

Abstract
Toll-like receptors (TLRs) are pattern recognition receptors that recognize conserved molecular patterns expressed by pathogens. Pneumolysin, an intracellular toxin found in all Streptococcus pneumoniae clinical isolates, is an important virulence factor of the pneumococcus that is recognized by TLR4. Although TLR2 is considered the most important receptor for Gram-positive bacteria, our laboratory previously could not demonstrate a decisive role for TLR2 in host defence against pneumonia caused by a serotype 3 S. pneumoniae. Here we tested the hypothesis that in the absence of TLR2, S. pneumoniae can still be sensed by the immune system through an interaction between pneumolysin and TLR4. C57BL/6 wild-type (WT) and TLR2 knockout (KO) mice were intranasally infected with either WT S. pneumoniae D39 (serotype 2) or the isogenic pneumolysin-deficient S. pneumoniae strain D39 PLN. TLR2 did not contribute to antibacterial defence against WT S. pneumoniae D39. In contrast, pneumolysin-deficient S. pneumoniae only grew in lungs of TLR2 KO mice. TLR2 KO mice displayed a strongly reduced early inflammatory response in their lungs during pneumonia caused by both pneumolysin-producing and pneumolysin-deficient pneumococci. These data suggest that pneumolysin-induced TLR4 signalling can compensate for TLR2 deficiency during respiratory tract infection with S. pneumoniae.
Introduction
Streptococcus pneumoniae is the most common cause of community-acquired pneumonia (Bernstein, 1999; Campbell, 1999). Infections caused by S. pneumoniae are increasingly difficult to treat due to the emergence of antibiotic-resistant strains (Pikis et al., 1995; Schreiber and Jacobs, 1995). Increased knowledge of the first interaction between S. pneumoniae and host immune cells may facilitate the development of novel prophylactic and therapeutic tools to combat pneumococcal infections. In this respect, Toll-like receptors (TLRs), a family of pattern recognition receptors that are capable of recognizing conserved molecular patterns expressed by pathogens, are of particular interest (Kawai and Akira, 2005; Paterson and Mitchell, 2006).
The pneumococcal cell wall consists of several proteins and enzymes that contribute to the virulence of the pathogen and the pathogenesis of pneumonia (Jedrzejas, 2001). Pneumolysin is an intracellular toxin found in S. pneumoniae, which is produced by all clinical isolates and is an important factor for the virulence of the pneumococcus (Hirst et al., 2004). Indeed, mice infected with a pneumolysin-deficient strain of S. pneumoniae showed a reduced lethality and a diminished inflammatory response compared with mice infected with a normal, pneumolysin-producing strain (Berry et al., 1989; Benton et al., 1995; Canvin et al., 1995; Rubins et al., 1995; Berry and Paton, 2000; Kadioglu et al., 2002). At sublytic dose, pneumolysin affects polymorphonuclear cell activity, including respiratory burst, degranulation, chemotaxis and bactericidal activity (Paton and Ferrante, 1983). Furthermore, pneumolysin activates the classical pathway of complement and induces cytokine production by macrophages and monocytes (Houldsworth et al., 1994; Paton, 1996; Cockeran et al., 2002). At lytic dose, pneumolysin forms ring-shaped pores in cholesterol-containing cell membranes, which results in cell death (Alouf, 2000; Gilbert, 2002). Recent work has suggested that the immune system recognizes pneumolysin through TLR4 (Malley et al., 2003; Srivastava et al., 2005). Both pneumolysin-induced cytokine production and pneumolysin-induced apoptosis are mediated through TLR4 (Malley et al., 2003; Srivastava et al., 2005). In a model of nasopharyngeal colonization by S. pneumoniae, the interaction between pneumolysin and TLR4 was found to be essential for preventing invasive disease (Malley et al., 2003). Our laboratory reported a protective role of TLR4 during infection of the lower respiratory tract by S. pneumoniae, demonstrating an enhanced growth of bacteria in lungs of TLR4-deficient mice (Branger et al., 2004).
Within the family of TLRs, TLR2 has been implicated as the major pattern recognition receptor for ligands derived from Gram-positive bacteria (Yoshimura et al., 1999; Han et al., 2003; Schroder et al., 2003; Kawai and Akira, 2005). However, our laboratory recently demonstrated that TLR2 does not play a key role in host resistance to pneumonia caused by a serotype 3 strain of S. pneumoniae (Knapp et al., 2004). We here hypothesized that TLR2 knockout (KO) mice have an intact protective immune response against S. pneumoniae, because they are still capable of activating their immune system through an interaction between pneumolysin and TLR4. If this assumption is true, TLR2 KO mice would display a reduced antibacterial defence against pneumolysin-deficient S. pneumoniae, considering that these modified bacteria, devoid of a major TLR4 ligand, would primarily express TLR2 ligands. Therefore, in the present study we compared the response of TLR2 KO and wild-type (WT) mice during respiratory tract infection with WT and pneumolysin-deficient S. pneumoniae.
Results
Toll-like receptor 2 does not contribute to host defence and pulmonary inflammation against pneumonia caused by WT S. pneumoniae D39
We previously showed that TLR2 KO mice are indistinguishable from WT mice with regard to bacterial outgrowth and mortality after intranasal infection with a serotype 3 S. pneumoniae strain (Knapp et al., 2004). Considering that the pneumolysin-deficient strain used here is a serotype 2 (derived from S. pneumoniae D39), we first investigated the impact of TLR2 deficiency on the course of pneumonia caused by WT S. pneumoniae D39 (Fig. 1). Mortality did not differ between TLR2 KO and WT mice after intranasal infection with S. pneumoniae D39; if anything, TLR2 KO mice displayed a slightly reduced mortality (62.5%) although the difference with WT mice (75% mortality) was not significant (P = 0.13; Fig. 1A). We next determined bacterial loads in whole lung homogenates at 24 and 48 h after infection, i.e. at time points just before the first mice started dying (Fig. 1B). At both 24 and 48 h, bacterial loads were identical in lungs of TLR2 KO and WT mice. Together these data extend our earlier study using a serotype 3 S. pneumoniae strain (Knapp et al., 2004), showing that TLR2 does not contribute to a protective immune response during pneumonia caused by a serotype 2 pneumococcus.
Fig. 1.

TLR2 does not contribute to host defence against WT S. pneumoniae. Survival (A) and bacterial outgrowth (B) of WT mice (closed symbols or bars) and TLR2 KO mice (open symbols or bars) with 5 × 107 cfu S. pneumoniae D39. Mortality was assessed four times daily for 14 days (n = 16 per group). Bacterial loads in WT mice and TLR2 KO mice were determined 24 and 48 h after infection. Data of bacterial loads are mean ± SEM (n = 7–8 per group at each time point).
Toll-like receptor 2 (TLR2) deficiency modestly attenuates the inflammatory response induced by WT S. pneumoniae D39
Cytokines and chemokines play an important role in the antibacterial defence against bacterial pneumonia (Moore and Standiford, 2001; Knapp et al., 2005). We therefore determined the concentrations of tumour necrosis factor (TNF)-α, interleukin (IL)-1β, IL-10, macrophage inflammatory protein (MIP)-2 and cytokine-induced neutrophil chemoattractant (KC) in whole lung homogenates obtained 24 and 48 h after inoculation (Table 1). Although in general the pulmonary concentrations of these mediators were lower in TLR2 KO mice, the differences with WT mice were statistically significant only for KC (P < 0.005 at 24 and 48 h post infection) and IL-1β (P < 0.05 at 48 h). To further investigate lung inflammation, we determined pulmonary myeloperoxidase (MPO) levels, reflecting the whole-organ neutrophil content, in TLR2 KO mice and WT mice (Table 1). Similar to cytokine and chemokine levels, MPO concentrations were modestly lower in TLR2 KO, significantly so at 48 h post infection (P < 0.01). Moreover, total lung inflammation scores, determined from lung tissue slides prepared 24 and 48 h after infection with S. pneumoniae D39, were similar in WT and TLR2 KO mice (Table 1). Together, these data obtained with a serotype 2 pneumococcus confirm our earlier data generated with a serotype 3 S. pneumoniae (Knapp et al., 2004), establishing that TLR2 plays a modest role in the induction of a pulmonary inflammatory response to respiratory tract infection with WT S. pneumoniae.
Table 1.
Parameters of lung inflammation in TLR2 KO and WT mice 24 and 48 h after infection with WT S. pneumoniae D39.
| 24 h | 48 h | |||
|---|---|---|---|---|
| WT | TLR2 KO | WT | TLR2 KO | |
| TNF-α | 1229 ± 351 | 1026 ± 212 | 500 ± 92 | 361 ± 63 |
| IL-1β | 4029 ± 599 | 3248 ± 495 | 2874 ± 594 | 1462 ± 329* |
| IL-10 | 62 ± 23 | 48 ± 14 | 123 ± 51 | 72 ± 13 |
| MIP-2 | 5912 ± 876 | 5689 ± 1039 | 1447 ± 177 | 1182 ± 243 |
| KC | 5943 ± 867 | 2244 ± 510** | 3481 ± 339 | 695 ± 90** |
| MPO | 7668 ± 1123 | 6158 ± 2317 | 9183 ± 2365 | 4057 ± 578*** |
| TLIS | 16.7 ± 0.9 | 16.8 ± 1.0 | 13.5 ± 0.6 | 15.5 ± 0.8 |
Mice were intranasally infected with 5 × 107 cfu WT S. pneumoniae D39; whole lung homogenates were obtained 24 and 48 h thereafter. Data are means ± SEM (n = 6–8 per group).
P < 0.05 versus WT mice.
P < 0.005 versus WT mice.
P < 0.01 versus WT mice.
TNF-α, IL-1β, IL-10, MIP-2 and KC values are in pg ml−1; MPO values are in ng ml−1; TLIS (total lung inflammation score) in arbitrary units.
Toll-like receptor 2 limits the outgrowth of pneumolysin-deficient S. pneumoniae PLN
Having established that TLR2 is not essential for host defence against WT S. pneumoniae D39, we next infected TLR2 KO and WT mice with the isogenic mutant S. pneumoniae PLN (Fig. 2). As expected (Berry et al., 1989), S. pneumoniae PLN was less virulent, in particular in WT mice. Only 23% of WT mice died during a 2 week follow-up, versus 38% of TLR2 KO mice (not significant for the difference between mouse strains; P = 0.29; Fig. 2A). Interestingly, TLR2 KO mice started to die after 3 days, whereas the first deaths among WT mice occurred after 5 days. To obtain insight in the growth of S. pneumoniae PLN during the infection (i.e. before the first mice started dying), we infected TLR2 KO and WT mice with S. pneumoniae PLN and determined bacterial loads in whole lung homogenates at 24, 48 and 72 h thereafter (Fig. 2B). Whereas the bacterial burdens were not significantly different between TLR2 KO and WT mice 24 h post infection, at 48 and 72 h TLR2 KO displayed significantly higher bacterial loads in their lungs than WT mice (both P < 0.05). Remarkably, whereas S. pneumoniae PLN did not further grow in the lungs of WT mice from 24 h after infection onward, which is in line with a previous investigation (Canvin et al., 1995), the bacterial load increased more than 10-fold in lungs of TLR2 KO mice between 24 and 72 h after inoculation. Hence, these data show that TLR2 serves to limit the growth of S. pneumoniae PLN during pneumonia.
Fig. 2.
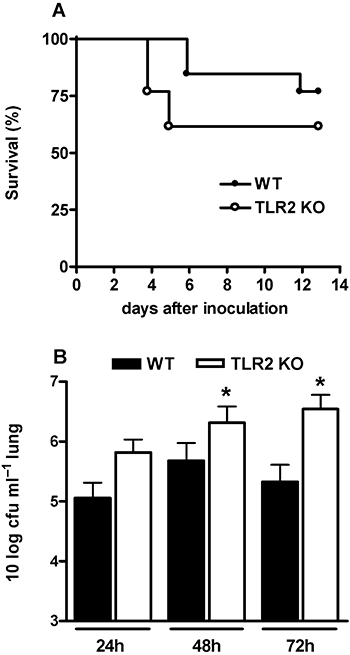
TLR2 limits outgrowth of pneumolysin-deficient S. pneumoniae PLN. Survival (A) and bacterial outgrowth (B) of WT mice (closed symbols or bars) and TLR2 KO mice (open symbols or bars) with 5 × 107 cfu S. pneumoniae PLN. Mortality was assessed four times daily for 14 days (n = 13 per group). Bacterial loads in WT mice and TLR2 KO mice were determined 24, 48 and 72 h after infection. Data of bacterial loads are mean ± SEM (n = 7–8 per group at each time point) *P < 0.05 versus WT mice.
Toll-like receptor 2 (TLR2) deficiency reduces lung inflammation induced by S. pneumoniae PLN
To further investigate the role of TLR2 during infection with S. pneumoniae PLN, we determined TNF-α, IL-1β, IL-10, MIP-2, KC and MPO levels in whole lung homogenates obtained 24, 48 and 72 h after inoculation (Table 2). At 24 h post infection, pulmonary cytokine and chemokine levels were lower in TLR2 KO mice, significantly so for IL-1β (P < 0.05). In addition, lung MPO levels were lower in TLR2 KO mice at this time point (P < 0.05). In contrast, at 48 and 72 h after infection, when TLR2 KO mice displayed higher bacterial burdens in their lungs, the pulmonary concentrations of cytokines, chemokines and MPO did not differ between TLR2 KO and WT mice. Histopathological analyses of lung tissue slides demonstrated reduced lung inflammation in TLR2 KO mice at 24 h, but increased lung inflammation at 48 and 72 h (Table 2). Representative lung tissue slides from WT and TLR2 KO mice 24, 48 and 72 h after infection with S. pneumoniae PLN are shown in Fig. 3.
Table 2.
Parameters of lung inflammation in TLR2 KO and WT mice 24, 48 and 72 h after infection with pneumolysin-deficient S. pneumoniae PLN.
| 24 h | 48 h | 72 h | ||||
|---|---|---|---|---|---|---|
| WT | TLR2 KO | WT | TLR2 KO | WT | TLR2 KO | |
| TNF-α | 325 ± 77 | 295 ± 87 | 195 ± 34 | 195 ± 34 | 506 ± 114 | 522 ± 163 |
| IL-1β | 2414 ± 519 | 903 ± 496* | 552 ± 215 | 802 ± 200 | 1724 ± 638 | 2092 ± 956 |
| IL-10 | BD | BD | BD | BD | 135 ± 27 | 153 ± 63 |
| MIP-2 | 2703 ± 1033 | 1956 ± 1001 | 1919 ± 194 | 2720 ± 649 | 1029 ± 589 | 1297 ± 692 |
| KC | 1781 ± 791 | 600 ± 158 | 1700 ± 980 | 610 ± 188 | 1385 ± 323 | 958 ± 391 |
| MPO | 3173 ± 391 | 1666 ± 411* | 1675 ± 424 | 2205 ± 226 | 2722 ± 251 | 2402 ± 500 |
| TLIS | 7.7 ± 2.0 | 3.1 ± 1.2 | 6.9 ± 1.6 | 11.9 ± 2.1 | 5.6 ± 0.9 | 8.7 ± 0.8* |
Mice were intranasally infected with 5 × 107 cfu S. pneumoniae PLN; whole lung homogenates were obtained 24, 48 and 72 h thereafter. Data are means ± SEM (n = 6–8 per group).
P < 0.05 versus WT mice.
TNF-α, IL-1β, IL-10, MIP-2 and KC values are in pg ml−1; MPO values are in ng ml−1; TLIS (total lung inflammation score) in arbitrary units.
BD, below detection limit.
Fig. 3.
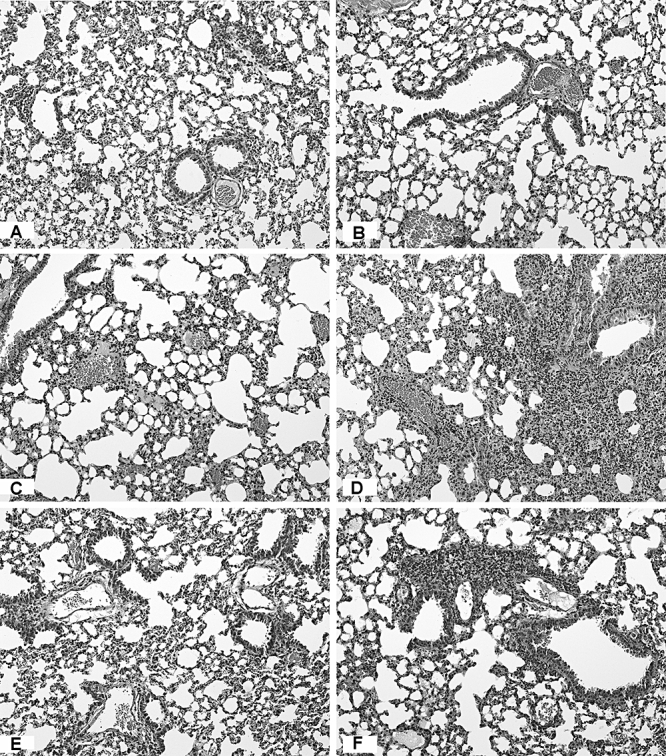
Lung histology in WT and TLR2 KO mice after infection with S. pneumoniae PLN. Representative lung tissue slides from WT mice (A, C and E) and TLR2 KO mice (B, D and F) after infection with 5 × 107 cfu S. pneumoniae PLN. Mice were sacrificed after 24 h (A and B), 48 h (C and D) or 72 h (E and F). HE staining: magnification 4×.
Toll-like receptor 2 (TLR2) deficiency strongly impairs the early inflammatory response to both S. pneumoniae D39 and S. pneumoniae PLN
The role of TLR2 in lung inflammation later in the course of pneumonia could have been obscured by the growing bacterial load in TLR2 KO mice (that is, at 48 and 72 h after infection, TLR2 deficiency could be compensated for by the higher bacterial load, providing a more potent proinflammatory stimulus via TLR2-independent pathways). An earlier study performed at our laboratory has shown an important role for TLR2 in the early host defence against S. pneumoniae pneumonia using serotype 3 (Knapp et al., 2004). TLR2 could also be more important for the early host inflammatory response to S. pneumoniae with serotype 2. Thus, we intranasally inoculated TLR2 KO and WT mice with S. pneumoniae D39 or S. pneumoniae PLN and evaluated their response to the infection 6 h later (Table 3). The bacterial loads in lungs of TLR2 KO mice and WT mice were similar at this early time point during infection with S. pneumoniae D39 or S. pneumoniae PLN. Interestingly, compared with WT mice, TLR2 KO mice showed a strongly reduced capacity to respond to both S. pneumoniae D39 and S. pneumoniae PLN; the lung concentrations of TNF-α, IL-1β, MIP-2 and KC were much lower in TLR2 KO mice (P < 0.05 to P < 0.001) (Table 3). In addition, histopathological analyses of lung tissue slides demonstrated a significantly reduced inflammation in lungs of TLR2 KO mice 6 h after infection with S. pneumoniae D39 or S. pneumoniae PLN (Fig. 4). Of note, some inflammatory responses to S. pneumoniae PLN were more strongly reduced in TLR2 KO mice than the inflammatory responses to S. pneumoniae D39. For KC, MIP-2 and TNF-α, there was a significant interaction (P < 0.001, P = 0.034 and P = 0.007, respectively); the effect of the pneumococcal strain used depended on the mouse strain. In particular, whereas in WT mice S. pneumoniae D39 and PLN induced a similar early TNF-α response in the lungs, the pulmonary levels of this crucially protective cytokine in the early response to pneumococcal pneumonia (van der Poll et al., 1997; Rijneveld et al., 2001) were more than 4-fold lower in TLR2 KO mice after infection with S. pneumoniae PLN versus 2-fold lower after inoculation with S. pneumoniae D39. In addition, whereas overall S. pneumoniae PLN elicited less profound histopathological alterations in lung tissue than S. pneumoniae D39, the difference in total lung histology scores between TLR2 KO and WT mice was especially clear after infection with the pneumolysin-deficient strain.
Table 3.
Role of TLR2 in the early inflammatory response in the lungs after infection with S. pneumoniae D39 or PLN.
| D39 | PLN | |||
|---|---|---|---|---|
| WT | TLR2 KO | WT | TLR2 KO | |
| cfu | 6.5 ± 1.4 × 106 | 5.5 ± 1.3 × 106 | 13.2 ± 1.3 × 106 | 10.6 ± 1.8 × 106 |
| TNF-α | 9 781 ± 780 | 4 656 ± 375** | 9 124 ± 1 203 | 2270 ± 236** |
| IL-1β | 5 754 ± 714 | 3 566 ± 348* | 18 963 ± 2 522 | 8716 ± 1939* |
| IL-10 | 31 ± 2 | 32 ± 3 | BD | BD |
| MIP-2 | 42 193 ± 2 529 | 23 855 ± 3 396*** | 9 114 ± 1 112 | 2643 ± 505** |
| KC | 61 120 ± 2 879 | 23 266 ± 3 124*** | 16 776 ± 2 314 | 4783 ± 1352** |
| MPO | 76.7 ± 9.3 | 41.6 ± 3.4* | 15.5 ± 3.4 | 12.4 ± 4.0 |
| TLIS | 18.4 ± 0.5 | 15.1 ± 0.8* | 9.5 ± 0.9 | 5.3 ± 1.3* |
Mice were intranasally infected with 5 × 107 cfu S. pneumoniae D39 or PLN, and whole lung homogenates were obtained 6 h later. Data are means ± SEM (n = 8 per group).
P < 0.05 versus WT mice.
P < 0.001 versus WT mice.
P < 0.01 versus WT mice.
TNF-α, IL-1β, IL-10, MIP-2 and KC values are in pg ml−1; cfu values are in cfu ml−1 lung; MPO levels are in μg ml−1; TLIS (total lung inflammation score) in arbitrary units.
BD, below detection limit.
Fig. 4.

Reduced lung inflammation in TLR2 KO mice early after infection with S. pneumoniae D39 or S. pneumoniae PLN. Representative lung tissue slides from WT mice (A and C) and TLR2 KO mice (B and D) 6 h after infection with 5 × 107 cfu S. pneumoniae D39 (A and B) or S. pneumoniae PLN (C and D). HE staining: magnification 4×. Insets show Ly-6G staining.
Discussion
Pneumolysin is an essential virulence factor of S. pneumoniae (Hirst et al., 2004). Recent studies have identified TLR4 as a recognition receptor for pneumolysin in the respiratory tract (Malley et al., 2003; Srivastava et al., 2005). The interaction between pneumolysin and TLR4 was found to contribute to a protective immune response to S. pneumoniae, in particular in a model of upper airway colonization (Malley et al., 2003; Srivastava et al., 2005) and to a lesser extent during experimental lower respiratory tract infection (Branger et al., 2004). Although the pneumococcus expresses several potent TLR2 ligands (Yoshimura et al., 1999; Han et al., 2003; Schroder et al., 2003), our laboratory previously could not demonstrate a decisive role for TLR2 in host defence against pneumococcal pneumonia (Knapp et al., 2004). We here hypothesized that in the absence of TLR2, S. pneumoniae could still be sensed by the immune system through an interaction between pneumolysin and TLR4. The experiments described herein support this hypothesis: whereas the growth of WT pneumococci occurred to a similar extent in TLR2 KO and WT mice, the pneumolysin-deficient S. pneumoniae PLN strain only grew out in TLR2 KO mice. These data suggest that pneumolysin-induced TLR4 signalling can compensate for TLR2 deficiency during respiratory tract infection with S. pneumoniae.
In a series of elegant experiments Malley and coworkers demonstrated that pneumolysin is a ligand for TLR4 (Malley et al., 2003; Srivastava et al., 2005). Purified pneumolysin was shown to activate cells via a TLR4-dependent, TLR2-independent pathway, accomplished by a physical interaction between pneumolysin and TLR4 (Malley et al., 2003; Srivastava et al., 2005). Interestingly, pneumolysin induced proinflammatory responses in primary macrophages in synergy with TLR2 ligands derived from S. pneumoniae, in particular peptidoglycan and whole pneumococcal cell walls (Malley et al., 2003; Srivastava et al., 2005), suggesting that during infection with intact pneumococci, the combined action of TLR4 and TLR2 may facilitate an optimal innate immune response. Such roles for these two distinct TLRs is further corroborated by findings that, in the human embryonic kidney cell line 293, transfection of either TLR2 or TLR4 conferred responsiveness to S. pneumoniae (Koedel et al., 2003). Thus far, the isolated roles of either TLR4 or TLR2 in host defence against S. pneumoniae in vivo have been investigated in a number of studies. The most dramatic phenotype was reported in the original publication by Malley et al. (Malley et al., 2003; Srivastava et al., 2005), showing that C3H/HeJ mice, which carry a loss-of-function tlr4 mutation, are more susceptible to pneumococcal colonization after nasopharyngeal challenge, eventually resulting in invasive infection, bacteremia and death. Our laboratory found a more modest protective role for TLR4 during lower respiratory tract infection by S. pneumoniae, as reflected by a reduced survival and a slightly enhanced bacterial outgrowth after intranasal infection of C3H/HeJ mice with a relatively low infectious dose (Branger et al., 2004). TLR2 KO mice demonstrated an increased disease severity together with a moderately enhanced bacterial growth in the central nervous system during meningitis induced by intracisternal injection of pneumococci (Echchannaoui et al., 2002; Koedel et al., 2003). In contrast, our group could not demonstrate a protective role for TLR2 in pneumonia caused by S. pneumoniae, showing similar bacterial multiplication and lethality after intranasal infection of TLR2 KO and WT mice (Knapp et al., 2004). A limited role for TLR2 during infection with WT S. pneumoniae is further supported by a recent study in which intact pneumococci were administered intraperitoneally (Khan et al., 2005), although TLR2 KO mice displayed a modestly slower clearance of S. pneumoniae from their nasopharynx in another investigation (van Rossum et al., 2005). Altogether these studies suggest that TLR2 at best plays a modest role in host defence against S. pneumoniae airway infection, and led us to hypothesize that intact TLR4 signalling through pneumolysin may balance the lack of TLR2 signalling. We tested this hypothesis by infecting TLR2 KO mice with pneumolysin-deficient S. pneumoniae, arguing that these bacteria, devoid of a major TLR4 ligand, predominantly express TLR2 ligands. Indeed, whereas antibacterial defence in TLR2 KO mice was unimpaired during infection with S. pneumoniae D39, infection with S. pneumoniae PLN resulted in enhanced outgrowth in these mice. If our hypothesis is correct, inoculation of WT S. pneumoniae D39 in TLR2 × 4 double KO mice should result in a comparable setting as pneumolysin-deficient S. pneumoniae in TLR2 KO mice, i.e. absence of TLR2 and TLR4 signalling. Our first preliminary results show that indeed this is the case: growth of WT S. pneumoniae D39 was significantly higher in the lungs of TLR2 × 4 double KO mice compared with WT mice 48 h after inoculation (data not shown). In line, Albiger et al. (2005) recently showed that mice deficient of the TLR2- and TLR4-common intracellular adaptor molecule MyD88 also displayed an enhanced bacterial outgrowth in MyD88 KO mice compared with WT mice.
The early inflammatory response is an essential component of host defence in this model of pneumococcal pneumonia, as documented by previous studies in which the early cytokine response was inhibited (van der Poll et al., 1997; Rijneveld et al., 2001). Although TLR2 KO mice displayed a reduced inflammatory response 6 h after infection with either S. pneumoniae D39 or PLN, some responses were more strongly diminished after infection with the pneumolysin-deficient strain. This was in particular true for the early TNF-α response. Considering that especially low TNF-α concentrations in the lungs early after induction of pneumococcal pneumonia are important for limiting the growth of S. pneumoniae (van der Poll et al., 1997; Rijneveld et al., 2001), this differential response may have contributed to the enhanced growth of S. pneumoniae PLN in TLR2 KO mice. In addition, mediators other than measured in this study could contribute to this finding. Of note, TLR2 KO mice still display an induction of cytokines and chemokines when infected with pneumolysin-deficient S. pneumoniae, suggesting that other pattern recognition receptors contribute to this response, In this respect, the recent finding that TLR9 can recognize pneumococcall DNA is of relevance (Mogensen et al., 2006). Moreover, although histopathological analysis of lung tissue showed diminished lung inflammation in TLR2 KO mice during the early course of infection with S. pneumoniae PLN, which is in line with a TLR2-dependent immune response, during the later phase of pneumonia lung inflammation of TLR2 KO mice was enhanced compared with WT mice, which corresponded with the higher bacterial loads. This finding suggests that, in the presence of a high bacterial burden, S. pneumoniae PLN is able to elicit significant lung inflammation via a TLR2-independent route. Hence, it is conceivable that the early recognition of S. pneumoniae and, thereby, the initial inflammatory response in the airways at least in part are mediated by TLR2, through an interaction between this receptor and the various TLR2 ligands expressed by the pneumococcus (Yoshimura et al., 1999; Han et al., 2003; Schroder et al., 2003), but that, during a more established infection with a higher bacterial burden, the TLR2-induced inflammation is ‘overruled’ by other pathways stimulated by TLR2-independent pneumococcal antigens.
Our results exemplify the complex interactions at play during the first encounter between the host, expressing multiple pattern recognition receptors, and an intact pathogen, expressing multiple virulence factors and pathogen-associated molecular patterns. Whereas during infection of TLR2 KO mice with WT pneumococci, the interaction between TLR4 and pneumolysin apparently is sufficient to maintain an adequate immune response, during infection of TLR2 KO mice with pneumolysin-deficient S. pneumoniae, the absence of the interaction between pneumococcal TLR2 ligands, such as lipoteichoic acid and peptidoglycan, cannot be compensated for by the TLR4 pneumolysin-mediated immune response. As such, our data demonstrate redundancy at both the microbial site and the site of the host during airway infection by S. pneumoniae.
Experimental procedures
Animals
C57BL/6 WT mice were purchased from Charles Rivers (Maastricht, the Netherlands). TLR2 KO mice (Takeuchi et al., 1999), backcrossed to a C57BL/6 genetic background six times, were bred in the animal facility of the Academic Medical Center in Amsterdam. Sex- and age-matched (10–12 weeks) mice were used in all experiments. All experiments were approved by the Animal Care and Use Committee of the University of Amsterdam.
Design
The experimental procedures to induce pneumonia have been described earlier (Branger et al., 2004; Knapp et al., 2004; Dessing et al., 2007). S. pneumoniae serotype 2 (strain D39) and isogenic pneumolysin-deficient S. pneumoniae (strain PLN) (Berry et al., 1989) were grown for 5 h to mid-logarithmic phase at 37°C using Todd–Hewitt broth (Difco, Detroit, MI), harvested by centrifugation at 1500 g for 15 min, and washed twice in sterile isotonic saline. A total of 50 μl containing 5 × 107 colony-forming units (cfu) were inoculated intranasally in mice which were lightly anesthetized by inhalation of isoflurane (Upjohn, Ede, the Netherlands). Mice were killed 6, 24 or 48 h after infection with S. pneumoniae D39, or 6, 24, 48 or 72 h after infection with S. pneumoniae PLN. In separate studies, survival of mice was determined during a 2 week follow-up.
Measurement of bacterial loads
Lung bacterial loads were determined as described earlier (Branger et al., 2004; Knapp et al., 2004; Dessing et al., 2007). Briefly, mice were sacrificed, and blood and lungs were collected. Lungs were homogenized at 4°C in 5 vols of sterile isotonic saline with a tissue homogenizer (Biospect Products, Bartlesville, OK). Serial 10-fold dilutions in sterile isotonic saline were made from these homogenates (and blood), and 50 μl vols were plated onto sheep-blood agar plates and incubated overnight at 37°C and 5% CO2.
Histology
Lungs for histology were fixed in 10% formalin and embedded in paraffin. Four millimetre sections were stained with haematoxylin and eosin (HE) and analysed by a pathologist who was blinded for groups. To score lung inflammation and damage, the entire lung surface was analysed with respect to the following parameters: bronchitis, oedema, interstitial inflammation, intra-alveolar inflammation, pleuritis and endothelialitis. Each parameter was graded on a scale of 0–4, with 0 as ‘absent’ and 4 as ‘severe’. The total ‘lung inflammation score’ was expressed as the sum of the scores for each parameter, the maximum being 24. Granulocyte staining was performed as described earlier by Ly-6G staining (Dessing et al., 2007).
Assays
Lung homogenates were prepared as described earlier (Knapp et al., 2004). MPO was measured by ELISA (HyCult, Uden, the Netherlands). TNF-α, IL-1β, IL-10, MIP-2 and KC were measured by ELISA (R and D Systems, Abingdon, UK).
Statistical analysis
Statistics were performed with GraphPad Prism version 4.00 for Windows (GraphPad Software, San Diego, CA). All data are given as means ± SEM. Differences between groups were analysed using Mann–Whitney U-test. For survival analyses, Kaplan-Meier analysis followed by log rank test was performed. To analyse the effect of the used bacterial strains and their interactions in more detail in WT and TLR2 KO mice 6 h after inoculation, results of the groups were compared using an anova. Data were checked for normal distribution and equal variances using the residuals. All variables were normally distributed, but MIP-2, IL-1β and TNF-α were ln-transformed to obtain equal variances. Later statistics was performed using spss statistical software version 12.0.1 (SPSS, Chicago, IL). A value of P < 0.05 was considered statistically significant.
Acknowledgments
We would like to thank Joost Daalhuisen and Marieke 10 Brink for technical assistance during the animal experiments, Regina de Beer for preparations of lung sections, and Michael Tanck for statistical advice.
References
- Albiger B, Sandgren A, Katsuragi H, Meyer-Hoffert U, Beiter K, Wartha F, et al. Myeloid differentiation factor 88-dependent signalling controls bacterial growth during colonization and systemic pneumococcal disease in mice. Cell Microbiol. 2005;7:1603–1615. doi: 10.1111/j.1462-5822.2005.00578.x. [DOI] [PubMed] [Google Scholar]
- Alouf JE. Cholesterol-binding cytolytic protein toxins. Int J Med Microbiol. 2000;290:351–356. doi: 10.1016/S1438-4221(00)80039-9. [DOI] [PubMed] [Google Scholar]
- Benton KA, Everson MP, Briles DE. A pneumolysin-negative mutant of Streptococcus pneumoniae causes chronic bacteremia rather than acute sepsis in mice. Infect Immun. 1995;63:448–455. doi: 10.1128/iai.63.2.448-455.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein JM. Treatment of community-acquired pneumonia – IDSA guidelines. Infectious Diseases Society of America. Chest. 1999;115:9S–13S. doi: 10.1378/chest.115.suppl_1.9s. [DOI] [PubMed] [Google Scholar]
- Berry AM, Paton JC. Additive attenuation of virulence of Streptococcus pneumoniae by mutation of the genes encoding pneumolysin and other putative pneumococcal virulence proteins. Infect Immun. 2000;68:133–140. doi: 10.1128/iai.68.1.133-140.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry AM, Yother J, Briles DE, Hansman D, Paton JC. Reduced virulence of a defined pneumolysin-negative mutant of Streptococcus pneumoniae. Infect Immun. 1989;57:2037–2042. doi: 10.1128/iai.57.7.2037-2042.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branger J, Knapp S, Weijer S, Leemans JC, Pater JM, Speelman P, et al. Role of Toll-like receptor 4 in gram-positive and gram-negative pneumonia in mice. Infect Immun. 2004;72:788–794. doi: 10.1128/IAI.72.2.788-794.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell GD., Jr Commentary on the 1993 American Thoracic Society guidelines for the treatment of community-acquired pneumonia. Chest. 1999;115:14S–18S. doi: 10.1378/chest.115.suppl_1.14s. [DOI] [PubMed] [Google Scholar]
- Canvin JR, Marvin AP, Sivakumaran M, Paton JC, Boulnois GJ, Andrew PW, Mitchell TJ. The role of pneumolysin and autolysin in the pathology of pneumonia and septicemia in mice infected with a type 2 pneumococcus. J Infect Dis. 1995;172:119–123. doi: 10.1093/infdis/172.1.119. [DOI] [PubMed] [Google Scholar]
- Cockeran R, Durandt C, Feldman C, Mitchell TJ, Anderson R. Pneumolysin activates the synthesis and release of interleukin-8 by human neutrophils in vitro. J Infect Dis. 2002;186:562–565. doi: 10.1086/341563. [DOI] [PubMed] [Google Scholar]
- Dessing MC, Knapp S, Florquin S, de Vos AF, van der Poll T. CD14 facilitates invasive respiratory tract infection by Streptococcus pneumoniae. Am J Respir Crit Care Med. 2007;175:604–611. doi: 10.1164/rccm.200606-824OC. [DOI] [PubMed] [Google Scholar]
- Echchannaoui H, Frei K, Schnell C, Leib SL, Zimmerli W, Landmann R. Toll-like receptor 2-deficient mice are highly susceptible to Streptococcus pneumoniae meningitis because of reduced bacterial clearing and enhanced inflammation. J Infect Dis. 2002;186:798–806. doi: 10.1086/342845. [DOI] [PubMed] [Google Scholar]
- Gilbert RJ. Pore-forming toxins. Cell Mol Life Sci. 2002;59:832–844. doi: 10.1007/s00018-002-8471-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SH, Kim JH, Martin M, Michalek SM, Nahm MH. Pneumococcal lipoteichoic acid (LTA) is not as potent as staphylococcal LTA in stimulating Toll-like receptor 2. Infect Immun. 2003;71:5541–5548. doi: 10.1128/IAI.71.10.5541-5548.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst RA, Kadioglu A, O'Callaghan C, Andrew PW. The role of pneumolysin in pneumococcal pneumonia and meningitis. Clin Exp Immunol. 2004;138:195–201. doi: 10.1111/j.1365-2249.2004.02611.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houldsworth S, Andrew PW, Mitchell TJ. Pneumolysin stimulates production of tumor necrosis factor alpha and interleukin-1 beta by human mononuclear phagocytes. Infect Immun. 1994;62:1501–1503. doi: 10.1128/iai.62.4.1501-1503.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedrzejas MJ. Pneumococcal virulence factors: structure and function. Microbiol Mol Biol Rev. 2001;65:187–207. doi: 10.1128/MMBR.65.2.187-207.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadioglu A, Taylor S, Iannelli F, Pozzi G, Mitchell TJ, Andrew PW. Upper and lower respiratory tract infection by Streptococcus pneumoniae is affected by pneumolysin deficiency and differences in capsule type. Infect Immun. 2002;70:2886–2890. doi: 10.1128/IAI.70.6.2886-2890.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. Pathogen recognition with Toll-like receptors. Curr Opin Immunol. 2005;17:338–344. doi: 10.1016/j.coi.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Khan AQ, Chen Q, Wu ZQ, Paton JC, Snapper CM. Both innate immunity and type 1 humoral immunity to Streptococcus pneumoniae are mediated by MyD88 but differ in their relative levels of dependence on toll-like receptor 2. Infect Immun. 2005;73:298–307. doi: 10.1128/IAI.73.1.298-307.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp S, Wieland CW, van 't Veer C, Takeuchi O, Akira S, et al. Toll-like receptor 2 plays a role in the early inflammatory response to murine pneumococcal pneumonia but does not contribute to antibacterial defense. J Immunol. 2004;172:3132–3138. doi: 10.4049/jimmunol.172.5.3132. [DOI] [PubMed] [Google Scholar]
- Knapp S, Schultz MJ, Poll TV. Pneumonia models and innate immunity to respiratory bacterial pathogens. Shock. 2005;24(Suppl. 1):12–18. doi: 10.1097/01.shk.0000191385.41689.f3. [DOI] [PubMed] [Google Scholar]
- Koedel U, Angele B, Rupprecht T, Wagner H, Roggenkamp A, Pfister HW, Kirschning CJ. Toll-like receptor 2 participates in mediation of immune response in experimental pneumococcal meningitis. J Immunol. 2003;170:438–444. doi: 10.4049/jimmunol.170.1.438. [DOI] [PubMed] [Google Scholar]
- Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, Thompson CM, et al. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci USA. 2003;100:1966–1971. doi: 10.1073/pnas.0435928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogensen TH, Paludan SR, Kilian M, Ostergaard L. Live Streptococcus pneumoniae, Haemophilus influenzae, and Neisseria meningitidis activate the inflammatory response through Toll-like receptors 2,4, and 9 in species-specific patterns. J Leukoc Biol. 2006;80:267–277. doi: 10.1189/jlb.1105626. [DOI] [PubMed] [Google Scholar]
- Moore TA, Standiford TJ. Cytokine immunotherapy during bacterial pneumonia: from benchtop to bedside. Semin Respir Infect. 2001;16:27–37. doi: 10.1053/srin.2001.22726. [DOI] [PubMed] [Google Scholar]
- Paterson GK, Mitchell TJ. Innate immunity and the pneumococcus. Microbiology. 2006;152:285–293. doi: 10.1099/mic.0.28551-0. [DOI] [PubMed] [Google Scholar]
- Paton JC. The contribution of pneumolysin to the pathogenicity of Streptococcus pneumoniae. Trends Microbiol. 1996;4:103–106. doi: 10.1016/0966-842X(96)81526-5. [DOI] [PubMed] [Google Scholar]
- Paton JC, Ferrante A. Inhibition of human polymorphonuclear leukocyte respiratory burst, bactericidal activity, and migration by pneumolysin. Infect Immun. 1983;41:1212–1216. doi: 10.1128/iai.41.3.1212-1216.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikis A, Akram S, Donkersloot JA, Campos JM, Rodriguez WJ. Penicillin-resistant pneumococci from pediatric patients in the Washington, DC, area. Arch Pediatr Adolesc Med. 1995;149:30–35. doi: 10.1001/archpedi.1995.02170130032007. [DOI] [PubMed] [Google Scholar]
- van der Poll T, Keogh CV, Buurman WA, Lowry SF. Passive immunization against tumor necrosis factor-alpha impairs host defense during pneumococcal pneumonia in mice. Am J Respir Crit Care Med. 1997;155:603–608. doi: 10.1164/ajrccm.155.2.9032201. [DOI] [PubMed] [Google Scholar]
- Rijneveld AW, Florquin S, Branger J, Speelman P, Van Deventer SJ, van der Poll T. TNF-alpha compensates for the impaired host defense of IL-1 type I receptor-deficient mice during pneumococcal pneumonia. J Immunol. 2001;167:5240–5246. doi: 10.4049/jimmunol.167.9.5240. [DOI] [PubMed] [Google Scholar]
- van Rossum AM, Lysenko ES, Weiser JN. Host and bacterial factors contributing to the clearance of colonization by Streptococcus pneumoniae in a murine model. Infect Immun. 2005;73:7718–7726. doi: 10.1128/IAI.73.11.7718-7726.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubins JB, Charboneau D, Paton JC, Mitchell TJ, Andrew PW, Janoff EN. Dual function of pneumolysin in the early pathogenesis of murine pneumococcal pneumonia. J Clin Invest. 1995;95:142–150. doi: 10.1172/JCI117631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber JR, Jacobs MR. Antibiotic-resistant pneumococci. Pediatr Clin North Am. 1995;42:519–537. doi: 10.1016/s0031-3955(16)38977-5. [DOI] [PubMed] [Google Scholar]
- Schroder NW, Morath S, Alexander C, Hamann L, Hartung T, Zahringer U, et al. Lipoteichoic acid (LTA) of Streptococcus pneumoniae and Staphylococcus aureus activates immune cells via Toll-like receptor (TLR) -2, lipopolysaccharide-binding protein (LBP), and CD14, whereas TLR-4 and MD-2 are not involved. J Biol Chem. 2003;278:15587–15594. doi: 10.1074/jbc.M212829200. [DOI] [PubMed] [Google Scholar]
- Srivastava A, Henneke P, Visintin A, Morse SC, Martin V, Watkins C, et al. The apoptotic response to pneumolysin is Toll-like receptor 4 dependent and protects against pneumococcal disease. Infect Immun. 2005;73:6479–6487. doi: 10.1128/IAI.73.10.6479-6487.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- Yoshimura A, Lien E, Ingalls RR, Tuomanen E, Dziarski R, Golenbock D. Cutting edge: recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J Immunol. 1999;163:1–5. [PubMed] [Google Scholar]