

Abstract
Evidence of oxidative stress and the accumulation of fibrillar amyloid β proteins (Aβ) in senile plaques throughout the cerebral cortex are consistent features in the pathology of Alzheimer disease. To define a mechanistic link between these two processes, various aspects of the relationship between oxidative lipid membrane damage and amyloidogenesis were characterized by chemical and physical techniques. Earlier studies of this relationship demonstrated that oxidatively damaged synthetic lipid membranes promoted amyloidogenesis. The studies reported herein specify that 4-hydroxy-2-nonenal (HNE) is produced in both synthetic lipids and human brain lipid extracts by oxidative lipid damage and that it can account for accelerated amyloidogenesis. Aβ promotes the copper-mediated generation of HNE from polyunsaturated lipids, and in turn, HNE covalently modifies the histidine side chains of Aβ. HNE-modified Aβ have an increased affinity for lipid membranes and an increased tendency to aggregate into amyloid fibrils. Thus, the prooxidant activity of Aβ leads to its own covalent modification and to accelerated amyloidogenesis. These results illustrate how lipid membranes may be involved in templating the pathological misfolding of Aβ, and they suggest a possible chemical mechanism linking oxidative stress with amyloid formation.
Alzheimer disease (AD)3 is an age-related neurodegenerative disorder characterized by misfolded and aggregated fibrillar amyloid β proteins (Aβ) in the brain. Among the factors associated with the pathogenesis of AD, oxidative stress is one of the most closely scrutinized (1, 2). It has been shown, for example, that the brain in AD has increased susceptibility to oxidative stress (3–5) and that isoprostanes, markers of oxidative stress, are specifically elevated (6–8). Isoprostanes are chemically stable and nonreactive compounds that arise nonenzymatically from the spontaneous decomposition of lipid hydroperoxides. These hydroperoxides may decompose along other pathways, however, yielding highly reactive short chain alkenals such as 4-oxo-2-nonenal and 4-hydroxy-2-nonenal (HNE) (9–17). HNE concentrations in human ventricular fluid are 8–15 μM and elevated in AD (3, 18, 19). HNE has a well known propensity to react with the side chains of various amino acid residues, and HNE-protein adducts have been used as biomarkers of oxidative stress (15).
In light of these observations, it is noteworthy that lipid oxidation products such as HNE modify Aβ and increase Aβ misfolding (20–25). Moreover, the immunoreactivity of antibodies to HNE-modified His residues localizes to amyloid plaques (26, 27). This suggests that not only does Aβ promote lipid oxidation but that there may also be a mechanistic link between the lipid oxidation products formed during oxidative stress and Aβ misfolding (21). Conversely, several lines of evidence suggest that Aβ contribute to oxidative stress. For example, the over-expression of Aβ in transgenic mice, in Caenorhabditis elegans, and in cell culture results in an increase in biomarkers of oxidative stress and in HNE production (28–30). The mechanism underlying this relationship is unknown, but Aβ complexed with Cu(II) promote the oxidation of diverse substances, including cholesterol and phospholipids (31–35). Furthermore, lipid oxidative products and lipid susceptibility to oxidative damage are increased in AD (1, 3, 19, 36–39).
We have demonstrated previously that oxidatively damaged lipid membranes promote the misfolding and aggregation of amyloid β proteins (Aβ) into fibrils, and that misfolded Aβ promote oxidative damage in synthetic lipid membranes. In further investigations of the phenomena described below, we have verified that Aβ promote oxidative damage in human brain lipids, identified HNE as an oxidation product that by itself mimics the effect of oxidatively damaged membranes on the misfolding and aggregation of Aβ, identified the nature of the chemical reaction between HNE and Aβ, and demonstrated that HNE modification of Aβ promotes misfolding, aggregation, and membrane association. Most significantly, we have demonstrated that Aβ are covalently modified by the HNE that they help produce. This represents a positive feedforward mechanism involving oxidative damage and aggregation, in which Aβ promotes oxidative damage, and the products of oxidative damage promote fibril formation.
EXPERIMENTAL PROCEDURES
Materials
Synthetic 1-stearoyl-2-arachidonyl-sn-glycero-3-phosphocholine (SAPC) in chloroform and 1,2-dimyristoyl-sn-glycero-phosphocholine (DMPC) powder were obtained from Avanti Polar Lipids (Alabaster, AL). SAPC was packaged as 10-mg aliquots in sealed glass ampoules, under argon, stored at −80 °C, and lyophilized overnight prior to use. Excision grade trypsin and the endoproteinase Asp-N were obtained from Calbiochem. The trypsin stock concentration was 1 μg/μl in 50 mM ammonium bicarbonate at pH 8.5, and the Asp-N stock was 0.04 μg/μl in H2O. 4-Hydroxy-2-nonenal (HNE) and deuterated HNE (d3-HNE) were purchased from Cayman Chemical (Ann Arbor, MI.). 3,5-Di-tert-butylhydroxytoluene (BHT), O-2,3,4,5,6-(pentafluorobenzyl)hydroxylamine hydrochloride (PFBHA), diethylenetriaminepentaacetic acid (DTPA), and bis(trimethylsilyl)trifluoroacetamide were obtained from Sigma. α-Cyano-4-hydroxycinnamic acid (CHCA) and 2,5-dimethoxy-4-hydroxycinnamic acid (sinapinic acid) (>99% pure) were also obtained from Sigma. Water was purified through an Elix and MilliQ A10 synthesis water purification system (Millipore, Bedford, MA). When needed, Cu(II) was added as CuSO4. Although several experiments were nominally free of Cu(II) ions, no special procedures were employed to remove trace amounts of Cu(II) except where noted.
Lyophilized Aβ-(1–40) (Aβ40) and Aβ-(1–42) (Aβ42), both at >95% purity, were obtained from rPeptide (Athens, GA). Aβ-(1–11), Aβ-(10–20), and Aβ-(22–35) were obtained from American Peptide Co. (Sunnyvale, CA). Aβ-(16–20) (KLVFF) (40) was obtained from Bachem Bioscience (King of Prussia, PA). Before use, proteins were stored desiccated at −20 °C. Stock solutions at a concentration of 0.5 mg/ml were prepared by dissolving Aβ40 and Aβ42 in hexafluoroisopropanol, dissolving Aβ-(1–11), Aβ-(10–20), and Aβ-(16–20) in H2O, and dissolving Aβ-(22–35) in 30% acetonitrile with 0.1% trifluoroacetic acid. Hexafluoroisopropanol in the Aβ40 and Aβ42 stocks was evaporated immediately prior to use, and the proteins were redissolved in aqueous buffer at concentrations of 1–5 μM. Aβ-(1–28), Aβ-(1–11), Aβ-(10–20), and Aβ-(22–35) stocks were diluted into aqueous buffer at concentrations of 1–5 μM.
Extraction of Brain Lipids
Frozen samples of normal human brain tissue from the temporal cortex were obtained from the Center for Neurodegenerative Disease Research Brain Bank, at the University of Pennsylvania. Lipids were extracted from ~4-mg specimens using a modified Folch method, described below (41, 42). To minimize lipid oxidation during the extraction process, samples and aqueous solvents were purged with argon. The tissue was homogenized with an ultrasonic tip dismembranator in 10 ml/g methanol two times for 10 s on ice. 20 ml/g of chloroform was added, and the tissue was further sonicated two times for 10 s at room temperature. After centrifugation at 5000 × g for 2 min, the supernatant was removed and kept. The pellet was resuspended in 30 ml/g of chloroform:methanol (2:1), sonicated two times for 10 s on ice, incubated at room temperature for 5 min, and centrifuged as before. This supernatant was added to that of the first extract. The combined supernatants were sequentially washed with 0.88% potassium chloride and then methanol:saline (1:1) to remove non-lipid contaminants such as salts, amino acids, sugars, and urea. Following low speed centrifugation after the final wash, the lower organic layer was removed, lyophilized, and extruded into lipid vesicles as described below.
Lipid Vesicle Preparation, Oxidation, and Analysis
SAPC, DMPC, and brain lipid extract vesicle suspensions were prepared by extrusion as described previously (32). Immediately prior to use, aliquots were mixed with 5 mM HEPES, pH 7.5, and the oxidation was initiated by the addition of ascorbate and Cu(II). Final concentrations in these suspensions were 10 μM SAPC and 25 μM DMPC or 10 μM brain lipid and 1 μM DMPC in a final volume of 400 μl. Where Aβ42 was used, it was premixed with Cu(II) and incubated for 30 min prior to the addition of other reactants as described previously (32). Quantitative determination of SAPC content relative to a DMPC internal standard was performed by multiple reaction monitoring (MRM) mass spectrometry (LC/MS/MS) as described previously (32).
HNE Assay
HNE concentrations were determined by following HNE derivatization, organic extraction, and gas chromatography mass/spectrometry (GC/MS) similar to the method described previously (43). The samples (400 μl) were analyzed 120 min after initiating oxidation. At this point 10 μM DTPA was added to stop the oxidation, 620 nM d3-HNE was added as an internal standard, and the HNE within the mixture was derivatized with 16 mM PFBHA for 1 h at room temperature. At this point, samples were extracted with 2 volumes of chloroform:methanol (2:1). Following low speed centrifugation, the lower organic layer was removed, passed through a 0.45-μm perfluorocarbon filter, and dried under nitrogen. The dried material was dissolved in 10 μl of pyridine and further derivatized by adding 10 μl of bis(trimethylsilyl)trifluoroacetamide. After 10 min, the samples were dried, dissolved in 100 μl of dodecane, and assayed with the GC/MS in negative ion electron capture ionization mode using ammonia as the collision gas. A temperature program increasing at a rate of 30 °C/min from 190 to 250 °C was used to separate the derivatized d3-HNE and HNE on a DB 35-MS capillary GC column (60 m × 0.25 mm, 0.25 μm coating). The ions monitored were m/z 286.3 for d3-HNE and m/z 283.3 for HNE. Peak areas quantified and expressed as 283.3:286.3 ratios.
Mass Spectrometric Analysis of Aβ-HNE Adducts
Aβ40, Aβ42, or a mixture of Aβ-(1–11), Aβ-(10–20), and Aβ-(22–35) (5 μM each) were incubated with 3.8 mM HNE in 10 mM phosphate buffer, pH 7.4, for 1–3 h at 37 °C. Prior to the addition of HNE, 2.5 mM DMPC lipid vesicles and 50 μM of the Aβ aggregation inhibitor Aβ-(16–20) were added to reduce aggregation of HNE-modified Aβ. The reaction of Aβ with HNE was terminated by addition of an equal volume of trifluoroacetic acid, and the samples were lyophilized. For samples subjected to Asp-N digestion, the reaction between HNE and Aβ was terminated by the addition of an equal volume of 100 mM ammonium bicarbonate solution, pH 8.5. The reaction of any excess HNE with ammonium ions allowed for subsequent digestion of the Aβ-HNE adducts with Asp-N. 30 μl of the HNE/Aβ reaction mixture (~1 μg of Aβ) was quenched with 30 μl of 100 mM ammonium bicarbonate, pH 8.5. 1 μl of Asp-N (0.04 μg) was added to 60 μl of this mixture (~1 μg of Aβ) and incubated overnight at 37 °C. The enzyme was inactivated by adding 60 μl of trifluoroacetic acid, and the sample was lyophilized. For MALDI-mass spectrometry, the Aβ-HNE adducts or Aβ-HNE adduct digests were dissolved in either 30 μl of a CHCA solution (5 mg of CHCA in 1 ml of 50% ACN with 0.3% trifluoroacetic acid) or sinapinic acid solution (10 mg sinapinic acid in 30% ACN with 0.3% trifluoroacetic acid). MALDI analysis was performed on TOF, Q-TOF, and TOF-Q-TOF instruments (Voyager DE, QStar, and model 4700 mass spectrometers, respectively; Applied Biosystems/MDS Sciex, Foster City, CA).
Immunoblot Analysis of Aβ and Aβ-HNE Adducts
Murine anti-His-HNE monoclonal antibody (HNEJ-2) was obtained from Genox Corp. (Baltimore, MD) (44). 6E10 antibodies with specificity for Aβ residues 4–9 and 4G8 antibodies with specificity for residues 18–22 were obtained from Senetek (Maryland Heights, MO) by a kind gift from Domenico Pratico. Donkey anti-mouse antibody conjugated to horseradish peroxidase (SC2314) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA).
Slot blots were used to detect the modification of Aβ by endogenous HNE generated during lipid oxidation. 200 μl of the Aβ oxidation reaction solution containing 4.5 μg of protein was mixed with 200 μl of 100 mM ammonium bicarbonate, pH 7.5. Samples were adsorbed onto 0.2-μm nitrocellulose membranes by slot blot microfiltration (Bio-Dot SF microfiltration apparatus), and the wells were washed twice with 50 mM Tris, 150 mM NaCl, pH 7.4. Membranes were blocked with a 5% solution of powdered fat-free milk (BLOTTO in TTBS; Bio-Rad). The slot blot membranes were incubated overnight with 15 μg/ml of the antibody to HNE-modified histidine (HNE-J) at 4 °C. Following washes in TTBS and incubation of the membranes with secondary antibody (diluted 1:1000) for 1 h, the blot was incubated with Renaissance Luminol reagents (PerkinElmer Life Sciences). X-Omat Blue XB-1 film (Eastman Kodak Co.) was exposed to blots of synthetic lipids for 10 s and to blots of brain-derived lipids for 10 min. Quantification of the slot blot was performed by digitally scanning the X-Omat film and measuring film background and slot blot image densities with ImageQuant TL software (Amersham Biosciences).
Western blots were used to detect Aβ and modification of Aβ with excesses of synthetic HNE. Both synthetic Aβ and HNE-modified Aβ were electrophoresed on a Tris-Tricine 10–20% polyacrylamide gel (72 ng of protein per well) and were transferred to 0.2-μm nitrocellulose membranes. The membranes were removed and washed with Tween in Tris-buffered saline (TTBS, 0.1% Tween 20 in 150 mM NaCl, 20 mM Tris, pH 7.6). Aβ epitope retrieval was performed by boiling the unblocked membrane in PBS for 5 min. The membranes were immunoblotted overnight at 4 °C with the HNEJ-2 antibody and Aβ antibodies (4G8, 1:200 and 6E10, 1:500). They were then washed, incubated with Luminol reagents, and exposed in the same manner as the slot blots.
Congo Red (CR) Binding Assay
Aβ40 (10 μM) or Aβ42 (2 μM) was incubated at a 1:1 mole ratio with DMPC vesicles containing 3 mol % HNE in PBS (150 mM NaCl, 10 mM sodium phosphate, pH 7.4) at room temperature with continuous gentle agitation. The HNE concentrations were therefore 0.3 and 0.06 μM for Aβ40 and Aβ42, respectively. At intervals, aliquots of Aβ40 (112 μl) and Aβ42 (98 μl) were added to 28 or 42 μl, respectively, of 10 μM CR in PBS and incubated for 30 min at room temperature. Fibril formation was assayed by measuring the ratio of sample absorption at 541 and 403 nm, which are the wavelengths of maximum difference and of an isosbestic point for fibril-bound CR and unbound CR, respectively.
Infrared Spectroscopy
Polarized attenuated total internal reflection Fourier transform infrared (PATIR-FTIR) spectra were collected in rapid-scanning mode as 1024 co-added inter-ferograms using a Bio-Rad FTS-60A spectrometer, a liquid nitrogen-cooled MCT detector, an aluminum wire grid polarizer, a resolution of 2 cm−1, scanning speed of 20 kHz, triangular apodization, and one level of zero filling. Supported lipid monolayers, composed of either DMPC or brain lipid extract, were prepared in a Langmuir trough by applying ~5 nmol of DMPC in a mixture of hexane:ethanol (9:1 by volume) to the surface of a subphase buffer. An enclosure around the Langmuir trough is filled with argon to avoid spontaneous air oxidation of lipids at the air-water interface, and all studies were performed at ~21 °C. The subphase buffer contained 30 mM HEPES buffer in D2O at pD 7.4. When indicated, an aliquot of HNE in ethanol was lyophilized, resuspended in subphase buffer, and injected into the subphase before forming the monolayer. For DMPC membranes, 5 μl of 28 mM HNE stock was injected into 6 ml of subphase to yield an HNE concentration of 23 μM in the subphase. For brain lipid extracts, 10 μl of a 32 mM HNE stock solution was injected into a 2.5-ml subphase volume to yield an HNE concentration of 128 μM in the subphase.
The monolayer was compressed to a surface pressure of 20 dynes/cm and applied onto a silane-treated germanium internal reflection crystal as described previously (45). At this point, 5 μl of 28 mM HNE stock in ethanol was lyophilized, resuspended in the subphase buffer, and injected into the subphase. After collecting a base-line spectrum, 500 ng of samples of Aβ40 or Aβ42 in 30 mM NaDPO4 buffer, pD 11.9, were injected into the continuously stirred subphase to yield a protein concentration of 300 nM in experiments with DMPC and 230 nM in experiments with brain lipid extracts. Sample spectra were collected over 90 and 125 min, as indicated. A flat base-line correction was performed on sample spectra assuming zero absorbance at 1800 cm−1, but no water vapor subtraction or smoothing manipulations were performed. The spectra were fitted using IRfit, a procedure that fits a limited set of component bands simultaneously to several spectra with a minimum number of adjustable parameters (46). In this study, one simultaneous fit was performed on 12 spectra from three independent experiments. Only the spectra region between 1700 and 1600 cm−1 was fitted.
RESULTS
Aβ42 and Brain Lipid Oxidation
The ability of Aβ42 to promote the oxidation of SAPC in human brain lipid extracts was measured using an MRM-LC/MS/MS assay as described previously (32). This assay is superior to many other assays of oxidative damage in that it yields precise and unambiguous quantitative information about oxidative loss of a specific substrate. Two adaptations of the conditions described previously for synthetic lipids were necessary for the application of this assay to human brain lipid extracts. First, brain vesicle suspensions with phosphate concentrations of 10 μM contained virtually no DMPC by MRM-LC/MS/MS. Therefore, 1 μM DMPC was added to all brain lipid vesicles as an internal standard. The SAPC concentration in these extracts was 0.34 ± 0.1 μM. Second, 1.5 mM MgSO4 was added to minimize sequestration of Cu(II) by anionic lipids.
Ascorbate (50 μM) and Cu(II) (0.5 μM) reduced SAPC to 79 ± 19% of original levels after 120 min (Fig. 1). The addition of 5 μM Aβ42 decreased it to 38 ± 16% of its original level over the same time interval. These results are consistent with previously reported results from synthetic SAPC and DMPC vesicles, although the variability of the data was considerably larger in these human lipid extracts (32). The data in Fig. 1 only represent four of the five extracts that were examined. Lipids from brain specimen 3 (Table 1) had only about half of the SAPC content as the other samples, and it was completely resistant to oxidation with or without Aβ42 present. Therefore, this sample was excluded from our analysis.
FIGURE 1. Prooxidant activity of Aβ in brain-derived lipid vesicles.
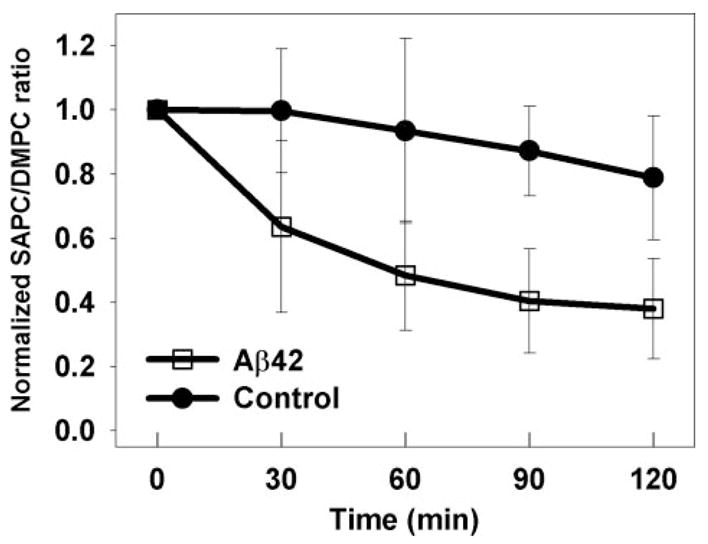
SAPC content was measured relative to added DMPC as an internal standard by MRM-LC/MS/MS. Vesicles made from human brain lipid extracts (10 μM) were treated with Cu(II) (0.5 μM), ascorbate (50 μM), and MgSO4 (1.5 mM), with and without Aβ42 (5.0 μM). The data represent the average of results from four of the five different brain lipid extracts listed in Table 1 (see explanation in text). t test of the differences at 60, 90, and 120 min yield p < 0.05.
TABLE 1. Characteristics of frozen human brain tissues from which brain lipid vesicles were prepared.
All specimens were designated clinically normal. PMI indicates post-mortem interval; date indicates year of death; age indicates years.
| Case no. | Date | PMI | Age at death |
|---|---|---|---|
| 1 | 2003 | 8 | 73 |
| 2 | 1996 | 9 | 92 |
| 3 | 1993 | 6 | 74 |
| 4 | 1990 | 4 | 72 |
| 5 | 1989 | 23 | 2 |
Endogenous HNE Generation during Aβ Promoted Lipid Oxidation
Endogenous HNE production during oxidation of synthetic lipids and brain lipid extracts was measured by GC/MS. In synthetic lipid vesicles the action of Cu(II) and ascorbate on 10 μM SAPC yields 6.3 ± 0.6 nM HNE, and the addition of Aβ42 increases this level to 13.4 ± 0.9 nM (Fig. 2). Chelation of Cu(II) by DTPA or inhibition of oxidation with BHT eliminates HNE production. The apparent yield of HNE from SAPC was 0.06% in the absence of Aβ42, and 0.13% in the presence of Aβ42. However, the true yield may be higher because of reactions between HNE and protein side chains (see below). In brain lipid extracts (10 μM total phosphate concentration), free HNE concentrations were below our limits of detection (<0.5 nM). This is expected because SAPC contains most of the ω-6 fatty acyl chains that can produce HNE in brain lipid, and a 0.13% yield from 0.34 μM SAPC would only produce 0.44 nM HNE. Nevertheless, immunoblot data presented below demonstrate that substantial amounts of HNE reaction products are produced.
FIGURE 2. HNE concentrations measured by GC/MS during the oxidation of synthetic SAPC (10 μM) by Cu(II) (0. 5 μM) and ascorbate (50 μM).
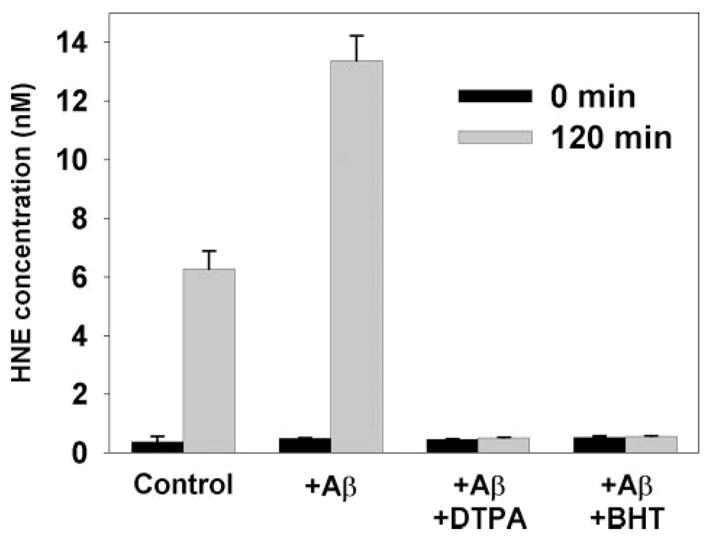
Aβ42 (5.0 μM), the copper chelator DTPA (10 μM), or the antioxidant BHT (7.5 μM) are present as indicated.
Covalent Modification of Aβ40 and Aβ42 by Exogenous HNE
Aβ40 and Aβ42 were treated with excess HNE to generate HNE-Aβ adducts for analysis by mass spectrometry. Simple incubation of 100 μM HNE with 5 μM Aβ40 or Aβ42 for 3 h at 37 °C yields insoluble amorphous aggregates by electron microscopy (data not shown). This physical state precluded study with electrospray ionization-mass spectrometry; however MALDI-Q-TOF-MS of HNE-modified Aβ40 detected unmodified protein at 4329.5 m/z and three HNE modified species at 4485.5, 4641.6, and 4797.6 m/z (Fig. 3). These peaks represent sequential mass shifts of 156 m/z and are characteristic of Michael addition reactions involving HNE.
FIGURE 3. MALDI-Q-TOF mass spectrometry analysis of covalent adducts formed by the reaction of Aβ40 (5 μM) with HNE (769 μM) at 37°C for 3 h.
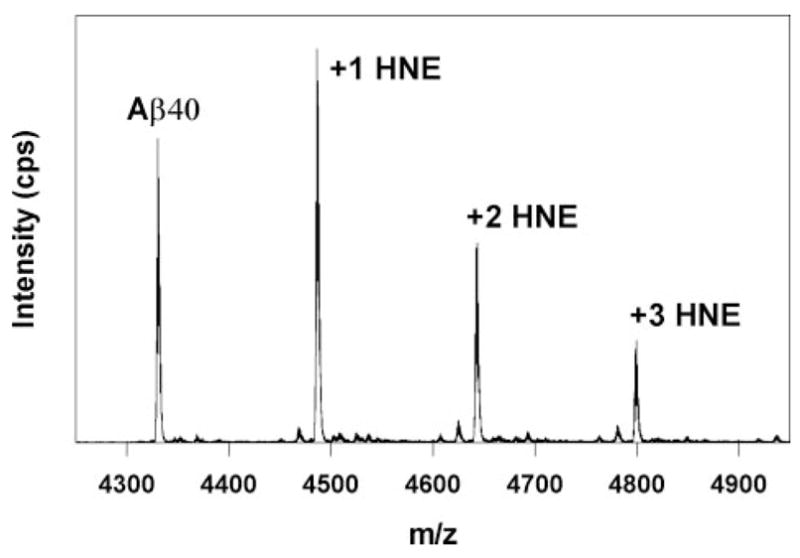
Small peaks to the left of each HNE adduct correspond to the loss of water (18 atomic mass units).
To determine the residues modified by HNE, 5.0 μM Aβ42 was incubated for 1 h in 10 mM phosphate buffer, pH 7.4, at 37 °C and then digested with Asp-N overnight. Despite any aggregation that ensued during the 1st h, the expected proteolytic fragments corresponding to Aβ-(1–6), Aβ-(7–22), and Aβ-(23–42) were identified by MALDI-TOF-Q-TOF-MS (Fig. 4A). The identities of peaks at 1483 and 1944 m/z are not clear, but the former may be Aβ-(11–22) resulting from N-side cleavage at Glu-11.
FIGURE 4. MALDI-TOF-Q-TOF mass spectrometry of samples digested overnight with endoproteinase Asp-N at 37°C.
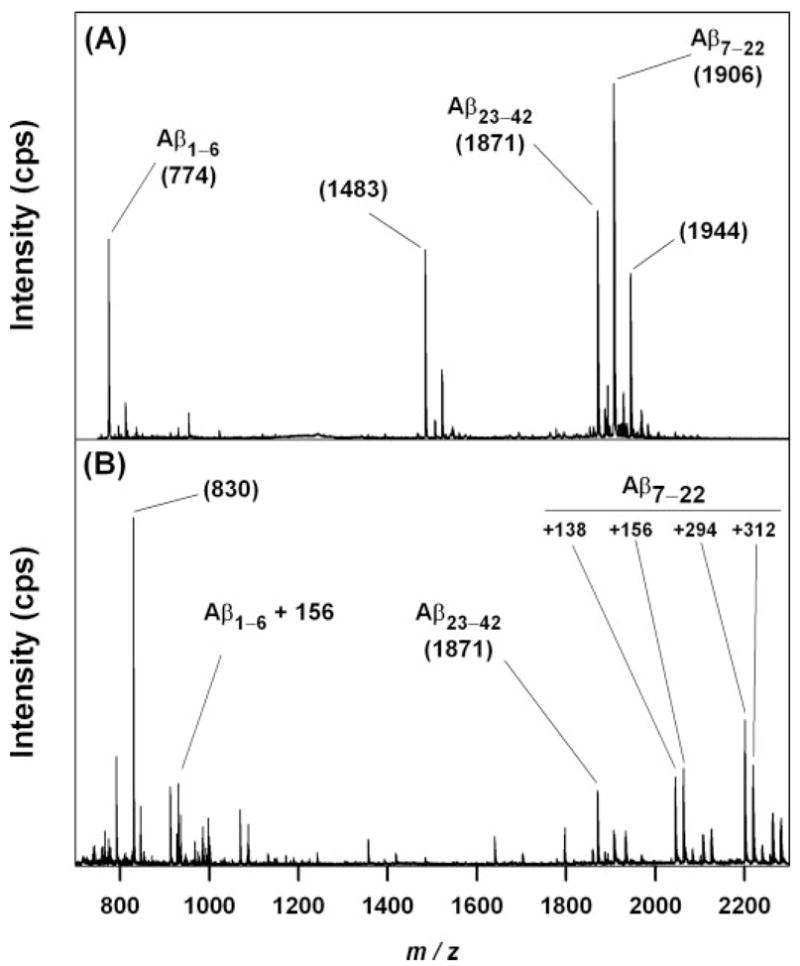
A, unmodified Aβ42 (5 μM). The three fragments expected from N-side cleavage at Asp-6 and Asp-22 are evident at m/z 774, 1871, and 1906. Other ions present are discussed in the text. B, Aβ42 (5 μM) treated with excess HNE (3.8 mM) in the presence of 2.5 mM DMPC and 50 μM Aβ-(16 –20). An ion at m/z 930 corresponds to Aβ-(1– 6) that has shifted 156 atomic mass units by Michael addition of HNE. Treatment with HNE also produced multiple ions between m/z 2000 and 2200, with several corresponding to the addition of 138, 156, 138, + 156 = 294, and 156 + 156 = 312 atomic mass unit to the ion at m/z 1906. The ion at m/z 1871 is unchanged by HNE treatment. Similar data were obtained following the digestion of Aβ40 with and without HNE treatment (data not shown).
When 5.0 μM Aβ42 was incubated for 1 h at 37 °C with 3.8 mM HNE, the insoluble aggregates that formed precluded mass spectrometric analysis. Therefore, 2.5 mM DMPC in the form of 100 nm lipid vesicles and 50 μM of the Aβ aggregation inhibitor Aβ-(16–20) (KLVFF) were added prior to the addition of HNE. The lipid vesicles provided a hydrophobic phase into which modified Aβ could partition before aggregating. Under these conditions, MALDI-TOF-Q-TOF-MS revealed a new ion at m/z 930, corresponding to a 156-atomic mass unit increase in mass of the Aβ-(1–6) fragment (Fig. 4B). Additional new ions appeared between m/z 2000 and 2400, including ions corresponding to A-(7–22) shifted by 138, 156, 294 (= 138 + 156), and 312 (=2 × 156) atomic mass units. The ion corresponding to Aβ-(23–42) at m/z 1871 was not shifted. The ion at 830 m/z only appeared when the KLVFF inhibitor was used and may represent a sodiated HNE adduct of this polypeptide. New ions with expected m/z values were observed following HNE treatment of Aβ40 (digested with endoproteinase Asp-N), Aβ-(1–28), Aβ-(1–11), and Aβ-(10–20) but not Aβ-(22–35) (data not shown).
Sequence analysis of the 774 m/z ion yielded a His immonium ion at 110 m/z and an assortment of y and b ions consistent with Aβ-(1–6) (Fig. 5A). Likewise, sequence analysis of the 1906 m/z ion yielded a His immonium ion at 110 m/z and a broad assortment of y and b ions consistent with Aβ-(7–22) (Fig. 6A). Sequence analysis of the new ion at 930 m/z revealed an ion at 266 m/z, consistent with a His immonium ion modified by HNE via Michael addition (Fig. 5B). It also yielded a b5 ion that was unshifted, and y2 and y5 ions that were shifted by 156 atomic mass unit. Neutral loss of 156 atomic mass units from the parent was also observed. These data indicate that HNE underwent Michael addition with His-6 in Aβ-(1–6). Sequence analysis of the new 2200 m/z ion also revealed an ion at 266 m/z, consistent with a His immonium ion modified by HNE via Michael addition (Fig. 6B). In addition, there was a broad array of b and y ions shifted by 138, 156, or 294 (= 138 + 156) atomic mass units, as well as neutral losses of 138, 156, and 294 from the parent. There was no evidence of unshifted b ions larger than b7 or y ions larger than y9. These data indicate that modifications occurred on His-13 and His-14. Similar analyses of several other new ions observed in Fig. 4 between 2000 and 2400 m/z and of Aβ-(1–11) and Aβ-(10–20) treated with HNE pointed to the same conclusions (data not shown).
FIGURE 5. MALDI-TOF-Q-TOF sequencing of Asp-N digestion fragments.
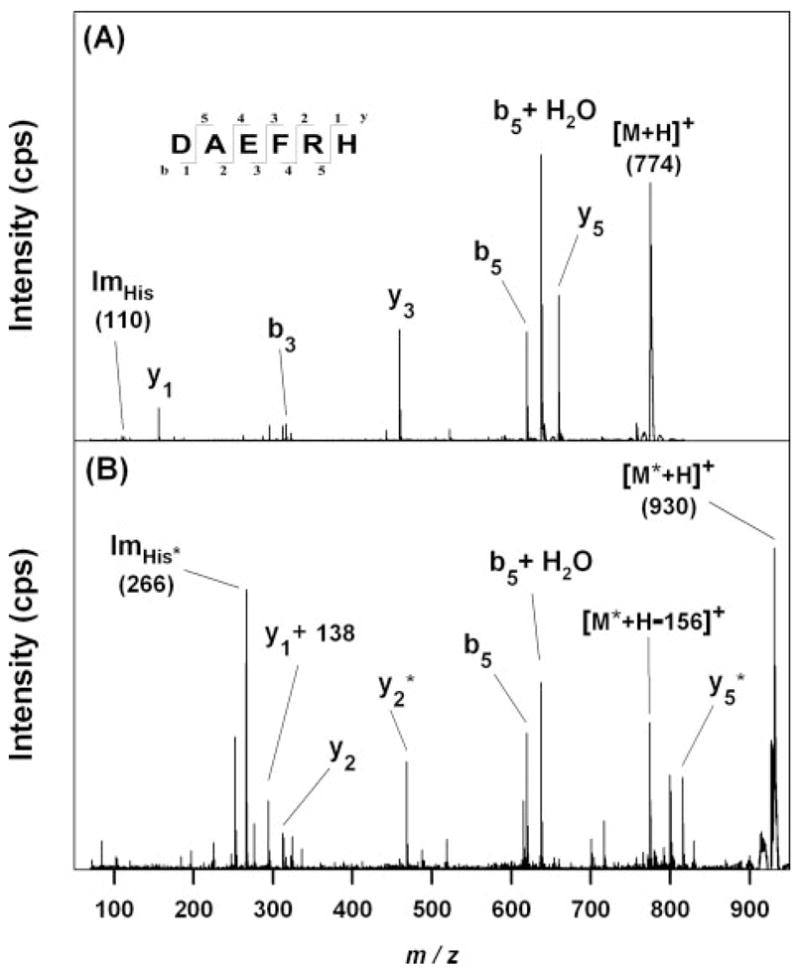
A, m/z 774 ion from Aβ42 (Fig. 4A) yielded product ions consistent with Aβ-(1–6), including a His immonium ion at m/z 110. B, m/z 930 ion from HNE-treated Aβ42 (Fig. 4B) yielded product ions suggesting that HNE has modified one residue in Aβ-(1– 6) by Michael addition and added 156 atomic mass unit to its mass. Ions that appear to be shifted by 156 atomic mass units for this reason are marked with an asterisk. The presence of an unmodified b5 ion and the appearance of a new ion at m/z 266 as expected for an HNE-His immonium ion indicate that the residue modified by HNE is His-6. The origin of the y1 + 138 ion is discussed in the text.
FIGURE 6. MALDI-TOF-Q-TOF sequencing of Asp-N digestion fragments.
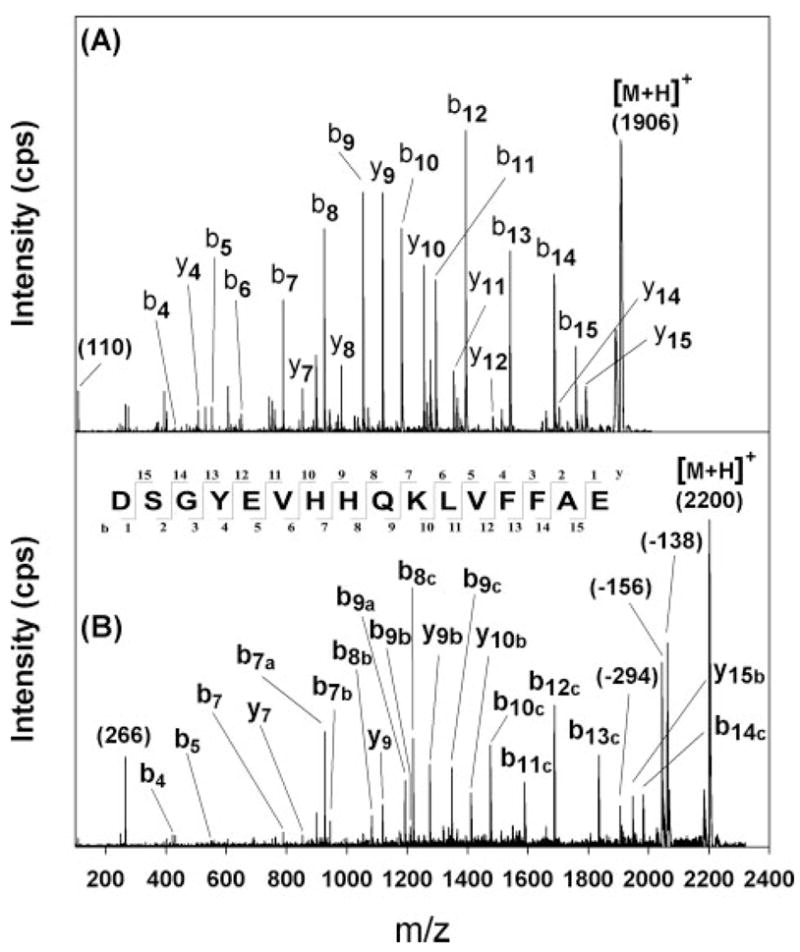
A, m/z 1906 ion from Aβ42 (Fig. 4A) yielded product ions consistent with Aβ-(7–22), including a His immonium ion at m/z 110. B, m/z 2200 ion from HNE-treated Aβ42 (Fig. 4B) yielded an ion at m/z 266 as expected for an HNE-His immonium ion. There is no evidence that ions smaller than b7 or y8 are shifted, but larger ions apparently shifted by 138 atomic mass unit are marked with an “a,” by 156 atomic mass unit with a “b,” and by 294 (= 138 + 156) with a “c.” These data suggest that His-13 and His-14 are both modified by HNE. The nature of these modifications is discussed in the text.
Mass shifts of 156 atomic mass units are likely to represent Michael adducts between HNE and the His residues at positions 6, 13, and 14. Mass shifts of 138 atomic mass units upon HNE treatment may indicate Schiff base formation with Lys residues; however, this modification would not occur on a His residue. Moreover, samples were not treated with a reducing agent to stabilize any Schiff bases that form. Michael adducts involving HNE may cyclize to form a hemiacetal (15, 16, 47, 48); however, the mass shifts would remain 156 atomic mass units in both cases. Therefore, the most likely modification by HNE that would account for a 138-atomic mass unit shift is formation of the hemiacetal from the Michael adduct followed by dehydration to form a dihydrofuran (14). This reaction may require catalysis by an adjacent His residue, however, because a mass shift of 276 (= 2 × 138) atomic mass units was not observed. The cyclic product known to form with His-X-Lys tripeptides and 4-oxy-2-nonenal would exhibit a mass shift of 118 m/z (49) and was not observed in our spectra.
Western Blot Characterization of His-HNE Adduct
HNEJ-2 antibodies identified HNE-modified Aβ as sharp bands migrating at ~100 kDa and several faint bands between 150–200 kDa (Fig. 7). These antibodies did not react with unmodified Aβ. 4G8 and 6E10 antibodies reacted with unmodified monomeric Aβ and higher molecular weight oligomeric species up to about 56 kDa. HNE modification reduced the amounts of monomeric and oligomeric Aβ that were observed and created high molecular weight smears. Most importantly, however, 4G8 and 6E10 antibodies did not react with the HNE-modified Aβ that migrated at ~100 kDa.
FIGURE 7. Western blot characterization His-HNE adducts.
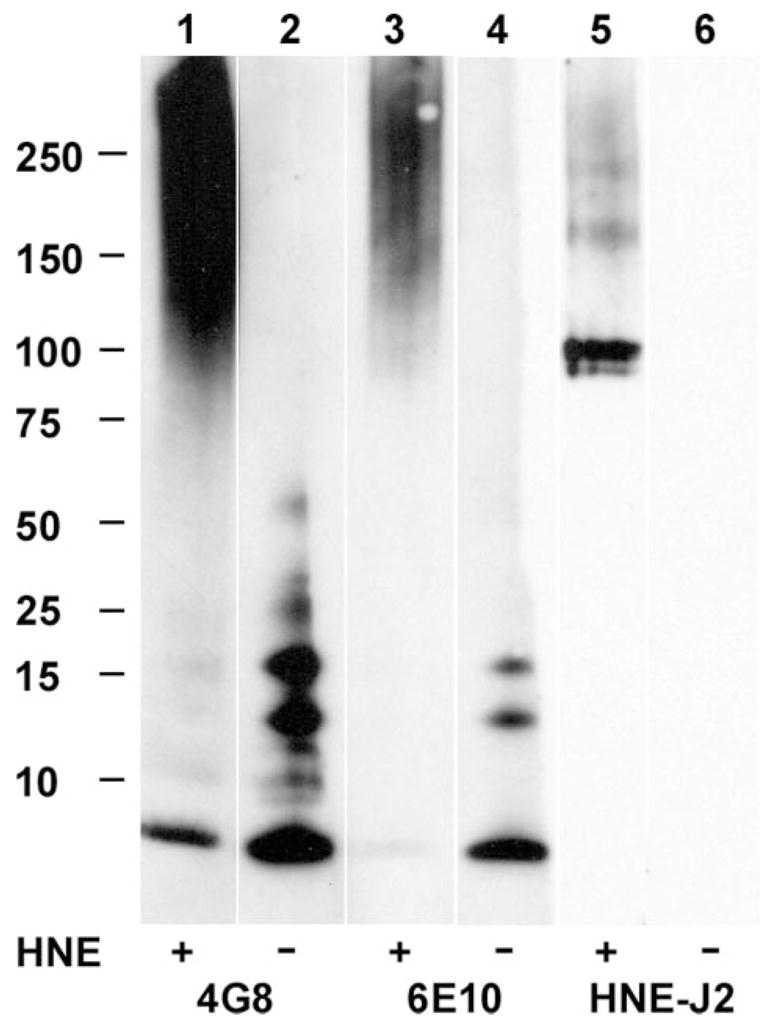
Aβ (lanes 2, 4, and 6) and HNE-modified Aβ (lanes 1, 3, and 5) were stained with antibodies to Aβ (4G8 and 6E10) and an antibody to His-HNE epitope (HNEJ-2). All lanes are from the same gel that was transferred to a nitrocellulose membrane, cut, and separated for staining.
Covalent Modification of Aβ42 by Endogenously Produced HNE
Compared with the millimolar concentrations of exogenous HNE used above to create HNE-Aβ adducts for mass spectrometric sequencing, endogenous HNE production can only be expected to produce nanomolar concentrations (Fig. 2). Given the insolubility of these adducts and the low concentrations expected, a mass spectrometry approach to their detection in brain tissue is not yet possible. Therefore, His-HNE antibodies were used to assess the effect of redox activity on the quantity of HNE-Aβ adducts formed during Aβ-mediated lipid oxidation. As described above, 50 μM ascorbate and 0.5 μM Cu(II) were added to mixtures containing 10 μM synthetic SAPC vesicles and 25 μM synthetic DMPC vesicles or 10 μM brain lipid extract vesicles containing 1 μM DMPC and 5 μM Aβ42 or a redox-inactive Aβ42 variant (32). After 120 min, unreacted HNE was quenched with an equal volume of 100 mM ammonium bicarbonate, pH 7.5, and the entire sample (400 μl) was adsorbed onto nitrocellulose.
Results indicate that immunoblotting with anti-His-HNE was twice as intense with Aβ42 than with redox-inactive variants (Fig. 8A). A similar increase was observed when vesicles made from human brain lipid extracts were substituted for synthetic lipid vesicles (Fig. 8B). The results demonstrate that HNE is indeed generated during brain lipid oxidation and that Aβ42 promotes its production, despite an inability to measure free HNE in brain lipids by GC/MS (see above). Therefore, our inability to detect unreacted HNE by GC/MS in human brain lipids was because of reactions that consumed HNE before it was assayed. Representative slot blot data used for the quantification of the anti-His-HNE immunoreactivity toward the adsorbed Aβ proteins are shown in Fig. 8C. Because Aβ42 was the only protein present in these experiments, this suggests that the nanomolar concentrations of HNE produced in the presence of Aβ42 have modified the His residues of Aβ42. This supports but also contrasts with the aforementioned mass spectrometric demonstration of Aβ40 modification by a molar excess of HNE.
FIGURE 8. Modification of Aβ42 by endogenously produced HNE.
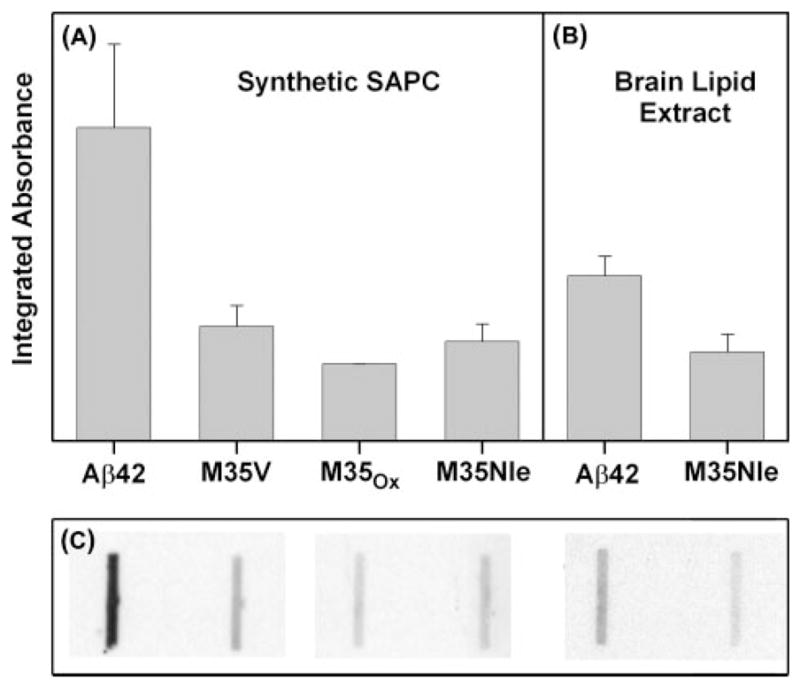
HNE was produced by oxidizing lipids with Cu(II) and ascorbate in the presence of Aβ42, or one of several redox incompetent forms of Aβ (32). Panels show the densitometric quantification of anti-HNE-His immunoreactivity arising from proteins exposed to synthetic lipid vesicles containing SAPC (A), and brain-derived lipid vesicles (B). The data in A and B are on the same (arbitrary) vertical scale. The data in A represent three separate determinations for Aβ42, and two determinations each for the redox incompetent forms. The data in B represent three separate determinations. C, representative slot-blot corresponding to the data in A and B.
Aβ Misfolding by HNE
Previously published studies demonstrated that oxidatively damaged polyunsaturated lipid membranes promote both the adsorption of Aβ42 onto lipid membranes and the formation of amyloid fibrils (45, 50). Because HNE was a likely component of these membranes, experiments with lipid vesicles and a CR binding assay to measure fibril formation were repeated using HNE and saturated lipid membranes instead of oxidatively damaged unsaturated lipid membranes. For Aβ40, protein and DMPC lipid concentrations were 11 μM; for Aβ42 they were both 2 μM. CR binding was measured at 0, 24, and 48 h after mixing, and the amount of binding at time 0 in each sample was subtracted from the amount measured at all three times. CR binding to both proteins increased substantially in the absence of HNE, as expected for the protein concentrations used. However, the presence of 3 mol % of HNE in DMPC vesicles increased CR binding to Aβ40 by 3-fold and to Aβ42 by 1.3-fold at 24 h. In the case of Aβ40, there was a slight decrease in CR binding from 24 to 48 h, which may have been because of precipitation of aggregated or fibrillized protein (see supplemental figures).
The same previously published studies also demonstrated that oxidatively damaged polyunsaturated lipid membranes promote a conformational change in Aβ that resembled the conformation of this protein in a mature amyloid fibril. Therefore, the effect of HNE on Aβ conformation was examined by PATIR-FTIR spectroscopy. The injection of 8 μg of Aβ40 or Aβ42 into the 6-ml subphase of a Langmuir trough yields a protein concentration of ~300 nM. After a 90-min exposure of a pure DMPC membrane to this subphase, the infrared spectrum of Aβ40 in the amide I′ region was broad and featureless (Fig. 9A), whereas that of Aβ42 exhibited a small peak at 1625 cm−1 superimposed on a similarly broad spectrum (Fig. 9B). The addition of sufficient HNE to yield a nominal subphase concentration of 23 μM caused significant increases in the amide I′ region absorption for both proteins and either the appearance or enhancement of spectral features characteristic of mature amyloid fibrils, especially enhanced absorption at 1625 cm−1.
FIGURE 9. Promotion of Aβ misfolding on synthetic lipid monolayers by HNE.
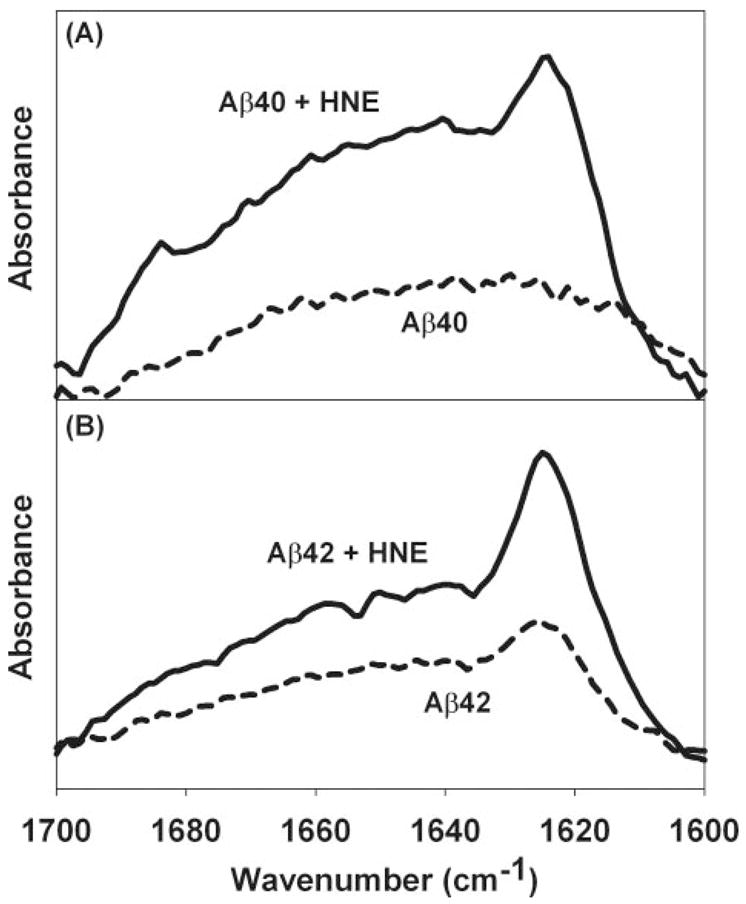
Representative PATIR-FTIR amide I′ spectra for 300 nM proteins in the subphase under a DMPC lipid monolayer after 90 min of incubation with and without 23 mM HNE. In both panels, dashed lines represent spectra collected in the absence of HNE, and solid lines are spectra collected in the presence of HNE. A, Aβ40. B, Aβ42. The increased absorption at ~1625 cm−1 in the presence of HNE suggests the formation of fibrillar β-structure.
For experiments with monolayers made from brain lipid extracts, upgraded instrumentation permitted the study of smaller quantities of protein at lower concentrations. Hence, ~2.5 μg of Aβ42 was injected into a 2.5-ml subphase to yield a protein concentration of ~230 nM. The amide I′ spectrum of Aβ42 that adsorbed to a brain lipid extract monolayer after 125 min exhibited small but clear features of mature amyloid fibrils with an absorption maximum at 1625 cm−1, and a distinct high frequency shoulder at 1685 cm−1 (Fig. 10A). The addition of 128 μM HNE to the subphase caused a marked increase in absorption at 1625–1630 cm−1, indicating an increase in membrane-adsorbed protein containing β-structure (Fig. 10B). Simultaneous analysis of three spectra collected in the absence of HNE and three spectra collected in the presence of HNE over 125 min demonstrates that these spectra may be fitted with components at 1625, 1645, 1664, and 1685 cm−1, shown as thin lines in Fig. 10, A and B. Quantitative analysis of the 1625 cm−1 component shows that it is significantly increased by HNE (Fig. 10C). This increase occurred at the expense of other components at 1645, 1664, and 1685 cm−1.
FIGURE 10. Promotion of Aβ42 misfolding on brain lipid monolayers by HNE.
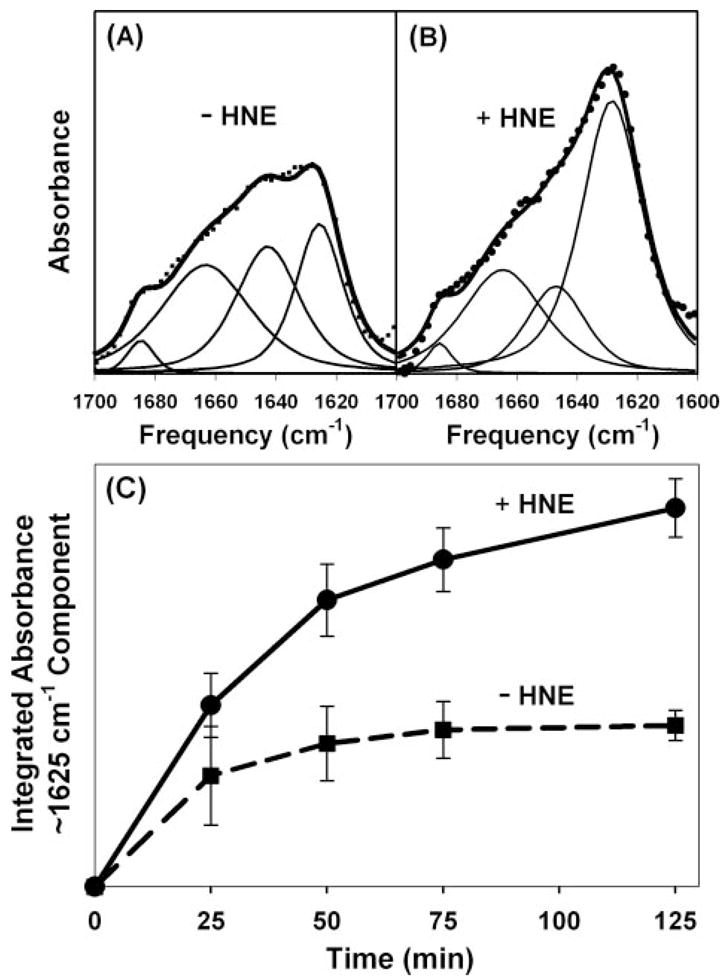
Representative PATIR-FTIR amide I′ spectra for Aβ42 (230 nM) binding to brain lipid monolayers after 125 mi of incubation in the absence of HNE (A) and in the presence of 128 mM HNE (B). The data in A and B are on the same arbitrary absorbance scale. Spectral data are represented by symbols, and thin lines represent component bands resulting from a simultaneous IRfit analysis of multiple spectra (46), and thick solid lines represent the sum of the fitted components. C, integrated absorbance of the 1625 cm−1 amide I′ components in three separate determinations obtained using three different brain lipid extracts. Error bars represent standard deviations.
DISCUSSION
Numerous in vivo studies have observed that Aβ promotes oxidative damage (28, 30, 31, 39, 51) and that oxidative damage promotes or at least precedes the aggregation of Aβ into fibrils (21, 23, 29). Taken together, these results suggest that positive feed-forward mechanisms may exist in which Aβ promotes its own conversion into fibrils. The results reported herein demonstrate that lipid membranes can support such a mechanism using physiologically reasonable reagent concentrations. In Fig. 8B, for example, brain-derived lipids were treated with 50 μM ascorbate and 0.5 μM copper. Ascorbate concentrations in brain tissue are reported to be 50–100 μM (53). Copper concentrations in normal brain tissue are also at least 50 μM and elevated in AD (54–56). Virtually all of this copper is protein-bound, and these conditions are mimicked in our experiments by a 10-fold molar excess of Aβ42, which has high affinity for copper (57). An HNE concentration of 60 nM was sufficient to accelerate fibril formation by Aβ42 in a CR binding assay, whereas 300 nM HNE was sufficient for Aβ40. These concentrations are far below the HNE concentrations of 8–15 μM found in human ventricular fluid (3).
The data show that Aβ increases the production of HNE as it promotes oxidative damage (Fig. 2), that HNE undergoes Michael addition with the three His residues in Aβ40 and Aβ42 (Figs. 3–6), and that HNE induces Aβ to form β-structure (Figs. 9 and 10) and amyloid fibrils (see CR binding data in supplemental figures). In a brain lipid extract, HNE most likely arises from arachidonoate (20:4) because it is the most abundant β-6 polyunsaturated fatty acyl chain (58). These fatty acyl chains undergo a constant low level of nonenzymatic oxidative damage producing isoprostanes (59) and HNE (3), as well as many other products (18). Because HNE and isoprostanes are both derived from the same precursor via similar chemical reactions, the association between isoprostanes and AD also implicates HNE (7).
HNE-Aβ may be a significant stimulus to fibril formation in AD, but HNE-Aβ may not ultimately be present in Aβ fibrils if it merely functions as a membrane-bound template for fibril formation. Fibrils that have started growing on such a template need not remain bound to the template, because fibrils can certainly seed continued fibrillogenesis on their own. Even if HNE-Aβ is incorporated into a fibril, there is no a priori reason to expect more than one HNE-Aβ molecule to do so, and methods of sufficient sensitivity have not yet been applied to detect such low concentrations. Likewise, techniques to identify oxidatively modified proteins in AD brain tissue appear to exclude proteins such as Aβ if they have aggregated before analysis (60). HNEJ-2 antibodies react with HNE-His epitopes, but these epitopes are not unique to HNE-Aβ. 4G8 and 6E10 antibodies with specificity for Aβ segments, on the other hand, clearly do not react with HNE-modified Aβ (Fig. 7). Therefore, techniques designed to detect extremely low concentrations of HNE-Aβ in fibrils, in a complex membrane environment, and in AD brain tissue in general are needed to define the role of HNE-Aβ in disease pathogenesis.
Our results provide evidence for the relationships between oxidative damage and amyloidogenesis illustrated in Fig. 11. Aggregated but nonfibrillar Aβ (AβI), with bound copper ions, have been shown previously to promote lipid peroxidation and HNE production in synthetic lipids (32), as well as oxidative damage to other compound classes (39, 51). The data presented herein show that Aβ also promote HNE production in human brain lipid extracts (Fig. 8B). The HNE that is produced modifies soluble Aβ (AβS) and promotes the formation of fibrillar Aβ (AβF). HNE is a highly reactive compound with a short half-life in a chemically diverse environment. A short half-life in brain-derived lipid extracts may explain why oxidative loss of SAPC could be demonstrated in both synthetic and brain-derived lipid vesicles, but free HNE was only detected with synthetic lipid vesicles (Fig. 2). Low SAPC concentrations in brain lipid extracts may also explain our inability to detect free HNE because the SAPC concentration was 10 μM in synthetic lipid preparations but only 0.34 μM in brain lipid extracts. With an HNE yield of only 0.13% from SAPC, we would expect less than our 0.5 nM limit of detection to be produced. Nonetheless, immunoblots with anti-HNE-His clearly demonstrate that HNE is produced in brain-derived lipid vesicles (Fig. 8B).
FIGURE 11. Schematic of inferred relationships between oxidative lipid damage and amyloidogenesis.
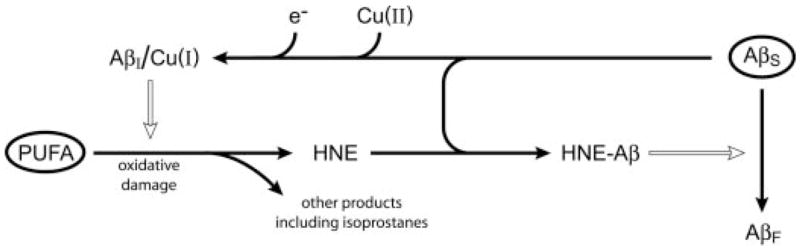
Solid arrows represent chemical conversions; open arrows represent positive kinetic effects. In the absence of biochemical pathology, polyunsaturated fatty acyl chains (PUFA) and soluble unaggregated Aβ (AβS) are present (the encircled species). Pathological cycles are entered when any type of oxidative damage causes lipid hydroperoxides to form. Lipid hydroperoxides decay spontaneously into HNE and other products, including isoprostanes. HNE and AβS combine to form various HNE-modified Aβ (HNE-Aβ). These species promote the conversion of AβS into fibrillar Aβ (AβF). AβS also forms a nonfibrillar intermediate species, AβI, and binds Cu(II) ions. When Aβ-bound Cu(II) is reduced, Cu(I) leads to hydroxyl radical formation and promotes oxidative lipid damage (32). It is not known whether AβI forms under the influence of external factors or if it is on path to the formation of AβF from AβS.
When Aβ are present, HNE spontaneously forms Michael adducts with one or more of the histidine residues at positions 6, 13, and 14. These results are consistent with an earlier report suggesting that HNE modified Aβ but did not identify the sites of modification (25). In principle, HNE may also react with the Lys residues of Aβ, forming either a Michael adduct or a Schiff base. Schiff base formation with a Lys side chain may indeed account for the small peaks of 18 atomic mass units below the 3 adduct ions in Fig. 3. However, numerous attempts to detect Schiff base formation in various proteins have concluded that the extent to which HNE modifies Lys side chains is insignificant compared with the extent to which His side chains are modified (25, 47, 61–66). Moreover, immunohistochemical studies have shown that anti-HNE-His antibodies bind to amyloid plaques in AD brain tissue (26), but anti-HNE-Lys antibodies do not (36). Thus, an abundance of data suggests that His residues are much more likely than Lys residues to be involved with HNE-mediated mechanisms of amyloidogenesis. The minor peaks in Fig. 3 most likely represent the loss of H2O from HNE-His adducts during desorption and ionization.
All three His residues appear to be essential for copper binding and thus for the prooxidant activity of Aβ (32). Therefore, we expect that the formation of HNE-Aβ eliminates the prooxidant activity of Aβ, or in terms of Fig. 11, HNE-Aβ adducts probably cannot form AβI/Cu(I). Yet HNE clearly promotes amyloid fibril formation in a manner that does not require modification of each protein molecule in the fibril. This may be inferred from the CR binding data because given the substoichiometric amounts of HNE present, it is unlikely that CR binding would have been detected if only HNE-modified Aβ had formed into fibrils (see supplemental figures). Thus, HNE-Aβ most likely promotes the formation of fibrils by unmodified Aβ, suggesting that HNE-Aβ acts as template. Gangliosides (67) and oxidized cholesterol (23) have also been implicated in such a mechanism.
HNE-Aβ has not been identified in vivo, but HNE and Aβ are present in normal human brain, and our experiments were performed with extracts from normal human brain tissue. Therefore, we should expect to find HNE-Aβ adducts in normal brain tissue, and a quantitative analysis of HNE-Aβ adducts will be required to establish whether normal and AD brain contain different amounts of these adducts. The observation that anti-HNE-His antibodies bind specifically to amyloid plaques in AD brain tissue (26) suggests that there is indeed a difference in either the quantity or distribution of HNE-Aβ adducts between human and AD brain. However, proving that these HNE-His epitopes represent HNE-Aβ adducts, and the quantitative analyses of these adducts, will be challenging. If HNE-Aβ adducts function as templates for amyloid fibril formation, they may have significant effects at very low concentrations, and need not be incorporated into any fibrils that form. These adducts are exceedingly hydrophobic, prone to aggregate, and exceedingly difficult to ionize for mass spectrometry. Immunoprecipitation is not feasible because modification by HNE appears to thwart recognition by common Aβ antibodies (Fig. 7). Therefore, it is not surprising that HNE-Aβ has not been detected by others using these antibodies for capture and mass spectrometry (68 –73). In principle, the proteomics approach of Butterfield (60) for identifying oxidatively modified proteins could identify HNE-Aβ; however, insoluble proteins are removed before analysis.
An important question remains unanswered at this point, namely whether HNE-Aβ promotes the formation of the prooxidant AβI-Cu complex. If so, then positive feedback may occur whereby HNE-Aβ promotes the formation of AβI-Cu(I), and this complex in turn promotes the formation of HNE-Aβ. The operation of a positive feedback loop in NT2 cells is supported by reports from other laboratories indicating that oligomeric but nonfibrillar Aβ42 increases HNE production, whereas HNE and fibrillar Aβ42 both increase Aβ42 production by inducing BACE-1 expression and activity (74–76). Oxidative stress also appears to alter levels of amyloid β precursor protein and increase Aβ production (77–80). Thus, additional relationships are likely to exist between the entities represented in Fig. 11.
An important related question is whether AβI is “on path” to the formation of fibrillar AβF from soluble AβS. If so, then AβI is likely to be a component of the aforementioned positive feedback loop. If not, then the diversion of AβS to the formation of AβF instead of AβI may protect cells from the formation of prooxidant activity of AβI (81). Therefore, further characterization of AβI and its role in vivo is of great interest. Yet another important question is whether lipid membranes have a role in amyloidogenesis apart from being a substrate for HNE production. We note that Aβ42 is more likely than Aβ40 to interact with oxidatively damaged membranes (82), and that Aβ42 promotes oxidative damage by concentrating redox-active metal ions in the vicinity of the membrane (32). Lipid membranes concentrate the molecular oxygen required for these reactions and may be required to stabilize HNE-modified Aβ in a conformation that seeds misfolding.
The scheme illustrated in Fig. 11 has components in common with “vicious” cycles implicated by others in the pathogenesis of AD (52, 83). However, our results only suggest how two conspicuous features of AD pathology, namely oxidative stress and amyloid fibril formation, may be mechanistically linked by lipid membranes. The role of Aβ fibrils or prefibrillar intermediate forms of Aβ in actually causing AD remains unclear.
Supplementary Material
Acknowledgments
The access to instrumentation in the Proteomics Core Facility of the Center for Cancer Pharmacology at the University of Pennsylvania, the tissue samples, and the processing assistance from Alzheimer Disease Core Center in the Center for Neurodegenerative Disease Research at the University of Pennsylvania were supported by National Institutes of Health Grant AG010124. We thank Jessica Cleck, John Hevko, Adam Zabell, and Omidreza Firuzi Fard Jahromi for their advice.
Footnotes
This work was supported by the NIA, National Institutes of Health, the American Health Assistance Foundation, and the Alzheimer Association.
The on-line version of this article (available at http://www.jbc.org) contains two supplemental figures.
The abbreviations used are: AD, Alzheimer disease; Aβ, amyloid β proteins; BHT, 3,5-di-tert-butylhydroxytoluene; CHCA, α-cyano-4-hydroxycinnamic acid; DMPC, 1,2-dimyristoyl-sn-glycerophosphocholine; DTPA, diethylenetriaminepentaacetic acid; HNE, 4-hydroxy-2-nonenal; SAPC, 1-stearoyl-2-arachidonyl-sn-glycero-3-phosphocholine; sinapinic acid, 2,5-dimethoxy-4-hydroxycinnamic acid; PBS, phosphate-buffered saline; Tricine, N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine; MALDI-TOF, matrix-assisted laser desorption ionization time-of-flight; MRM, multiple reaction monitoring; LC/MS/MS, liquid chromatography/tandem mass spectrometry; PATIR-FTIR, polarized attenuated total internal reflection-Fourier transform infrared; GC/MS, gas chromatography mass/spectrometry; CR, Congo Red; PFBHA, O-2,3,4,5,6-(pentafluorobenzyl)hydroxylamine hydrochloride.
References
- 1.Markesbery WR. Free Radic Biol Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 2.Butterfield DA, Boyd-Kimball D. Brain Pathol. 2004;14:426–432. doi: 10.1111/j.1750-3639.2004.tb00087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lovell MA, Ehmann WD, Mattson MP, Markesbery WR. Neurobiol Aging. 1997;18:457–461. doi: 10.1016/s0197-4580(97)00108-5. [DOI] [PubMed] [Google Scholar]
- 4.Schippling S, Kontush A, Arlt S, Buhmann C, Sturenburg HJ, Mann U, Muller-Thomsen T, Beisiegel U. Free Radic Biol Med. 2000;28:351–360. doi: 10.1016/s0891-5849(99)00247-6. [DOI] [PubMed] [Google Scholar]
- 5.Montine TJ, Neely MD, Quinn JF, Beal MF, Markesbery WR, Roberts LJ, Morrow JD. Free Radic Biol Med. 2002;33:620–626. doi: 10.1016/s0891-5849(02)00807-9. [DOI] [PubMed] [Google Scholar]
- 6.Pratico D, Lee VMY, Trojanowski JQ, Rokach J, FitzGerald GA. FASEB J. 1998;12:1777–1783. doi: 10.1096/fasebj.12.15.1777. [DOI] [PubMed] [Google Scholar]
- 7.Pratico D, Clark CM, Lee VMY, Trojanowski JQ, Rokach J, FitzGerald GA. Ann Neurol. 2000;48:809–812. [PubMed] [Google Scholar]
- 8.Yao Y, Zhukareva V, Sung S, Clark CM, Rokach J, Lee VMY, Trojanowski JQ, Pratico D. Neurology. 2003;61:475–478. doi: 10.1212/01.wnl.0000070185.02546.5d. [DOI] [PubMed] [Google Scholar]
- 9.Schneider C, Tallman KA, Porter NA, Brash AR. J Biol Chem. 2001;276:20831–20838. doi: 10.1074/jbc.M101821200. [DOI] [PubMed] [Google Scholar]
- 10.Morrow JD, Awad JA, Boss HJ, Blair IA, Roberts LJ., II Proc Natl Acad Sci U S A. 1992;89:10721–10725. doi: 10.1073/pnas.89.22.10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lawson JA, Rokach J, FitzGerald GA. J Biol Chem. 1999;274:24441–24444. doi: 10.1074/jbc.274.35.24441. [DOI] [PubMed] [Google Scholar]
- 12.Halliwell B, Gutteridge JMC. Free Radic Biol Med. 1999 doi: 10.1016/0891-5849(95)91457-3. [DOI] [PubMed] [Google Scholar]
- 13.Boutaud O, Li JY, Chaurand P, Brame CJ, Marnett LJ, Roberts JL, Oates JA. Adv Exp Med Biol. 2001;500:133–137. doi: 10.1007/978-1-4615-0667-6_16. [DOI] [PubMed] [Google Scholar]
- 14.Liu ZF, Minkler PE, Sayre LA. Chem Res Toxicol. 2003;16:901–911. doi: 10.1021/tx0300030. [DOI] [PubMed] [Google Scholar]
- 15.Uchida K. Prog Lipid Res. 2003;42:318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 16.Carini M, Aldini G, Facino RM. Mass Spectrom Rev. 2004;23:281–305. doi: 10.1002/mas.10076. [DOI] [PubMed] [Google Scholar]
- 17.Schaur RJ. Mol Aspects Med. 2006;24:149–159. doi: 10.1016/s0098-2997(03)00009-8. [DOI] [PubMed] [Google Scholar]
- 18.Markesbery WR, Lovell MA. Neurobiol Aging. 1998;19:33–36. doi: 10.1016/s0197-4580(98)00009-8. [DOI] [PubMed] [Google Scholar]
- 19.McGrath LT, McGleenon BM, Brennan S, McColl D, McIlroy S, Passmore AP. Q J Med. 2001;94:485–490. doi: 10.1093/qjmed/94.9.485. [DOI] [PubMed] [Google Scholar]
- 20.Boutaud O, Ou JJ, Chaurand P, Caprioli RM, Montine TJ, Oates JA. J Neurochem. 2002;82:1003–1006. doi: 10.1046/j.1471-4159.2002.01064.x. [DOI] [PubMed] [Google Scholar]
- 21.Bieschke J, Zhang Q, Powers ET, Lerner RA, Kelly JW. Biochemistry. 2005;44:4977–4983. doi: 10.1021/bi0501030. [DOI] [PubMed] [Google Scholar]
- 22.Lee EB, Leng LZ, Zhang B, Kwong L, Trojanowski JQ, Abel T, Lee VMY. J Biol Chem. 2006;281:4292–4299. doi: 10.1074/jbc.M511018200. [DOI] [PubMed] [Google Scholar]
- 23.Zhang QH, Powers ET, Nieva J, Huff ME, Dendle MA, Bieschke J, Glabe CG, Eschenmoser A, Wentworth P, Lerner RA, Kelly JW. Proc Natl Acad Sci U S A. 2004;101:4752–4757. doi: 10.1073/pnas.0400924101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shringarpure R, Grune T, Sitte N, Davies KJA. Cell Mol Life Sci. 2000;57:1802–1809. doi: 10.1007/PL00000660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Magni F, Galbusera C, Tremolada L, Ferrarese C, Kienle MG. Rapid Commun Mass Spectrom. 2002;16:1485–1493. doi: 10.1002/rcm.743. [DOI] [PubMed] [Google Scholar]
- 26.Ando Y, Brannstrom T, Uchida K, Nyhlin N, Nasman B, Suhr O, Yamashita T, Olsson T, El Salhy M, Uchino M, Ando M. J Neurol Sci. 1998;156:172–176. doi: 10.1016/s0022-510x(98)00042-2. [DOI] [PubMed] [Google Scholar]
- 27.Rofina JE, Singh K, Skoumalova-Vesela A, van Ederen AM, van Asten AJAM, Wilhelm J, Gruys E. Amyloid. 2004;11:90–100. doi: 10.1080/13506120412331285779. [DOI] [PubMed] [Google Scholar]
- 28.Wu Y, Luo Y. Curr Alzheimer Res. 2005;2:37–47. doi: 10.2174/1567205052772768. [DOI] [PubMed] [Google Scholar]
- 29.Pratico D, Uryu K, Leight S, Trojanowswki JQ, Lee VMY. J Neurosci. 2001;21:4183–4187. doi: 10.1523/JNEUROSCI.21-12-04183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Behl C, Davis JB, Lesley R, Schubert D. Cell. 1994;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 31.Opazo C, Huang XD, Cherny RA, Moir RD, Roher AE, White AR, Cappai R, Masters CL, Tanzi RE, Inestrosa NC, Bush AI. J Biol Chem. 2002;277:40302–40308. doi: 10.1074/jbc.M206428200. [DOI] [PubMed] [Google Scholar]
- 32.Murray IVJ, Sindoni ME, Axelsen PH. Biochemistry. 2005;44:12606–12613. doi: 10.1021/bi050926p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nelson TJ, Alkon DL. J Biol Chem. 2005;280:7377–7387. doi: 10.1074/jbc.M409071200. [DOI] [PubMed] [Google Scholar]
- 34.Puglielli L, Friedlich AL, Setchell KDR, Nagano S, Opazo C, Cherny RA, Barnham KJ, Wade JD, Melov S, Kovacs DM, Bush AI. J Clin Investig. 2005;115:2556–2563. doi: 10.1172/JCI23610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshimoto N, Tasaki M, Shimanouchi T, Umakoshi H, Kuboi R. J Biosci Bioeng. 2005;100:455–459. doi: 10.1263/jbb.100.455. [DOI] [PubMed] [Google Scholar]
- 36.Sayre LM, Zelasko DA, Harris PLR, Perry G, Salomon RG, Smith MA. J Neurochem. 1997;68:2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- 37.Bassett CN, Neely MD, Sidell KR, Markesbery WR, Swift LL, Montine TJ. Lipids. 1999;34:1273–1280. doi: 10.1007/s11745-999-0478-1. [DOI] [PubMed] [Google Scholar]
- 38.Galbusera C, Facheris M, Magni F, Galimberti G, Sala G, Tremolada L, Isella V, Guerini FR, Appollonio I, Galli-Kienle M, Ferrarese C. Curr Alzheimer Res. 2004;1:103–109. doi: 10.2174/1567205043332171. [DOI] [PubMed] [Google Scholar]
- 39.Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, Troncoso JC, Mattson MP. Proc Natl Acad Sci U S A. 2004;101:2070–2075. doi: 10.1073/pnas.0305799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tjernberg LO, Naslund J, Lindqvist F, Johansson J, Karlstrom AR, Thyberg J, Terenius L, Nordstedt C. J Biol Chem. 1996;271:8545–8548. doi: 10.1074/jbc.271.15.8545. [DOI] [PubMed] [Google Scholar]
- 41.Folch J, Lees M, Stanley GHS. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 42.Christie WW. Lipid Analysis. 3. Oily Press Lipid Library; Bridgewater, UK: 2003. pp. 96–102. [Google Scholar]
- 43.Meagher EA, Barry OP, Lawson JA, Rokach J, FitzGerald GA. J Am Med Assoc. 2001;285:1178–1182. doi: 10.1001/jama.285.9.1178. [DOI] [PubMed] [Google Scholar]
- 44.Toyokuni S, Miyake N, Hiai H, Hagiwara M, Kawakishi S, Osawa T, Uchida K. FEBS Lett. 1995;359:189–191. doi: 10.1016/0014-5793(95)00033-6. [DOI] [PubMed] [Google Scholar]
- 45.Koppaka V, Axelsen PH. Biochemistry. 2000;39:10011–10016. doi: 10.1021/bi000619d. [DOI] [PubMed] [Google Scholar]
- 46.Silvestro L, Axelsen PH. Biochemistry. 1999;38:113–121. doi: 10.1021/bi981289o. [DOI] [PubMed] [Google Scholar]
- 47.Uchida K, Stadtman ER. Proc Natl Acad Sci U S A. 1992;89:4544–4548. doi: 10.1073/pnas.89.10.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nadkarni DV, Sayre LM. Chem Res Toxicol. 1995;8:284–291. doi: 10.1021/tx00044a014. [DOI] [PubMed] [Google Scholar]
- 49.Oe T, Arora JS, Lee SH, Blair IA. J Biol Chem. 2003;278:42098–42105. doi: 10.1074/jbc.M308167200. [DOI] [PubMed] [Google Scholar]
- 50.Koppaka V, Axelsen PH. Langmuir. 2001;17:6309–6316. [Google Scholar]
- 51.Boyd-Kimball D, Castegna A, Sultana R, Poon HF, Petroze R, Lynn BC, Klein JB, Butterfield DA. Brain Res. 2005;1044:206–215. doi: 10.1016/j.brainres.2005.02.086. [DOI] [PubMed] [Google Scholar]
- 52.Standridge JB. Curr Alzheimer Res. 2006;3:95–107. doi: 10.2174/156720506776383068. [DOI] [PubMed] [Google Scholar]
- 53.Rice ME. Trends Neurosci. 2000;23:209–216. doi: 10.1016/s0166-2236(99)01543-x. [DOI] [PubMed] [Google Scholar]
- 54.Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. J Neurol Sci. 1998;158:47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 55.Loeffler DA, Lewitt PA, Juneau PL, Sima AAF, Nguyen HU, DeMaggio AJ, Brickman CM, Brewer GJ, Dick RD, Troyer MD, Kanaley L. Brain Res. 1996;738:265–274. doi: 10.1016/s0006-8993(96)00782-2. [DOI] [PubMed] [Google Scholar]
- 56.Strausak D, Mercer JFB, Dieter HH, Stremmel W, Multhaup G. Brain Res Bull. 2001;55:175–185. doi: 10.1016/s0361-9230(01)00454-3. [DOI] [PubMed] [Google Scholar]
- 57.Atwood CS, Scarpa RC, Huang XD, Moir RD, Jones WD, Fairlie DP, Tanzi RE, Bush AI. J Neurochem. 2000;75:1219–1233. doi: 10.1046/j.1471-4159.2000.0751219.x. [DOI] [PubMed] [Google Scholar]
- 58.White DA. The Phospholipid Composition of Mammalian Tissue. 3. Elsevier Science Publishing Co. Inc.; New York: 1973. pp. 441–483. [Google Scholar]
- 59.Morrow JD, Minton TA, Mukundan CR, Campbell MD, Zackert WE, Daniel VC, Badr KF, Blair IA, Roberts LJ., II J Biol Chem. 1994;269:4317–4326. [PubMed] [Google Scholar]
- 60.Butterfield DA. Brain Res. 2004;1000:1–7. doi: 10.1016/j.brainres.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 61.Bruenner BA, Jones AD, German JB. Chem Res Toxicol. 1995;8:552–559. doi: 10.1021/tx00046a009. [DOI] [PubMed] [Google Scholar]
- 62.Bolgar MS, Yang CY, Gaskell SJ. J Biol Chem. 1996;271:27999–28001. doi: 10.1074/jbc.271.45.27999. [DOI] [PubMed] [Google Scholar]
- 63.Bolgar MS, Gaskell SJ. Anal Chem. 1996;68:2325–2330. [Google Scholar]
- 64.Requena JR, Fu MX, Ahmed MU, Jenkins AJ, Lyons TJ, Baynes JW, Thorpe SR. Biochem J. 1997;322:317–325. doi: 10.1042/bj3220317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alderton AL, Faustman C, Liebler DC, Hill DW. Biochemistry. 2003;42:4398–4405. doi: 10.1021/bi0271695. [DOI] [PubMed] [Google Scholar]
- 66.Fenaille F, Guy PA, Tabet JC. J Am Soc Mass Spectrom. 2003;14:215–226. doi: 10.1016/S1044-0305(02)00911-X. [DOI] [PubMed] [Google Scholar]
- 67.Hayashi H, Kimura N, Yamaguchi H, Hasegawa K, Yokoseki T, Shibata M, Yamamoto N, Michikawa M, Yoshikawa Y, Terao K, Matsuzaki K, Lemere CA, Selkoe DJ, Naiki H, Yanagisawa K. J Neurosci. 2004;24:4894–4902. doi: 10.1523/JNEUROSCI.0861-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McGowan E, Sanders S, Iwatsubo T, Takeuchi A, Saido T, Zehr C, Yu X, Uljon S, Wang R, Mann D, Dickson D, Duff K. Neurobiol Dis. 1999;6:231–244. doi: 10.1006/nbdi.1999.0243. [DOI] [PubMed] [Google Scholar]
- 69.Murphy MP, Hickman LJ, Eckman CB, Uljon SN, Wang R, Golde TE. J Biol Chem. 1999;274:11914–11923. doi: 10.1074/jbc.274.17.11914. [DOI] [PubMed] [Google Scholar]
- 70.Pype S, Moechars D, Dillen L, Mercken M. J Neurochem. 2003;84:602–609. doi: 10.1046/j.1471-4159.2003.01556.x. [DOI] [PubMed] [Google Scholar]
- 71.Lewczuk P, Esselmann H, Groemer TW, Bibl M, Maler JM, Steinacker P, Otto M, Kornhuber J, Wiltfang J. Biol Psychiatry. 2004;55:524–530. doi: 10.1016/j.biopsych.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 72.Lewis HD, Beher D, Smith D, Hewson L, Cookson N, Reynolds DS, Dawson GR, Jiang M, Van der Ploeg JHX, Qian S, Rosahl TW, Kalaria RN, Shearman MS. Neurobiol Aging. 2004;25:1175–1185. doi: 10.1016/j.neurobiolaging.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 73.Bradbury LE, LeBlanc JF, McCarthy DB. Methods Mol Biol. 2004;264:245–257. doi: 10.1385/1-59259-759-9:245. [DOI] [PubMed] [Google Scholar]
- 74.Tong Y, Zhou W, Fung V, Christensen MA, Qing H, Sun X, Song W. J Neural Trans. 2005;112:455–469. doi: 10.1007/s00702-004-0255-3. [DOI] [PubMed] [Google Scholar]
- 75.Tamagno E, Parola M, Bardini P, Piccini A, Borghi R, Guglielmotto M, Santoro G, Davit A, Danni O, Smith MA, Perry G, Tabaton M. J Neurochem. 2005;92:628–636. doi: 10.1111/j.1471-4159.2004.02895.x. [DOI] [PubMed] [Google Scholar]
- 76.Tamagno E, Bardini P, Guglielmotto M, Danni O, Tabaton M. Free Radic Biol Med. 2006;41:202–212. doi: 10.1016/j.freeradbiomed.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 77.Yan SD, Yan SF, Chen X, Fu J, Chen M, Kuppusamy P, Smith MA, Perry G, Godman GC, Nawroth P, Zweiter JL, Stern D. Nat Med. 1995;1:693–699. doi: 10.1038/nm0795-693. [DOI] [PubMed] [Google Scholar]
- 78.Misonou H, Morishima-Kawashima M, Ihara Y. Biochemistry. 2000;39:6951–6959. doi: 10.1021/bi000169p. [DOI] [PubMed] [Google Scholar]
- 79.Schuessel K, Schafer S, Bayer TA, Czech C, Pradier L, Muller-Spahn F, Muller WE, Eckert A. Neurobiol Dis. 2005;18:89–99. doi: 10.1016/j.nbd.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 80.Li F, Calingasan NY, Yu F, Mauck WM, Toidze M, Almeida CG, Takahashi RH, Carlson GA, Beal MF, Lin MT, Gouras GK. J Neurochem. 2004;89:1308–1312. doi: 10.1111/j.1471-4159.2004.02455.x. [DOI] [PubMed] [Google Scholar]
- 81.Smith MA, Casadesus G, Joseph JA, Perry G. Free Radic Biol Med. 2002;33:1194–1199. doi: 10.1016/s0891-5849(02)01021-3. [DOI] [PubMed] [Google Scholar]
- 82.Koppaka V, Paul C, Murray IVJ, Axelsen PH. J Biol Chem. 2003;278:36277–36284. doi: 10.1074/jbc.M301334200. [DOI] [PubMed] [Google Scholar]
- 83.Bayer TA, Schafer S, Breyhan H, Wirths O, Treiber C, Multhaup G. Clin Neuropathol. 2006;25:163–171. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.