of their dimerization partners DP1/2 and the inhibition of E2F/DP DNA binding activity (Dynlacht et al., 1994). A third mechanism involves the p45SKP2/F-box-dependent degradation of the three E2F activators during S phase (Leone et al., 1998; Marti et al., 1999). Here we described a fourth repressive mechanism to keep E2F activity in control that involves direct transcriptional repression of E2f1 by E2F7 and E2F8. In contrast to other repressor E2Fs (E2F3b, E2F4-6), which play a predominant role in silencing gene expression in G0- G1, these two novel E2Fs may have a particularly important role in repressing E2F targets as cells transit through S phase and into G2. Gene expression analysis in synchronized E2f7-/-E2f8-/- MEFs revealed that E2f1 mRNA levels, as well as that of many other E2F-target genes, increased acutely during S and G2. Thus, by participating in the repression of E2F-target genes as cells transit through S-G2, E2F7 and E2F8 represent the first transcriptional mechanism described in mammals that contributes directly to the downswing in the oscillating pattern of cell cycle-specific gene expression.
In vitro and in vivo experiments described here provide clear-cut evidence for a role of E2F7 and E2F8 in the control of apoptosis that involves, at least in part, the regulation of E2f1 expression. Genetic inactivation of E2f7 and E2f8 resulted in the accumulation of E2f1 mRNA and a corresponding increase in its protein product. Consistent with a direct role in repression, ChIP experiments showed that E2F7 and E2F8 are recruited to the E2f1 promoter and that this requires intact DNA binding activity. The increase of E2F1 protein levels in E2f7-/-E2f8-/- cells coincided with the accumulation of p53 protein. Several mechanisms of how E2F1 may lead to the accumulation and activation of p53 have been described (Hsieh et al., 2002; Pomerantz et al., 1998; Rogoff et al., 2002; Russell et al., 2002). The increase of E2F1 and p53 in E2f7-/-E2f8-/- cells is of physiological significance since ablation of either E2f1 or p53 suppressed the widespread apoptosis observed in DKO embryos. The fact that these TKO embryos still died suggests that apoptosis in E2f7-/-E2f8-/- embryos is not simply due to an indirect consequence associated with embryonic lethality but rather due to a specific signal emanating from deregulated E2F1. These results also indicate that additional targets repressed by E2F7 and E2F8 must be involved in the many pathologies that likely underlie the lethality of DKO embryos. Indeed, profiling of global gene expression in DKO embryos confirmed the deregulation of many additional genes. Interestingly, a substantial portion of these included gene products known to be involved in various responses to stress such as hypoxia, nutrient deprivation and apoptosis. We view these results to mean that physiological stress, whether induced by vascular or other defects in DKO embryos or by the addition of DNA damaging agents in DKO MEFs, exacerbates the activation of the E2F1-p53 axis and unleashes a massive apoptotic response.
In summary, we show that E2F7 and E2F8 are essential for embryonic development. Their synergistic function may be viewed as a distinct arm of the E2F network involved in repression of transcription during S-G2, where E2f1 represents a particularly critical target that if not appropriately repressed can illicit widespread apoptosis in developing embryos.
Experimental Procedures
Generation of E2f7 and E2f8 knockout mice
E2f7 and E2f8 specific probes were used to screen a 129Sv/Ev bacterial artificial chromosome library. A 7.0 kb XbaI fragment of E2f7 spanning exon 4 and 5 was isolated from RPCI 22 431 H17 and a 6.6 kb fragment of E2f8 containing exon 3 and 4 was isolated from RPCI 22 539 P23. Standard cloning techniques were used to generate E2f7 and E2f8 targeting vectors, which were confirmed by direct sequencing. Targeting vectors were linearized with NotI and electroporated into TC1 129Sv/Ev embryonic stem (ES) cells. ES cells were selected in medium containing G418 plus ganciclovir and correct recombination was confirmed by Southern blots using the indicated probes in Figure 1A. Selected ES clones were injected into C57BL/6 blastocysts to generate chimeric mice, which were bred with Black Swiss females to obtain germline transmission. Appropriate offspring were then bred to EIIa-Cre mice (Lakso et al., 1996) to produce mice with conventional and conditional null alleles as illustrated in Figure 1A. Genotypic analysis of offspring was performed by Southern blot analysis and multiplex PCR using allele-specific primers described in Figure S7.
Real-time RT-PCR
Total RNA was isolated using Qiagen RNA miniprep columns as described by the manufacturer, which included the optional DNase treatment before elution from columns. Reverse transcription of total RNA was performed using Superscript III reverse transcriptase (Invitrogen) and RNAse Inhibitor (Roche) according to the manufacturer's protocol. Real-time PCR was performed using a BioRad iCycler and reactions were performed in triplicate and relative amounts of cDNA were normalized to GAPDH. The E2f7 and E2f8 real-time primers are located within the deleted regions of E2f7 and E2f8 respectively. Primer sequences for the indicated genes are included in Figure S7.
Whole mount in situ hybridization
Wild type E9.5 day-old embryos were harvested and fixed in 4% paraformaldehyde. Whole mounts were performed as described (Riddle et al., 1993). Probes for E2f7 and E2f8 were generated by PCR amplification (see Figure S7). The resulting PCR products were subcloned into pBluescript KS (Stratagene) and pGEM-TEasy vector (Promega), respectively. Sense and antisense RNA probes were transcribed from the appropriate promoters using T3 and T7 RNA polymerase (Roche).
Affimetrix microarray analysis
Total RNA was prepared from E10.5 embryos using the RNasy Kit (Qiagen) and included the optional DNase treatment before elution from purification columns. RNA was processed as described in www.osuccc.osu.edu/microarray/ and hybridized to oligonucleotide microarrays (Mouse Genome 430 2.0 array, Affymetrix, Santa Clara, CA). Scanned image files were quantified with GENECHIP 3.2 (Affymetrix). The Raw gene expression data is presented in Supplemental Figure S8. Genes that increased or decreased at least 3-fold (p< 0.008) in DKO samples relative to wild type samples were used to generate the heatmaps illustrated in Figure 6A.
Co-immunoprecipitation assays (Co-IP)
Human embryonic kidney (HEK) 293 cells were cultured in DMEM medium supplemented with 15% FBS and used for co-immunoprecipitation assays. Transient transfections with the indicated constructs were performed by standard calcium chloride techniques. Cells were harvested and washed in PBS at 4°C and cell pellets were lysed in 10 volumes of lysis buffer (0.05M sodium phosphate pH7.3, 0.3M NaCl, 0.1% NP40, 10% glycerol with protease inhibitor cocktail, Roche). Co-IP was performed as described before (Maiti et al., 2005).
Immuno-affinity purification (IAP)
HeLa cells were transduced with retroviruses expressing a bicistronic mRNA encoding human E2F7 or E2F8 (flag-HA-7 and flag-HA-8) and interleukin-2 receptor (IL-2R)-α (Ogawa et al., 2002). The transduced population was incubated with magnetic beads linked to IL-2R specific antibodies. E2F7/8-IL-2R expressing cells were then enriched by separating magnetic beads-bound cells from non-bound cells using a magnetic plate. Nuclear extracts from enriched E2F7/8 expressing cells were prepared by a modified Dignam preparation protocol using Buffer C-450 (20mM HEPES, pH 7.9, 450 mM KCl, 1.5 mM MgCl2, 25% glycerol, 0.2mM EDTA) (Dignam et al., 1983). Resulting extracts were incubated with pre-equilibrated flag M2 affinity resin (A2220, Sigma) for 3h. Resin/extract mixtures were decanted onto Bio-Spin columns (BioRad) and allowed to empty by gravity. After extensive washing with Wash Buffer (20mM HEPES, pH 7.9, 150mM KCl; 10% glycerol, 0.2mM EDTA), complexes were eluted by incubation with 200μg/mL of flag peptide (F3290, Sigma). The flag eluents were subsequently subjected to Western blot analysis using anti-E2F8 antibody (M01, Abnova). All nuclear extraction and immuno-affinity purification steps were peformed at 4°C and fresh protease inhibitors were added to all working solutions.
Chromatin immunoprecipitation (ChIP) and sequential ChIP assays
For ChIP assays, the EZ CHIP™ assay kit (Upstate) was used as described by the manufacturer. Briefly, harvested HEK 293 cells overexpressing flag-E2F7 and/or flag-E2F8 were crosslinked and chromatin was sonicated to an average size of 200-1000 bp. Lysates were subsequently pre-cleared with Salmon Sperm DNA/Protein G agarose slurry. Antibodies specific to flag, HA, or normal mouse IgG (Oncogene) were then added to each sample and incubated overnight at 4°C. Antibody-protein-DNA complexes were recovered by addition of 30 μl of Salmon Sperm DNA/Protein G agarose slurry and incubation for 1h at 4°C. Following extensive washing, the complexes were eluted and decrosslinked at 65°C for 4h. Finally, samples were treated with Proteinase K (Roche) and Rnase A (Roche) and purified through Qiaquick columns (Qiagen). Real-time PCR quantification of immunoprecipitated DNA was performed using the Biorad iCycler machine with primers specific for the indicated promoter regions. All primer sequences are listed in Figure S7. Reactions were performed in triplicate and normalized to 1% of the total input.
Sequential ChIP-PCR was carried out similarly using the same EZ CHIP™ protocol, except two rounds of chromatin immunoprecipitation steps were performed. The first round of immunoprecipitation was performed by incubation of extracts with M2 anti-flag antibody and Salmon Sperm DNA/Protein G agarose slurry. Precipitated DNA-protein complexes were eluted with excess flag peptide (200μg/mL) for 1h at 4°C and then subjected to a second round of precipitation with primary antibodies as indicated in Figure 3H-J. After extensive washing, sequentially precipitated complexes were recovered by addition of 30μl of Salmon Sperm DNA/Protein G agarose slurry and incubation for another 1h at 4°C. Protein-DNA complexes collected from sequential ChIP were treated and subjected to real-time PCR analysis as described above for ChIP experiments.
Western blot and antibodies
Immunoblot analyses were performed by standard procedures using ECL reagents as described by the manufacturer (Amersham Biosciences). The following commercial antibodies were used as indicated in the figures: flag (M2, Sigma), HA (12C5A, Roche), Myc (9E10, Santa Cruz), E2F1 (C-20, Santa Cruz), tubulin (T6199, Sigma), caspase-3 (9662, Cell Signaling), p53 (Ab-1, Oncogene).
Cell culture and viability assay
E2f7loxp/loxpE2f8loxp/loxp mouse embryo fibroblasts (MEFs) were isolated from E13.5 embryos and maintained in DMEM medium containing 15% fetal bovine serum (FBS). Immortalized cell lines were generated from primary MEFs using the standard 3T9 protocol. Immortalized MEFs were treated with retrovirus expressing cre recombinase using standard methods (Wu et al., 2001).
Apoptosis was measured in MEFs at the indicated times following treatment with 10μm cisplatin (Sigma) or 20μm camptothecin (Sigma) for 18h. Cell viability was determined by trypan blue exclusion.
Reporter assay
Luciferase reporter assays were performed in triplicate as described previously (de Bruin et al., 2003). Briefly, cre-treated wild type and E2f7loxP/loxPE2f8loxP/loxP MEFs were transfected with E2f1-Luc reporter plasmid (Araki et al., 2003), along with a thymidine kinase renilla luciferase construct as an internal control. Cells were harvested and luciferase activity was detected using dual luciferase system (Promega) following the manufacturer's protocol. E2f1-Luc reporter plasmid has been described previously.
FACS analysis
Cre-treated wild type and E2f7loxP/loxPE2f8loxP/loxP MEFs were synchronized by starvation in DMEM containing 0.2% FBS for 48h and blocked at the G1/S transition by the addition of DMEM containing 15% FBS with 1mM hydroxyurea (HU) for 18h. Cells were then washed 3 times with PBS and incubated with fresh medium containing 15% FBS. Cells were harvested at the indicated time points and fixed in 70% ethanol, followed by incubation in propidium iodide and analyzed by flow cytometry using standard methods.
BrdU and TUNEL assays
Pregnant females at 9.5 days postcoitum were injected intraperitoneally with BrdU (100ug/grams of body weight) 30min prior to harvesting. Embryos were fixed in formalin and 5μm paraffin embedded-sections were used for immunohistochemistry. After deparaffinization, anti-BrdU antibody (MO-0744, DAKO) and Vectastain Elite ABC reagents (Vector labs) were used to detect BrdU incorporation according to the manufacturer's instructions. Apoptotic cells were detected using TUNEL (S7101, Chemican) assays, performed according to the manufacturer's protocol. All slides were counterstained with hematoxylin.
Supplementary Material
Figure S1. Inactivation of E2f7 and E2f8 induces massive apoptosis in E9.5 embryos. (A) H&E staining of E9.5 embryo mesenchymal tissues. The right panel highlights the nuclear morphology of mesenchymal cells in E2f7-/-E2f8-/- embryos; black arrows point to examples of pyknotic nuclei. (B) Formalin fixed sections of embryos with the indicated genotypes were analyzed by TUNEL assays. Far left panels: low magnification pictures of whole embryos. Right panel: high magnification images of representative areas demarcated by the box in the low magnification images. (C) Quantification of TUNEL-positive cells in the branchial arch areas is presented as the average ± SD percentage of cells that are TUNEL-positive. Three sections per embryo and three different embryos for each genotype were analyzed.
Figure S2. Inactivation of E2f7 and E2f8 does not affect BrdU incorporation in vivo. (A) BrdU staining of embryos with the indicated genotypes. Far left panels: low magnification pictures of whole embryos. Right three panels: high magnification images of representative areas demarcated by the box in the low magnification images. (B) Quantification of proliferation in different tissue areas of embryos is presented as the average ± SD percentage of cells that are BrdU-positive. Three sections per embryo and two different embryos were counted for each genotype group.
Figure S3. DNA binding mutations abolish the DNA binding activity of E2F7. (A) EMSAs of in vitro translated myc-tagged wild type or mutant forms of E2F7 containing amino acid substitutions (Maiti et al., 2005) in the indicated DNA binding domains (DBD1, DBD2, DBD1,2) using a biotin-labeled E2 DNA probe. In vitro translated reactions of wild type E2F7 were incubated with mock buffer (-), anti-myc or IgG antibodies as indicated, or incubated with an unlabeled wild type (wt com.) or mutant E2 (mut com.) probe. (B) Western blot of in vitro translated wild type or mutant forms of myc-tagged E2F7 using anti-myc antibodies.
Figure S4. E2F7 and E2F8 bind to the cdc6 promoter. (A-E) The same chromatin-immunoprecipitated DNA that was used and described in Figure 4F-J was amplified using primers specific for the cdc6 promoter. Real-time PCR was performed in triplicate and cycle numbers were normalized to 1% of the input DNA.
Figure S5. MEFs deficient in E2f7 and E2f8 are hypersensitive to DNA damage induced apoptosis. (A) Total RNA isolated from same synchronized MEFs samples as in Figure 4F was analyzed by real-time RT-PCR assays specific for cdc6 expression. (B) Growth curve of cre-treated E2f7+/+E2f8+/+ and E2f7loxP/loxPE2f8loxP/loxP MEFs. Cells were plated and viable cells counted daily in triplicate. For convenience, cre-deleted loxP alleles were labeled as (-/-). (C) Light microscopy images of MEFs treated as in Figure 4G at 72h. (D) Lysates derived from MEFs treated as in Figure 4G were analyzed by Western blotting using E2F1 and p53 specific antibodies. Tubulin-specific antibodies were used as an internal loading control.
Figure S6. Functional annotation of gene expression. Genes are indicated with their gene symbols, medium Log2-differentials between wild type and E2f7-/-E2f8-/- samples, and gene functions (www.ncbi.nim.nih.gov/entrez). Based on their function annotations, 88 up-regulated genes are grouped in 3 categories and sorted in the descending order of Log2-differentails. References indicate previous reports for these genes function. For complete references list see Supplemental References.
Figure S7. Primer sequence information for PCR procedures.
Figure S8. Raw gene expression data for Affimetrix arrays. P_SET, probe set identification. NLP, probe-level “Negative Log10 P-value” for inter-group differentials. Columns 3-18, all 16 individual expression estimates. Columns 19-22, gene annotation.
Acknowledgments
We are grateful for technical assistance provided by J. Moffitt and J. Opavska. We also thank members of the Leone lab for helpful suggestions and Pam Wenzel for critical reading of the manuscript. This work was funded by NIH grants to G.L. (R01CA85619, R01CA82259, R01HD047470, P01CA097189), NIH grant to M.W. (R01HD42619) and DoD award to A.d.B. (BC030892). G.L. is the recipient of The Pew Charitable Trust Scholar Award and the Leukemia & Lymphoma Society Scholar Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests statement: The authors declare that they have no competing financial interests.
References
- Araki K, Nakajima Y, Eto K, Ikeda MA. Distinct recruitment of E2F family members to specific E2F-binding sites mediates activation and repression of the E2F1 promoter. Oncogene. 2003;22:7632–7641. doi: 10.1038/sj.onc.1206840. [DOI] [PubMed] [Google Scholar]
- Attwooll C, Lazzerini Denchi E, Helin K. The E2F family: specific functions and overlapping interests. EMBO J. 2004;23:4709–4716. doi: 10.1038/sj.emboj.7600481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken AP, Ciro M, Cocito A, Helin K. E2F target genes: unraveling the biology. Trends Biochem Sci. 2004;29:409–417. doi: 10.1016/j.tibs.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Christensen J, Cloos P, Toftegaard U, Klinkenberg D, Bracken AP, Trinh E, Heeran M, Di Stefano L, Helin K. Characterization of E2F8, a novel E2F-like cell-cycle regulated repressor of E2F-activated transcription. Nucleic Acids Res. 2005;33:5458–5470. doi: 10.1093/nar/gki855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin A, Maiti B, Jakoi L, Timmers C, Buerki R, Leone G. Identification and characterization of E2F7, a novel mammalian E2F family member capable of blocking cellular proliferation. J Biol Chem. 2003;278:42041–42049. doi: 10.1074/jbc.M308105200. [DOI] [PubMed] [Google Scholar]
- DeGregori J, Johnson DG. Distinct and overlapping roles for E2F family members in transcription, proliferation and apoptosis. Curr Mol Med. 2006;6:739–748. doi: 10.2174/1566524010606070739. [DOI] [PubMed] [Google Scholar]
- DeGregori J, Leone G, Miron A, Jakoi L, Nevins JR. Distinct roles for E2F proteins in cell growth control and apoptosis. Proc Natl Acad Sci U S A. 1997;94:7245–7250. doi: 10.1073/pnas.94.14.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stefano L, Jensen MR, Helin K. E2F7, a novel E2F featuring DP-independent repression of a subset of E2F-regulated genes. EMBO J. 2003;22:6289–6298. doi: 10.1093/emboj/cdg613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimova DK, Dyson NJ. The E2F transcriptional network: old acquaintances with new faces. Oncogene. 2005;24:2810–2826. doi: 10.1038/sj.onc.1208612. [DOI] [PubMed] [Google Scholar]
- Dynlacht BD, Flores O, Lees JA, Harlow E. Differential regulation of E2F transactivation by cyclin/cdk2 complexes. Genes Dev. 1994;8:1772–1786. doi: 10.1101/gad.8.15.1772. [DOI] [PubMed] [Google Scholar]
- Field SJ, Tsai FY, Kuo F, Zubiaga AM, Kaelin WG, Jr, Livingston DM, Orkin SH, Greenberg ME. E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell. 1996;85:549–561. doi: 10.1016/s0092-8674(00)81255-6. [DOI] [PubMed] [Google Scholar]
- Gaubatz S, Lindeman GJ, Ishida S, Jakoi L, Nevins JR, Livingston DM, Rempel RE. E2F4 and E2F5 play an essential role in pocket protein-mediated G1 control. Mol Cell. 2000;6:729–735. doi: 10.1016/s1097-2765(00)00071-x. [DOI] [PubMed] [Google Scholar]
- Giangrande PH, Zhu W, Schlisio S, Sun X, Mori S, Gaubatz S, Nevins JR. A role for E2F6 in distinguishing G1/S- and G2/M-specific transcription. Genes Dev. 2004;18:2941–2951. doi: 10.1101/gad.1239304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh JK, Yap D, O'Connor DJ, Fogal V, Fallis L, Chan F, Zhong S, Lu X. Novel function of the cyclin A binding site of E2F in regulating p53-induced apoptosis in response to DNA damage. Mol Cell Biol. 2002;22:78–93. doi: 10.1128/MCB.22.1.78-93.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert PO, Rogers C, Ganiatsas S, Landsberg RL, Trimarchi JM, Dandapani S, Brugnara C, Erdman S, Schrenzel M, Bronson RT, et al. E2F4 is essential for normal erythrocyte maturation and neonatal viability. Mol Cell. 2000a;6:281–291. doi: 10.1016/s1097-2765(00)00029-0. [DOI] [PubMed] [Google Scholar]
- Humbert PO, Verona R, Trimarchi JM, Rogers C, Dandapani S, Lees JA. E2f3 is critical for normal cellular proliferation. Genes Dev. 2000b;14:690–703. [PMC free article] [PubMed] [Google Scholar]
- Kosugi S, Ohashi Y. E2Ls, E2F-like repressors of Arabidopsis that bind to E2F sites in a monomeric form. J Biol Chem. 2002;277:16553–16558. doi: 10.1074/jbc.M200913200. [DOI] [PubMed] [Google Scholar]
- Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, Westphal H. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci U S A. 1996;93:5860–5865. doi: 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone G, DeGregori J, Yan Z, Jakoi L, Ishida S, Williams RS, Nevins JR. E2F3 activity is regulated during the cell cycle and is required for the induction of S phase. Genes Dev. 1998;12:2120–2130. doi: 10.1101/gad.12.14.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeman GJ, Dagnino L, Gaubatz S, Xu Y, Bronson RT, Warren HB, Livingston DM. A specific, nonproliferative role for E2F-5 in choroid plexus function revealed by gene targeting. Genes Dev. 1998;12:1092–1098. doi: 10.1101/gad.12.8.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan N, Delavaine L, Graham A, Reilly C, Wilson J, Brummelkamp TR, Hijmans EM, Bernards R, La Thangue NB. E2F-7: a distinctive E2F family member with an unusual organization of DNA-binding domains. Oncogene. 2004;23:5138–5150. doi: 10.1038/sj.onc.1207649. [DOI] [PubMed] [Google Scholar]
- Logan N, Graham A, Zhao X, Fisher R, Maiti B, Leone G, La Thangue NB. E2F-8: an E2F family member with a similar organization of DNA-binding domains to E2F-7. Oncogene. 2005;24:5000–5004. doi: 10.1038/sj.onc.1208703. [DOI] [PubMed] [Google Scholar]
- Maiti B, Li J, de Bruin A, Gordon F, Timmers C, Opavsky R, Patil K, Tuttle J, Cleghorn W, Leone G. Cloning and characterization of mouse E2F8, a novel mammalian E2F family member capable of blocking cellular proliferation. J Biol Chem. 2005;280:18211–18220. doi: 10.1074/jbc.M501410200. [DOI] [PubMed] [Google Scholar]
- Mariconti L, Pellegrini B, Cantoni R, Stevens R, Bergounioux C, Cella R, Albani D. The E2F family of transcription factors from Arabidopsis thaliana. Novel and conserved components of the retinoblastoma/E2F pathway in plants. J Biol Chem. 2002;277:9911–9919. doi: 10.1074/jbc.M110616200. [DOI] [PubMed] [Google Scholar]
- Marti A, Wirbelauer C, Scheffner M, Krek W. Interaction between ubiquitin-protein ligase SCFSKP2 and E2F-1 underlies the regulation of E2F-1 degradation. Nat Cell Biol. 1999;1:14–19. doi: 10.1038/8984. [DOI] [PubMed] [Google Scholar]
- Muller H, Helin K. The E2F transcription factors: key regulators of cell proliferation. Biochim Biophys Acta. 2000;1470:M1–12. doi: 10.1016/s0304-419x(99)00030-x. [DOI] [PubMed] [Google Scholar]
- Murga M, Fernandez-Capetillo O, Field SJ, Moreno B, Borlado LR, Fujiwara Y, Balomenos D, Vicario A, Carrera AC, Orkin SH, et al. Mutation of E2F2 in mice causes enhanced T lymphocyte proliferation, leading to the development of autoimmunity. Immunity. 2001;15:959–970. doi: 10.1016/s1074-7613(01)00254-0. [DOI] [PubMed] [Google Scholar]
- O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- Ogawa H, Ishiguro K, Gaubatz S, Livingston DM, Nakatani Y. A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science. 2002;296:1132–1136. doi: 10.1126/science.1069861. [DOI] [PubMed] [Google Scholar]
- Pomerantz J, Schreiber-Agus N, Liegeois NJ, Silverman A, Alland L, Chin L, Potes J, Chen K, Orlow I, Lee HW, et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2's inhibition of p53. Cell. 1998;92:713–723. doi: 10.1016/s0092-8674(00)81400-2. [DOI] [PubMed] [Google Scholar]
- Reis T, Edgar BA. Negative regulation of dE2F1 by cyclin-dependent kinases controls cell cycle timing. Cell. 2004;117:253–264. doi: 10.1016/s0092-8674(04)00247-8. [DOI] [PubMed] [Google Scholar]
- Rempel RE, Saenz-Robles MT, Storms R, Morham S, Ishida S, Engel A, Jakoi L, Melhem MF, Pipas JM, Smith C, et al. Loss of E2F4 activity leads to abnormal development of multiple cellular lineages. Mol Cell. 2000;6:293–306. doi: 10.1016/s1097-2765(00)00030-7. [DOI] [PubMed] [Google Scholar]
- Resnitzky D, Gossen M, Bujard H, Reed SI. Acceleration of the G1/S phase transition by expression of cyclins D1 and E with an inducible system. Mol Cell Biol. 1994;14:1669–1679. doi: 10.1128/mcb.14.3.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle RD, Johnson RL, Laufer E, Tabin C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell. 1993;75:1401–1416. doi: 10.1016/0092-8674(93)90626-2. [DOI] [PubMed] [Google Scholar]
- Rogoff HA, Pickering MT, Debatis ME, Jones S, Kowalik TF. E2F1 induces phosphorylation of p53 that is coincident with p53 accumulation and apoptosis. Mol Cell Biol. 2002;22:5308–5318. doi: 10.1128/MCB.22.15.5308-5318.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell JL, Powers JT, Rounbehler RJ, Rogers PM, Conti CJ, Johnson DG. ARF differentially modulates apoptosis induced by E2F1 and Myc. Mol Cell Biol. 2002;22:1360–1368. doi: 10.1128/mcb.22.5.1360-1368.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saavedra HI, Wu L, de Bruin A, Timmers C, Rosol TJ, Weinstein M, Robinson ML, Leone G. Specificity of E2F1, E2F2, and E2F3 in mediating phenotypes induced by loss of Rb. Cell Growth Differ. 2002;13:215–225. [PubMed] [Google Scholar]
- Seville LL, Shah N, Westwell AD, Chan WC. Modulation of pRB/E2F functions in the regulation of cell cycle and in cancer. Curr Cancer Drug Targets. 2005;5:159–170. doi: 10.2174/1568009053765816. [DOI] [PubMed] [Google Scholar]
- Stevens C, La Thangue NB. The emerging role of E2F-1 in the DNA damage response and checkpoint control. DNA Repair (Amst) 2004;3:1071–1079. doi: 10.1016/j.dnarep.2004.03.034. [DOI] [PubMed] [Google Scholar]
- Storre J, Elsasser HP, Fuchs M, Ullmann D, Livingston DM, Gaubatz S. Homeotic transformations of the axial skeleton that accompany a targeted deletion of E2f6. EMBO Rep. 2002;3:695–700. doi: 10.1093/embo-reports/kvf141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Timmers C, Maiti B, Saavedra HI, Sang L, Chong GT, Nuckolls F, Giangrande P, Wright FA, Field SJ, et al. The E2F1-3 transcription factors are essential for cellular proliferation. Nature. 2001;414:457–462. doi: 10.1038/35106593. [DOI] [PubMed] [Google Scholar]
- Yamasaki L, Jacks T, Bronson R, Goillot E, Harlow E, Dyson NJ. Tumor induction and tissue atrophy in mice lacking E2F-1. Cell. 1996;85:537–548. doi: 10.1016/s0092-8674(00)81254-4. [DOI] [PubMed] [Google Scholar]
- Ziebold U, Reza T, Caron A, Lees JA. E2F3 contributes both to the inappropriate proliferation and to the apoptosis arising in Rb mutant embryos. Genes Dev. 2001;15:386–391. doi: 10.1101/gad.858801. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Inactivation of E2f7 and E2f8 induces massive apoptosis in E9.5 embryos. (A) H&E staining of E9.5 embryo mesenchymal tissues. The right panel highlights the nuclear morphology of mesenchymal cells in E2f7-/-E2f8-/- embryos; black arrows point to examples of pyknotic nuclei. (B) Formalin fixed sections of embryos with the indicated genotypes were analyzed by TUNEL assays. Far left panels: low magnification pictures of whole embryos. Right panel: high magnification images of representative areas demarcated by the box in the low magnification images. (C) Quantification of TUNEL-positive cells in the branchial arch areas is presented as the average ± SD percentage of cells that are TUNEL-positive. Three sections per embryo and three different embryos for each genotype were analyzed.
Figure S2. Inactivation of E2f7 and E2f8 does not affect BrdU incorporation in vivo. (A) BrdU staining of embryos with the indicated genotypes. Far left panels: low magnification pictures of whole embryos. Right three panels: high magnification images of representative areas demarcated by the box in the low magnification images. (B) Quantification of proliferation in different tissue areas of embryos is presented as the average ± SD percentage of cells that are BrdU-positive. Three sections per embryo and two different embryos were counted for each genotype group.
Figure S3. DNA binding mutations abolish the DNA binding activity of E2F7. (A) EMSAs of in vitro translated myc-tagged wild type or mutant forms of E2F7 containing amino acid substitutions (Maiti et al., 2005) in the indicated DNA binding domains (DBD1, DBD2, DBD1,2) using a biotin-labeled E2 DNA probe. In vitro translated reactions of wild type E2F7 were incubated with mock buffer (-), anti-myc or IgG antibodies as indicated, or incubated with an unlabeled wild type (wt com.) or mutant E2 (mut com.) probe. (B) Western blot of in vitro translated wild type or mutant forms of myc-tagged E2F7 using anti-myc antibodies.
Figure S4. E2F7 and E2F8 bind to the cdc6 promoter. (A-E) The same chromatin-immunoprecipitated DNA that was used and described in Figure 4F-J was amplified using primers specific for the cdc6 promoter. Real-time PCR was performed in triplicate and cycle numbers were normalized to 1% of the input DNA.
Figure S5. MEFs deficient in E2f7 and E2f8 are hypersensitive to DNA damage induced apoptosis. (A) Total RNA isolated from same synchronized MEFs samples as in Figure 4F was analyzed by real-time RT-PCR assays specific for cdc6 expression. (B) Growth curve of cre-treated E2f7+/+E2f8+/+ and E2f7loxP/loxPE2f8loxP/loxP MEFs. Cells were plated and viable cells counted daily in triplicate. For convenience, cre-deleted loxP alleles were labeled as (-/-). (C) Light microscopy images of MEFs treated as in Figure 4G at 72h. (D) Lysates derived from MEFs treated as in Figure 4G were analyzed by Western blotting using E2F1 and p53 specific antibodies. Tubulin-specific antibodies were used as an internal loading control.
Figure S6. Functional annotation of gene expression. Genes are indicated with their gene symbols, medium Log2-differentials between wild type and E2f7-/-E2f8-/- samples, and gene functions (www.ncbi.nim.nih.gov/entrez). Based on their function annotations, 88 up-regulated genes are grouped in 3 categories and sorted in the descending order of Log2-differentails. References indicate previous reports for these genes function. For complete references list see Supplemental References.
Figure S7. Primer sequence information for PCR procedures.
Figure S8. Raw gene expression data for Affimetrix arrays. P_SET, probe set identification. NLP, probe-level “Negative Log10 P-value” for inter-group differentials. Columns 3-18, all 16 individual expression estimates. Columns 19-22, gene annotation.