

Abstract
It is generally recognized that drug discovery and development are very time and resources consuming processes. There is an ever growing effort to apply computational power to the combined chemical and biological space in order to streamline drug discovery, design, development and optimization. In biomedical arena, computer-aided or in silico design is being utilized to expedite and facilitate hit identification, hit-to-lead selection, optimize the absorption, distribution, metabolism, excretion and toxicity profile and avoid safety issues. Commonly used computational approaches include ligand-based drug design (pharmacophore, a 3-D spatial arrangement of chemical features essential for biological activity), structure-based drug design (drug-target docking), and quantitative structure-activity and quantitative structure-property relationships. Regulatory agencies as well as pharmaceutical industry are actively involved in development of computational tools that will improve effectiveness and efficiency of drug discovery and development process, decrease use of animals, and increase predictability. It is expected that the power of CADDD will grow as the technology continues to evolve.
Keywords: Drug discovery, Drug development, Molecular modeling, Virtual screening, Computational modeling, In silico drug design, QSAR/QSPR, Predictive toxicology
INTRODUCTION
Use of computational techniques in drug discovery and development process is rapidly gaining in popularity, implementation and appreciation. Different terms are being applied to this area, including computer-aided drug design (CADD), computational drug design, computer-aided molecular design (CAMD), computer-aided molecular modeling (CAMM), rational drug design, in silico drug design, computer-aided rational drug design. Term Computer-Aided Drug Discovery and Development (CADDD) will be employed in this overview of the area to cover the entire process. Both computational and experimental techniques have important roles in drug discovery and development and represent complementary approaches. CADDD entails:
Use of computing power to streamline drug discovery and development process
Leverage of chemical and biological information about ligands and/or targets to identify and optimize new drugs
Design of in silico filters to eliminate compounds with undesirable properties (poor activity and/or poor Absorption, Distribution, Metabolism, Excretion and Toxicity, ADMET) and select the most promising candidates.
Fast expansion in this area has been made possible by advances in software and hardware computational power and sophistication, identification of molecular targets, and an increasing database of publicly available target protein structures. CADDD is being utilized to identify hits (active drug candidates), select leads (most likely candidates for further evaluation), and optimize leads i.e. transform biologically active compounds into suitable drugs by improving their physicochemical, pharmaceutical, ADMET/PK (pharmacokinetic) properties. Virtual screening is used to discover new drug candidates from different chemical scaffolds by searching commercial, public, or private 3-dimensional chemical structure databases. It is intended to reduce the size of chemical space and thereby allow focus on more promising candidates for lead discovery and optimization. The goal is to enrich set of molecules with desirable properties (active, drug-like, lead-like) and eliminate compounds with undesirable properties (inactive, reactive, toxic, poor ADMET/PK). In another words, in silico modeling is used to significantly minimize time and resource requirements of chemical synthesis and biological testing (Fig. 1). The rapid growth of virtual screening is evidenced by increase in the number of citations matching keywords “virtual screening” from 4 in 1997 to 302 in 2004 [1]. In his 2003 review article, Green of GlaxoSmithKline concluded that: “The future is bright. The future is virtual” [2].
Figure 1.

Comparison of traditional and virtual screening in terms of expected cost and time requirements.
PriceWaterhouseCoopers Pharma 2005: An Industrial Revolution in R&D report [3] stressed the reality that pharmaceutical industry needs to find means of improving efficiency and effectiveness of drug discovery and development in order to sustain itself. This was recently echoed at the 2006 Drug Discovery Technology Conference in Boston, MA by Dr. Steven Paul, head of science and technology at Eli Lilly & Co. who stated that the current business model will become fundamentally untenable unless there is a significant improvement in efficiency and effectiveness of the process. The PriceWaterhouseCoopers report emphasized growth and value of in silico approaches to address this issue and projected that in silico methods will become dominant from drug discovery through marketing. It was suggested that we are in a transitional period where the roles of primary (laboratory and clinical studies) and secondary (computational) science are in process of reversal [4].
Estimates of time and cost of currently bringing a new drug to market vary, but 7–12 years and $ 1.2 billion are often cited [5]. Furthermore, five out of 40,000 compounds tested in animals reach human testing and only one of five compounds reaching clinical studies is approved. This represents an enormous investment in terms of time, money and human and other resources. It includes chemical synthesis, purchase, curation, and biological screening of hundreds of thousands of compounds to identify hits followed by their optimization to generate leads which requiring further synthesis. In addition, predictability of animal studies in terms of both efficacy and toxicity is frequently suboptimal. Therefore, new approaches are needed to facilitate, expedite and streamline drug discovery and development, save time, money and resources, and as per pharma mantra “fail fast, fail early”. It is estimated that computer modeling and simulations account for ~ 10% of pharmaceutical R&D expenditure and that they will rise to 20% by 2016 [6].
Role of computational models is to increase prediction based on existing knowledge [7]. Computational methods are playing increasingly larger and more important role in drug discovery and development [7–15] (Fig. 2) and are believed to offer means of improved efficiency for the industry [7]. They are expected to limit and focus chemical synthesis and biological testing and thereby greatly decrease traditional resource requirements.
Figure 2.

Modern drug discovery and development process including prominent role of computational modeling.
Figure was reprinted from Drug Discovery Today 11: 326–333 (2006), “Integrating molecular design resources within modern drug discovery research: the Roche experience”, by M. Stahl, W. Guba and M. Kansy [15], with permission from Elsevier.
Growing presence, prominence and importance of CADDD is seen by multiple scientific sessions dedicated to it at major scientific conferences, e.g. 45th Annual Meeting of SOT in San Diego 2006 (http://www.toxicology.org/ai/meet/am2006/index.asp), 97th Annual Meeting of AACR in DC (http://www.aacr.org/page6029.aspx), PharmaDiscovery2006 in Bethesda, MD (http://www.pharmadiscoveryevent.com/app/homepage.cfm?appname=100304&moduleid=451&campaignid=32872&iUserCampaignID=23386619) as well as conferences dedicated to the subject such as Drug Design conference in London, UK (http://www.smi-online.co.uk/event_media/overview.asp?is=4&ref=2062) and bi-annual Gordon Research Conference-Computer-Aided Drug Design conference that dates back to 1970’s when it was known as Quantitative Structure Activity Relationships (QSAR) (http://www.grc.org/programs/2005/cadd.htm). At the 2005 London Drug Design conference, aspiration and expectation were expressed that computational methods will achieve similar role and utility in pharmaceutical industry as already exist in automotive and airplane industries.
This represents a brief overview, rather than an exhaustive review, of CADDD and the following commonly used computational approaches will be discussed: ligand-based design (e.g. pharmacophore)[16], structure (target)-based design (e.g. docking)[17], and quantitative structure-activity/property relationships (QSAR/QSPR) (e.g. computational predictive toxicology)[18].
IUPAC defines pharmacophore as: “the ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target structure and to trigger (or to block) its biological response. A pharmacophore does not represent a real molecule or a real association of functional groups, but a purely abstract concept that accounts for the common molecular interaction capacities of a group of compounds towards their target structure. The pharmacophore can be considered as the largest common denominator shared by a set of active molecules” (http://www.chem.qmul.ac.uk/iupac/medchem/ix.html#p7). Pharmacophoric descriptors include H-bond donors, H-bond acceptors, hydrophobic, aromatic, positive ionizable groups, negative ionizable groups. They represent chemical feature complimentarity to the receptor in the 3-dimensional space. Further enhancement of a pharmacophore can be obtained by combining it with shape and exclusion volumes (steric) constraints [19, 20]. These enhancements decrease likelihood of finding molecules with a suitable 3-dimensional arrangement of functional groups but wrong shape that could prevent them from fitting into the receptor binding site. Pharmacophore requires knowledge of active ligands and/or target receptor. They are number of ways to build a pharmacophore. It can be done based on chemical structure of 3 or 4 known active compounds from different chemical scaffolds (http://www.accelrys.com/products/catalyst/catalystproducts/cathypo.html#hiphop) [21, 22]. Alternately, diverse chemical structures for about 15 compounds along with the corresponding IC50 or Ki50 values ranging over more than 3 orders of magnitude can be used (http://www.accelrys.com/products/catalyst/catalystproducts/cathypo.html) [21, 22]. Statistical validation of the pharmacophore model may be done using Fischer’s randomization test based on a random reassignment of activity values among the molecules of the training set. Further validation may be performed with a set of known active ligands, or a separate set of test compounds with known properties which were not used for model training, and ultimately by biological testing. Using published IC50 data [23–27] with values ranging over more than 3 orders of magnitude from several different chemical scaffolds, including phytoestrogens, we have derived an ERβ agonist pharmacophore (Fig. 3). Thirty compound training and twenty-two compound test set yielded correlation of 0.94 and 0.82, respectively. Resveratrol, compound with cancer chemopreventive activities, showed a reasonable fit to this pharmacophore (Fig. 3), but not quite as good as another naturally occurring chemopreventive compound genistein. In addition, pharmacophore can be designed de novo based on complimentarity to a known ligand binding site. Most commonly used pharmacophore software includes Catalyst (http://www.accelrys.com/products/catalyst/), Phase (http://www.schrodinger.com/ProductDescription.php?mID=6&sID=16&cID=0), Sybyl including Galahad, GASP, DISCOtech, and UNITY 3D (http://www.tripos.com/index.php?family=Modules,SimplePage,discovery_info), and MOE (http://www.chemcomp.com/software.htm).
Figure 3.
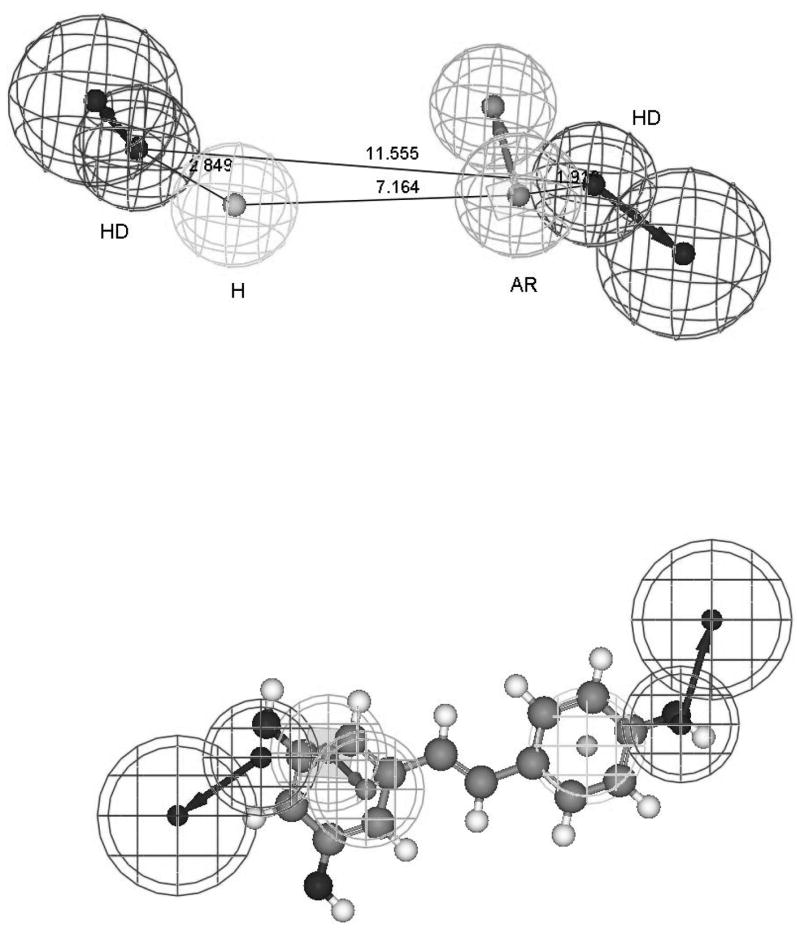
Estrogen receptor beta (ERβ) agonist pharmacophore.
Pharmacophore model of ERβ agonist consisting of two hydrogen bond donor (HD), one aromatic (AR) and one hydrophobic (H) descriptors, was developed using Catalyst 4.11 (http://www.accelrys.com/products/catalyst/) based on published IC50 data from Chemical Sciences Group at Wyeth Research [23–27]. Descriptors are represented by tolerance spheres around a centroid. Some descriptors (HD, AR) have directionality and are thus represented as vectors with two tolerance spheres each. Top figure represents the derived pharmacophore with inter-descriptor distances in Angstroms. Values for angles were omitted for visual clarity. Bottom figure represents an overlay of resveratrol with the derived pharmacophore. Goodness of fit of chemical structure to the pharmacophore is based on its having the functional moities complimentary to the pharmacophore descriptors and their closeness to the centroid and tolerance spheres of the latter in the 3-dimensional space.
Structure (target)-based drug design represents docking i.e. ligand binding to its receptor, target protein. Docking is used to identify and optimize drug candidates by examining and modeling molecular interactions between ligands and target macromolecules. An example of ligand binding and the associated van der Waals, hydrogen bonding and electrostatic energies as a function of the interatomic distance is shown in Fig. 4. Based on the X-ray structure of ERβ receptor co-crystallized with various ligands and ERα Met421 →ERβ Ile373 and ERα Leu384 →ERβ Met336 substitution in the ligand binding pocket and computational modeling, Wyeth group has designed a selective ERβ agonist, ERB-041 with similar affinity but more than 200-fold greater selectivity for ERβ than that of 17β-estradiol (Fig. 5). Structure (target)-based design requires structural information for the receptor which can be obtained from X-ray crystallography, NMR or homology modeling. The latter being another computational technique used to predict unknown protein structure from a sequence similarity to known protein structure(s). In the process of docking, multiple ligand conformations and orientations are generated and the most appropriate ones are selected. Scoring functions are applied to evaluate tightness of interaction i.e. estimate binding free energy. General observation is that consensus (combination of different scoring algorithms) scoring yields better results than individual scoring [28]. Validations may be perfomed with known active and inactive ligands, comparisons to crystallographic data and prediction of rank-ordering and binding affinities. Several recent publications have compared different docking methods [29–33]. One of these recent studies evaluated 10 docking programs and 37 scoring functions against eight proteins of seven protein types in terms of binding mode prediction, virtual screening for lead identification, and rank-ordering by affinity for lead optimization [33]. While these programs were able to identify active ligands, none of them performed well for all the targets. In general, the programs were also less effective in rank-ordering and predicting of ligand binding affinity. Most commonly used docking software includes Autodock (http://www.scripps.edu/mb/olson/doc/autodock/), Gold (http://www.ccdc.cam.ac.uk/products/life_sciences/gold/), Dock (http://dock.compbio.ucsf.edu/), Insight II Affinity and Cerius2 LigandFit (http://www.accelrys.com/), Sybyl including FlexE and FlexX (http://tripos.com/index.php?family=modules,SimplePage,,,&page=comp_informatics), Glide (http://www.schrodinger.com/ProductDescription.php?mID=6&sID=6), and MOE (http://www.chemcomp.com/software.htm).
Figure 4. Docking.
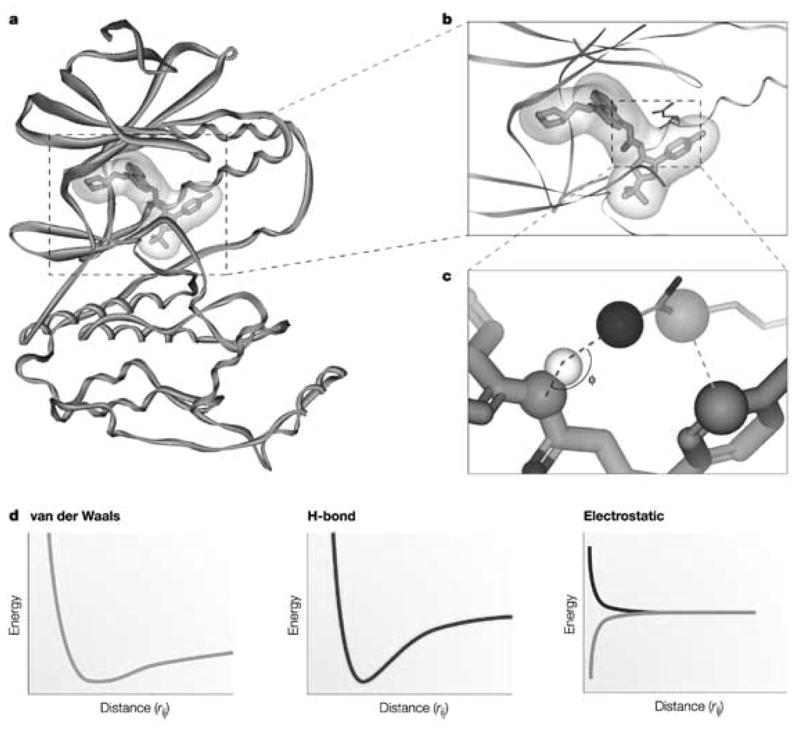
a.) Ligand binding of p38 mitogen-activated protein kinase with inhibitor BIRB796 (PDB code: 1KV2), including its electrostatic potential surface and b.) enlarged view. c.) hydrogen bonding and van der Waals interactions are depicted as darker and lighter dashed lines, respectively. d.) van der Waals, hydrogen bonding and electrostatic (same or opposite charges) energies as a function of inter-atomic distance (rij).
Figure was reproduced from Nature Reviews Drug Discovery 3: 935–949 (2004), “Docking and scoring in virtual screening for drug discovery: methods and applications”, by D.B. Kitchen, H. Decornez, J.R. Furr, and J. Bajorath [28], with permission from Macmillan Magazines Ltd.
Figure 5.
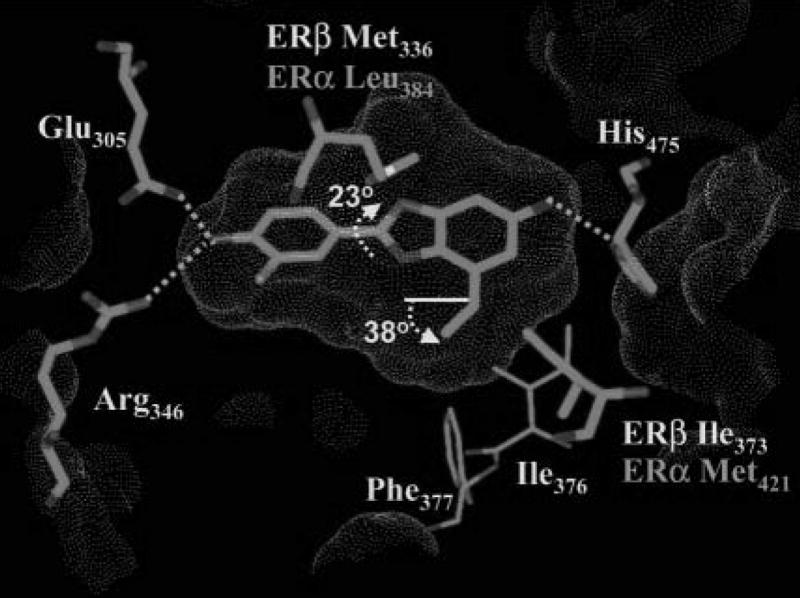
Binding of ERβ selective agonist, ERB-041 in the ligand binding domain of ERβ receptor. Shape of the binding site is represented by a Connolly surface. Two amino acid substitutions between ERα and ERβ ligand binding cavity are labeled accordingly.
Figure was reprinted from Journal of Medicinal Chemistry 47: 5021–5040 (2004), “Design and synthesis of aryl diphenolic azoles as potent and selective estrogen receptor-β ligands”, by M.S. Malamas, E.S. Manas, R.E. McDevitt, I. Gunawan, Z.B. Xu, M.D. Collini, C.P. Miller, T. Dinh, R.A. Henderson, J.C. Keith Jr., and H.A. Harris [25], with permission from the American Chemical Society.
Applications and benefits of CADDD have been reviewed and demonstrated in growing number of publications and supported by examples of drugs derived from the in silico approach [9, 34–39]. Virtual screening has been shown more efficient than commonly used empirical screening. Shoichet reported that ligand discovery i.e. hit rates (number of compounds binding to a target divided by number of compounds tested) is greater in virtual screening by 2 or 3 orders of magnitude than in empirical screening [40]. Others have reported similar results [41–43]. The “receiver operating characteristic (ROC)” curves have also been used as a metric to evaluate the ability of virtual screening in discriminating between active and inactive compounds [44, 45]. Evaluations using ROC curves have shown that virtual screening can exhibit reasonable sensitivity and specificity by minimizing false negatives and false positives, respectively [44, 45].
Number of reports citing successful application of CADDD in developing specific drugs in different therapeutic areas is expanding rapidly. A very interesting example which can also serve as a proof of principle of the in silico approach involves a type I TGF β receptor kinase inhibitor. The same molecule (HTS-466284/LY-364947), a 27 nM inhibitor, was discovered independently using virtual screening by Biogen IDEC [46] and traditional enzyme and cell-based high-throughput screening by Eli Lilly [47]. Another in silico modeling drug development program led to clinical trials of a novel, potent, and selective anti-anxiety, anti-depression 5-HT1A agonist in less than 2 years from the start and requiring less than 6 months of lead optimization and synthesis of only 31 compounds [48].
Pharmacophore library screening followed by docking represent complimentary screening methods with the combination providing optimum results [49]. Commonly, this screening approach is preceded by a prior filtering of virtual databases (e.g. physicochemical, ADMET/PK, stability, reactivity, toxicity, drug-like properties, etc.) [9, 50–54]. This combination of screening methods has been successfully employed in designing new hits and leads, e.g. PPARγ ligand [55], dopamine D3 receptor agonist [56], antibiotics [57], c-Src/Abl kinase inhibitors [58], checkpoint-1 kinase inhibitor [52], MDM2-p53 inhibitor [51], integrin αvβ3 antagonist [59]. Typically, this approach involves virtual screening (pharmacophore plus docking) of virtual chemical structure libraries containing hundreds of thousands of compounds and necessitating chemical synthesis and biological screening of less than 100 compounds to yield a handful of drug candidates with good receptor affinities. Recently, application and utility of this virtual screening approach in combination with activity-guided fractionation of medicinal plants was also demonstrated and coined “in combo screening” [60–64].
While computational techniques have already provided significant benefits, they hold a great promise for future progress in drug discovery and development. However, CADDD is still an evolving technology and has number of limitations [9, 28, 65]. Like high-throughput screening, virtual screening accepts limited accuracy (false-positives and false-negatives) in exchange for a list of few promising candidates for further evaluation. There are number of the issues that currently are not adequately addressed in pharmacophore modeling [16]. For example, receptor may have more than a single active site, receptor may adapt to different ligands and multiple pharmacophores may be possible for a single site. Docking also has its limitations [28, 30, 65, 66]. Sampling of molecular conformations to account for both ligand and receptor flexibility and selection of appropriate force fields is not straight forward or simple. Number of possible conformations mushrooms with increasing molecular mass and number of rotational bonds. This presents severe demands on computational hardware and software. Assumption of structural rigidity as an approximation may have severe entropy repercussions. In addition, binding may lead to protein adaptability and additional conformational changes that are not normally considered. Another issue raised is the importance of the crystallization process and how representative is a single crystal structure [67]. Role of solvent molecules is difficult to ascertain. Solvent molecules can play an important role in binding by serving as bridging hydrogen bonds between a ligand and its binding site or via entropy effects. Appropriate treatment of ionization and tautomerization of ligand and protein is also very important. There may be multiple binding sites and calculating ligand-receptor affinities (scoring functions) leaves lot to be desired. It is estimated that docking programs currently dock 70 – 80% of ligands correctly [34]. One recent study proposed that false positives, a significant issue in structure-based virtual screening, stems from inability of the current docking and scoring algorithms to identify key interactions and treat them appropriately [66]. Recognizing the drawbacks of the present state-of-art of docking and scoring, several workshops, including representatives from academia, industry and government, were held to address the issues [68]. The outcome of the workshops and the resultant proposed plan of action can be found in that reference and the cited references therein [68].
Extensive computational power is required to screen millions of compounds and take into the consideration their and receptors’ flexibilities. In order to address this need, several major virtual screening efforts utilizing grid or distributed computing have been initiated. One of these involves National Foundation for Cancer Research (NFCR) Center for Computational Drug Discovery established in conjunction with Oxford University as an effort of “finding the right key to open a special lock--from billions molecular keys” (http://www.nfcr.org/Default.aspx?tabid=399) [69]. It utilizes over 2 millions personal computers connected world wide via the Internet, and is capable of screening a library of 3.5 billion molecules against a dozen of targets in months. It has generated over 100,000 drug candidate molecules, or “hits” in the first phase of the project. Another similar project allowed scientist to screen over 35 million compounds against several smallpox proteins and returned over 100,000 hits in the first 72 hours http://www.worldcommunitygrid.org/projects_showcase/viewProjectArchives.do). Overall, 44 strong candidates were identified. Similar approach is also being utilized in the fight against AIDS by the Olson laboratory at the Scripps Research Institute (http://fightaidsathome.scripps.edu/discovery.html).
In addition to being able to efficiently identify drug hits and leads, it is also important to avoid drug attrition. Toxicity has almost doubled as a cause of drug attrition from year 1991 to 2000 and is one of the main causes of drug failure [70]. Preclinical safety studies using animals are lengthy, expensive and frequently of limited predictability for human outcomes. In addition, there is a strong push to develop alternative in silico methods of predictive evaluation of drug toxicity in order to minimize animal testing. The expectation is that predictive toxicology will help avoid resource waste, reduce regulatory review burden and expedite review, reduce animal use, avoid need for interspecies uncertainty factors, increase accuracy, sensitivity, and specificity, and predict adverse effects not detectable in animals (e.g. nausea, dizziness, headache, cognitive impairment, etc.). European policy for the evaluation of chemicals (REACH: Registration, Evaluation, and Authorization of Chemicals) has been a strong advocate of alternative in silico methods of predictive evaluation of chemical toxicity in order to minimize animal testing and conserve time and resources [71]. QSAR and QSPR are commonly used computational methods in predictive toxicology. In a strict sense, these two terms are not synonymous even though the term QSAR tends to be used for both QSAR and QSPR. The principle behind them is the same, but they have a different context in terms of the dependent variable, biological activity (QSAR) vs. bio-physico-chemical property (QSPR). Independent variables represent molecular descriptors, e.g. electronic, spatial, topological, conformational, thermodynamic, quantum mechanical, etc. The idea of structure-activity relationship dates back to 1868 [72] when Crum Brown and Frazer reported on the correlation of paralyzing activity to the nature of quarternary group of a collection of strychnine-like compounds. More recently, studies of Corwin Hansch in the 1960’s demonstrated applicability and usefulness of QSAR/QSPR approach and led to its growing use [73, 74]. Interest in the use of QSAR in the regulatory arena has been growing and is being evaluated [75]. Informatics and Computational Safety Analysis Staff (ICSAS) within CDER at FDA is actively evaluating the potential of predictive toxicology (http://www.fda.gov/cder/offices/ops_io/default.htm). They have constructed databases of toxicological and clinical endpoints and, in collaboration with software companies, are developing and evaluating data mining and QSAR computational techniques for predictive toxicology. Applying QSAR algorithms to toxicity data and corresponding chemical structures, they have developed tools for toxicity response (mutagenicity, carcinogenicity) and toxicity dosing (No Observed Effect Level, NOEL; Maximum Recommended Starting Dose, MRSD) predictions [76]. Their carcinogenicity QSAR model with 53 descriptors and data from 2 year rodent study FDA database exhibited 76% sensitivity and 84% specificity [77]. The anticipation is that it could avoid unexpected rodent carcinogenicity results from very costly and lengthy studies ($ 2 to $ 5 million and over 2 years) on top of the cost and time associated with a successful Phase 1 study. Positive predictability of 92.5 % and false positive rate of 4.8% were achieved using QSAR model to estimate the No Observed Effect Level (NOEL) of chemicals in man based on a database of Maximum Recommended Therapeutic Doses (MRTD) of marketed pharmaceuticals [78]. This represents a marked improvement over the poor correlation (R2 = 0.2005) between human MRTD and rodent Maximum Tolerated Dose (MTD) as reported by the same authors [78]. ICSAS has also developed a predictive model to estimate the Maximum Recommended Starting Dose (MRSD) for Phase 1 clinical trials based on the human Maximum Recommended Daily Dose (MRDD) [79](Fig. 6). QSAR validations are commonly done using internal (leave group out from training set for testing) or external (test compounds not present in the training set). Criteria for validation include accuracy and proper rank ordering. However, concerns still remain about predictability of these QSAR models for new chemical entities and need to be addressed in the future. Nevertheless, the present results are encouraging and it is hoped that predictions based on human data should decrease reliance on lengthy and expensive animal studies and limited predictability of interspecies extrapolations.
Figure 6.
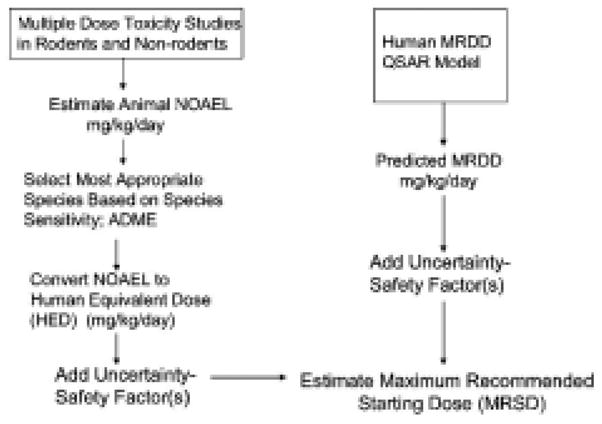
Traditional and computational approaches to selection of the Maximum Recommended Starting Dose (MRSD) for Phase 1 clinical trials.
Figure was reprinted from Regulatory Toxicology and Pharmacology 40: 185–206 (2004), “Estimating the safe and staring dose in phase I clinical trials and no observed effect level based on QSAR modeling of the human maximum recommended daily dose”, by J.F. Contrera, E.J. Matthews, N.L. Kruhlak, and R. D. Benz [79], with permission from Elsevier.
Number of open-source and free molecular modeling resources are available (http://www.chemoinf.com/) [80], including databases like Pubchem (http://pubchem.ncbi.nlm.nih.gov/) and Zinc (http://blaster.docking.org/zinc/). There is even simple, web-based software that allows drawing of chemical structures and estimation of some physicochemical, biological, and drug-like properties (http://www.organic-chemistry.org/prog/peo/index.html and http://www.molinspiration.com/cgi-bin/properties).
Other computational approaches are also being utilized but will not be discussed in this review. One of these, de novo or fragment-based technique [81–83] is based on local optimization and provides means of identifying new chemotypes and chemical scaffolds. In addition, new emerging fields like systems biology are expected to play important role in drug discovery and development [84–93]. Systems biology employs in silico techniques to integrate and analyze disparate chemical and biochemical data in a parallel as opposed to sequential fashion. Fueled by ‘omic’ technologies, it applies principles and mathematical tools of electrical engineering and networks to dynamic modeling and simulation of complex biological systems in a holistic manner.
Therefore, while it should be apparent that CADDD has a great potential, one should not rely on computational techniques in a black box manner and beware of the Garbage In-Garbage Out (GIGO) phenomenon. The in cerebro element is an essential and critical part of the process [7, 94]. CADDD should be based on the in cerebro-in silico-chemico-biological approach.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pozzan A. Molecular descriptors and methods for ligand based virtual high throughput screening in drug discovery. Curr Pharm Des. 2006;12(17):2099–2110. doi: 10.2174/138161206777585247. [DOI] [PubMed] [Google Scholar]
- 2.Green DV. Virtual screening of virtual libraries. Prog Med Chem. 2003;41:61–97. doi: 10.1016/s0079-6468(02)41002-8. [DOI] [PubMed] [Google Scholar]
- 3.PriceWaterhouseCoopers. PriceWaterhouseCoopers Pharma 2005: An Industrial Revolution in R&D. 2005 http://www.pwc.com/gx/eng/about/ind/pharma/industrial_revolution.pdf.
- 4.Contrera JF. Application of toxicology databases in drug development. (Estimating potential toxicity) 2004 http://www.lib.uchicago.edu/cinf/220nm/slides/220nm18/220nm18.ppt#285,24,Slide.
- 5.Shankar R, Frapaise X, Brown B. LEAN drug development in R&D. Drug Discov Development. 2006:57–60. [Google Scholar]
- 6.van de Waterbeemd H, Gifford E. ADMET in silico modelling: towards prediction paradise? Nat Rev Drug Discov. 2003;2(3):192–204. doi: 10.1038/nrd1032. [DOI] [PubMed] [Google Scholar]
- 7.Kumar N, Hendriks BS, Janes KA, de Graaf D, Lauffenburger DA. Applying computational modeling to drug discovery and development. Drug Discovery Today. 2006;11(17–18):806–811. doi: 10.1016/j.drudis.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 8.Hann MM, Oprea TI. Pursuing the leadlikeness concept in pharmaceutical research. Curr Opin Chem Biol. 2004;8(3):255–263. doi: 10.1016/j.cbpa.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 9.Oprea TI, Matter H. Integrating virtual screening in lead discovery. Curr Opin Chem Biol. 2004;8(4):349–358. doi: 10.1016/j.cbpa.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 10.Roche O, Guba W. Computational chemistry as an integral component of lead generation. Mini Rev Med Chem. 2005;5(7):677–683. doi: 10.2174/1389557054368826. [DOI] [PubMed] [Google Scholar]
- 11.Ekins S, Wang B. Computer Applications in Pharmaceutical Research and Development. Wiley-Interscience; 2006. [Google Scholar]
- 12.Chin DN, Chuaqui CE, Singh J. Integration of virtual screening into the drug discovery process. Mini Rev Med Chem. 2004;4(10):1053–1065. doi: 10.2174/1389557043403044. [DOI] [PubMed] [Google Scholar]
- 13.Jain AN. Virtual screening in lead discovery and optimization. Curr Opin Drug Discov Devel. 2004;7(4):396–403. [PubMed] [Google Scholar]
- 14.Stoermer MJ. Current status of virtual screening as analysed by target class. Med Chem. 2006;2(1):89–112. doi: 10.2174/157340606775197750. [DOI] [PubMed] [Google Scholar]
- 15.Stahl M, Guba W, Kansy M. Integrating molecular design resources within modern drug discovery research: the Roche experience. Drug Discovery Today. 2006;11(7–8):326–333. doi: 10.1016/j.drudis.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 16.Dror O, Shulman-Peleg A, Nussinov R, Wolfson HJ. Predicting molecular interactions in silico: I. A guide to pharmacophore identification and its applications to drug design. Curr Med Chem. 2004;11(1):71–90. doi: 10.2174/0929867043456287. [DOI] [PubMed] [Google Scholar]
- 17.Schneidman-Duhovny D, Nussinov R, Wolfson HJ. Predicting molecular interactions in silico: II. Protein-protein and protein-drug docking. Curr Med Chem. 2004;11(1):91–107. doi: 10.2174/0929867043456223. [DOI] [PubMed] [Google Scholar]
- 18.Matthews EJ, Kruhlak NL, Cimino MC, Benz RD, Contrera JF. An analysis of genetic toxicity, reproductive and developmental toxicity, and carcinogenicity data: II. Identification of genotoxicants, reprotoxicants, and carcinogens using in silico methods. Regulatory Toxicology and Pharmacology. 2006;44(2):97–110. doi: 10.1016/j.yrtph.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 19.Pandit D, So SS, Sun H. Enhancing Specificity and Sensitivity of Pharmacophore-Based Virtual Screening by Incorporating Chemical and Shape Features-A Case Study of HIV Protease Inhibitors. J Chem Inf Model. 2006 doi: 10.1021/ci050511a. [DOI] [PubMed] [Google Scholar]
- 20.Toba S, Srinivasan J, Maynard AJ, Sutter J. Using Pharmacophore Models To Gain Insight into Structural Binding and Virtual Screening: An Application Study with CDK2 and Human DHFR. J Chem Inf Model. 2006 doi: 10.1021/ci050410c. [DOI] [PubMed] [Google Scholar]
- 21.Funk OF, Kettmann V, Drimal J, Langer T. Chemical function based pharmacophore generation of endothelin-A selective receptor antagonists. J Med Chem. 2004;47(11):2750–2760. doi: 10.1021/jm031041j. [DOI] [PubMed] [Google Scholar]
- 22.Krovat EM, Langer T. Non-peptide angiotensin II receptor antagonists: chemical feature based pharmacophore identification. J Med Chem. 2003;46(5):716–726. doi: 10.1021/jm021032v. [DOI] [PubMed] [Google Scholar]
- 23.Collini MD, Kaufman DH, Manas ES, Harris HA, Henderson RA, Xu ZB, Unwalla RJ, Miller CP. 7-Substituted 2-phenyl-benzofurans as ER beta selective ligands. Bioorg Med Chem Lett. 2004;14(19):4925–4929. doi: 10.1016/j.bmcl.2004.07.029. [DOI] [PubMed] [Google Scholar]
- 24.Edsall RJ, Jr, Harris HA, Manas ES, Mewshaw RE. ERbeta ligands. Part 1: the discovery of ERbeta selective ligands which embrace the 4-hydroxy-biphenyl template. Bioorg Med Chem. 2003;11(16):3457–3474. doi: 10.1016/s0968-0896(03)00303-1. [DOI] [PubMed] [Google Scholar]
- 25.Malamas MS, Manas ES, McDevitt RE, Gunawan I, Xu ZB, Collini MD, Miller CP, Dinh T, Henderson RA, Keith JC, Jr, Harris HA. Design and synthesis of aryl diphenolic azoles as potent and selective estrogen receptor-beta ligands. J Med Chem. 2004;47(21):5021–5040. doi: 10.1021/jm049719y. [DOI] [PubMed] [Google Scholar]
- 26.Miller CP, Collini MD, Harris HA. Constrained phytoestrogens and analogues as ERbeta selective ligands. Bioorg Med Chem Lett. 2003;13(14):2399–2403. doi: 10.1016/s0960-894x(03)00394-9. [DOI] [PubMed] [Google Scholar]
- 27.Yang C, Edsall R, Jr, Harris HA, Zhang X, Manas ES, Mewshaw RE. ERbeta ligands. Part 2: Synthesis and structure-activity relationships of a series of 4-hydroxy-biphenyl-carbaldehyde oxime derivatives. Bioorg Med Chem. 2004;12(10):2553–2570. doi: 10.1016/j.bmc.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 28.Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3(11):935–949. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- 29.Cummings MD, DesJarlais RL, Gibbs AC, Mohan V, Jaeger EP. Comparison of automated docking programs as virtual screening tools. J Med Chem. 2005;48(4):962–976. doi: 10.1021/jm049798d. [DOI] [PubMed] [Google Scholar]
- 30.Kellenberger E, Rodrigo J, Muller P, Rognan D. Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins. 2004;57(2):225–242. doi: 10.1002/prot.20149. [DOI] [PubMed] [Google Scholar]
- 31.Mohan V, Gibbs AC, Cummings MD, Jaeger EP, DesJarlais RL. Docking: successes and challenges. Curr Pharm Des. 2005;11(3):323–333. doi: 10.2174/1381612053382106. [DOI] [PubMed] [Google Scholar]
- 32.Perola E, Walters WP, Charifson PS. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins. 2004;56(2):235–249. doi: 10.1002/prot.20088. [DOI] [PubMed] [Google Scholar]
- 33.Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS. A Critical Assessment of Docking Programs and Scoring Functions. J Med Chem. 2006;49(20):5912–5931. doi: 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- 34.Congreve M, Murray CW, Blundell TL. Structural biology and drug discovery. Drug Discov Today. 2005;10(13):895–907. doi: 10.1016/S1359-6446(05)03484-7. [DOI] [PubMed] [Google Scholar]
- 35.Jorgensen WL. The many roles of computation in drug discovery. Science. 2004;303(5665):1813–1818. doi: 10.1126/science.1096361. [DOI] [PubMed] [Google Scholar]
- 36.Kubinyi H. Success stories of computer-aided design. In: Ekins S, Wang B, editors. Computer Applications in Pharmaceutical Research and Development. Wiley-Interscience; 2006. pp. 377–424. [Google Scholar]
- 37.Alvarez J, Shoichet B. Virtual Screening in Drug Discovery. CRC Press; Boca Raton, FL: 2005. [Google Scholar]
- 38.Hou T, Xu X. Recent development and application of virtual screening in drug discovery: an overview. Curr Pharm Des. 2004;10(9):1011–1033. doi: 10.2174/1381612043452721. [DOI] [PubMed] [Google Scholar]
- 39.Keri G, Orfi L, Eros D, Hegymegi-Barakonyi B, Szantai-Kis C, Horvath Z, Waczek F, Marosfalvi J, Szabadkai I, Pato J, Greff Z, Hafenbradl D, Daub H, Muller G, Klebl B. UA, Signal transduction therapy with rationally designed kinase inhibitors. Current Signal Transduction Therapy. 2006;1:67–95. [Google Scholar]
- 40.Shoichet BK. Virtual screening of chemical libraries. Nature. 2004;432(7019):862–865. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boehm HJ, Boehringer M, Bur D, Gmuender H, Huber W, Klaus W, Kostrewa D, Kuehne H, Luebbers T, Meunier-Keller N, Mueller F. Novel Inhibitors of DNA Gyrase: 3D Structure Based Biased Needle Screening, Hit Validation by Biophysical Methods, and 3D Guided Optimization. A Promising Alternative to Random Screening. J Med Chem. 2000;43(14):2664–2674. doi: 10.1021/jm000017s. [DOI] [PubMed] [Google Scholar]
- 42.Doman TN, McGovern SL, Witherbee BJ, Kasten TP, Kurumbail R, Stallings WC, Connolly DT, Shoichet BK. Molecular Docking and High-Throughput Screening for Novel Inhibitors of Protein Tyrosine Phosphatase-1B. J Med Chem. 2002;45(11):2213–2221. doi: 10.1021/jm010548w. [DOI] [PubMed] [Google Scholar]
- 43.Paiva AM, Vanderwall DE, Blanchard JS, Kozarich JW, Williamson JM, Kelly TM. Inhibitors of dihydrodipicolinate reductase, a key enzyme of the diaminopimelate pathway of Mycobacterium tuberculosis. Biochim Biophys Acta. 2001;1545(1–2):67–77. doi: 10.1016/s0167-4838(00)00262-4. [DOI] [PubMed] [Google Scholar]
- 44.Meagher KL, Lerner MG, Carlson HA. Refining the Multiple Protein Structure Pharmacophore Method: Consistency across Three Independent HIV-1 Protease Models. J Med Chem. 2006 doi: 10.1021/jm050755m. [DOI] [PubMed] [Google Scholar]
- 45.Triballeau N, Acher F, Brabet I, Pin JP, Bertrand HO. Virtual screening workflow development guided by the “receiver operating characteristic” curve approach. Application to high-throughput docking on metabotropic glutamate receptor subtype 4. J Med Chem. 2005;48(7):2534–2547. doi: 10.1021/jm049092j. [DOI] [PubMed] [Google Scholar]
- 46.Singh J, Chuaqui CE, Boriack-Sjodin PA, Lee WC, Pontz T, Corbley MJ, Cheung HK, Arduini RM, Mead JN, Newman MN, Papadatos JL, Bowes S, Josiah S, Ling LE. Successful shape-based virtual screening: the discovery of a potent inhibitor of the type I TGFbeta receptor kinase (TbetaRI) Bioorg Med Chem Lett. 2003;13(24):4355–4359. doi: 10.1016/j.bmcl.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 47.Sawyer JS, Anderson BD, Beight DW, Campbell RM, Jones ML, Herron DK, Lampe JW, McCowan JR, McMillen WT, Mort N, Parsons S, Smith EC, Vieth M, Weir LC, Yan L, Zhang F, Yingling JM. Synthesis and activity of new aryl- and heteroaryl-substituted pyrazole inhibitors of the transforming growth factor-beta type I receptor kinase domain. J Med Chem. 2003;46(19):3953–3956. doi: 10.1021/jm0205705. [DOI] [PubMed] [Google Scholar]
- 48.Becker OM, Dhanoa DS, Marantz Y, Chen D, Shacham S, Cheruku S, Heifetz A, Mohanty P, Fichman M, Sharadendu A, Nudelman R, Kauffman M, Noiman S. An Integrated in Silico 3D Model-Driven Discovery of a Novel, Potent, and Selective Amidosulfonamide 5-HT1A Agonist (PRX-00023) for the Treatment of Anxiety and Depression. J Med Chem. 2006;49(11):3116–3135. doi: 10.1021/jm0508641. [DOI] [PubMed] [Google Scholar]
- 49.Steindl TM, Crump CE, Hayden FG, Langer T. Pharmacophore modeling, docking, and principal component analysis based clustering: combined computer-assisted approaches to identify new inhibitors of the human rhinovirus coat protein. J Med Chem. 2005;48(20):6250–6260. doi: 10.1021/jm050343d. [DOI] [PubMed] [Google Scholar]
- 50.Lipinski C, Hopkins A. Navigating chemical space for biology and medicine. Nature. 2004;432(7019):855–861. doi: 10.1038/nature03193. [DOI] [PubMed] [Google Scholar]
- 51.Lu Y, Nikolovska-Coleska Z, Fang X, Gao W, Shangary S, Qiu S, Qin D, Wang S. Discovery of a Nanomolar Inhibitor of the Human Murine Double Minute 2 (MDM2)-p53 Interaction through an Integrated, Virtual Database Screening Strategy. J Med Chem. 2006 doi: 10.1021/jm060023+. [DOI] [PubMed] [Google Scholar]
- 52.Lyne PD, Kenny PW, Cosgrove DA, Deng C, Zabludoff S, Wendoloski JJ, Ashwell S. Identification of compounds with nanomolar binding affinity for checkpoint kinase-1 using knowledge-based virtual screening. J Med Chem. 2004;47(8):1962–1968. doi: 10.1021/jm030504i. [DOI] [PubMed] [Google Scholar]
- 53.Langer T, Krovat EM. Chemical feature-based pharmacophores and virtual library screening for discovery of new leads. Curr Opin Drug Discov Devel. 2003;6(3):370–376. [PubMed] [Google Scholar]
- 54.Lloyd DG, Golfis G, Knox AJS, Fayne D, Meegan MJ, Oprea TI. Oncology exploration: charting cancer medicinal chemistry space. Drug Discovery Today. 2006;11(3–4):149–159. doi: 10.1016/S1359-6446(05)03688-3. [DOI] [PubMed] [Google Scholar]
- 55.Lu IL, Huang CF, Peng YH, Lin YT, Hsieh HP, Chen CT, Lien TW, Lee HJ, Mahindroo N, Prakash E, Yueh A, Chen HY, Goparaju CMV, Chen X, Liao CC, Chao YS, Hsu JTA, Wu SY. Structure-Based Drug Design of a Novel Family of PPARγ Partial Agonists: Virtual Screening, X-ray Crystallography, and in Vitro/in Vivo Biological Activities. J Med Chem. 2006;49(9):2703–2712. doi: 10.1021/jm051129s. [DOI] [PubMed] [Google Scholar]
- 56.Varady J, Wu X, Fang X, Min J, Hu Z, Levant B, Wang S. Molecular modeling of the three-dimensional structure of dopamine 3 (D3) subtype receptor: discovery of novel and potent D3 ligands through a hybrid pharmacophore- and structure-based database searching approach. J Med Chem. 2003;46(21):4377–4392. doi: 10.1021/jm030085p. [DOI] [PubMed] [Google Scholar]
- 57.Olsen L, Jost S, Adolph HW, Pettersson I, Hemmingsen L, Jorgensen FS. New leads of metallo-[beta]-lactamase inhibitors from structure-based pharmacophore design. Bioorganic & Medicinal Chemistry. 2006;14(8):2627–2635. doi: 10.1016/j.bmc.2005.11.046. [DOI] [PubMed] [Google Scholar]
- 58.Manetti F, Locatelli GA, Maga G, Schenone S, Modugno M, Forli S, Corelli F, Botta M. A Combination of Docking/Dynamics Simulations and Pharmacophoric Modeling To Discover New Dual c-Src/Abl Kinase Inhibitors. J Med Chem. 2006 doi: 10.1021/jm060236z. [DOI] [PubMed] [Google Scholar]
- 59.Dayam R, Aiello F, Deng J, Wu Y, Garofalo A, Chen X, Neamati N. Discovery of Small Molecule Integrin avβ3 Antagonists as Novel Anticancer Agents. J Med Chem. 2006;49(15):4526–4534. doi: 10.1021/jm051296s. [DOI] [PubMed] [Google Scholar]
- 60.Rollinger JJM, Mocka PP, Zidorn CC, Ellmerer EEP, Langer TT, Stuppner HH. Application of the in combo screening approach for the discovery of non-alkaloid acetylcholinesterase inhibitors from Cichorium intybus. Current drug discovery technologies. 2005;2(3):185–193. doi: 10.2174/1570163054866855. [DOI] [PubMed] [Google Scholar]
- 61.Rollinger JM. Strategies for efficient lead structure discovery from natural products. Current medicinal chemistry. 2006;13(13):1491–1507. doi: 10.2174/092986706777442075. [DOI] [PubMed] [Google Scholar]
- 62.Rollinger JM, Bodensieck A, Seger C, Ellmerer EP, Bauer R, Langer T, Stuppner H. Discovering COX-inhibiting constituents of Morus root bark: activity-guided versus computer-aided methods. Planta Med. 2005;71(5):399–405. doi: 10.1055/s-2005-864132. [DOI] [PubMed] [Google Scholar]
- 63.Rollinger JM, Haupt S, Stuppner H, Langer T. Combining ethnopharmacology and virtual screening for lead structure discovery: COX-inhibitors as application example. J Chem Inf Comput Sci. 2004;44(2):480–488. doi: 10.1021/ci030031o. [DOI] [PubMed] [Google Scholar]
- 64.Rollinger JM, Hornick A, Langer T, Stuppner H, Prast H. Acetylcholinesterase inhibitory activity of scopolin and scopoletin discovered by virtual screening of natural products. J Med Chem. 2004;47(25):6248–6254. doi: 10.1021/jm049655r. [DOI] [PubMed] [Google Scholar]
- 65.Klebe G. Virtual ligand screening: strategies, perspectives and limitations. Drug Discovery Today. 2006;11(13–14):580–594. doi: 10.1016/j.drudis.2006.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Perola E. Minimizing false positives in kinase virtual screens. Proteins. 2006;64(2):422–435. doi: 10.1002/prot.21002. [DOI] [PubMed] [Google Scholar]
- 67.Steuber H, Zentgraf M, Gerlach C, Sotriffer CA, Heine A, Klebe G. Expect the unexpected or caveat for drug designers: multiple structure determinations using aldose reductase crystals treated under varying soaking and co-crystallisation conditions. J Mol Biol. 2006;363(1):174–187. doi: 10.1016/j.jmb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 68.Leach AR, Shoichet BK, Peishoff CE. Prediction of Protein-Ligand Interactions. Docking and Scoring: Successes and Gaps. J Med Chem. 2006;49(20):5851–5855. doi: 10.1021/jm060999m. [DOI] [PubMed] [Google Scholar]
- 69.Richards WG. Virtual screening using grid computing: the screensaver project. Nat Rev Drug Discov. 2002;1(7):551–555. doi: 10.1038/nrd841. [DOI] [PubMed] [Google Scholar]
- 70.Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3(8):711–715. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 71.Simon-Hettich B, Rothfuss A, Steger-Hartmann T. Use of computer-assisted prediction of toxic effects of chemical substances. Toxicology. 2006;224(1–2):156–162. doi: 10.1016/j.tox.2006.04.032. [DOI] [PubMed] [Google Scholar]
- 72.Crum Brown A, Frazer T. On the connection between chemical constitution and physiological action. Part I. On the physiological action of the salts of the ammonia bases derived from Strycnia, Brucia, Thebaia, Codeia, Morphia, and Nicotinia. Trans Royal Soc Edinburgh. 1868;25:151–203. [PMC free article] [PubMed] [Google Scholar]
- 73.Hansch C, Fujita T. Rho-sigma-pi analysis. A method for the correlation of biological activity and chemical structure. J Am Chem Soc. 1964;86:1616–1626. [Google Scholar]
- 74.Hansch C, Steward AR. The Use of Substituent Constants in the Analysis of the Structure--Activity Relationship in Penicillin Derivatives. J Med Chem. 1964;44:691–694. doi: 10.1021/jm00336a001. [DOI] [PubMed] [Google Scholar]
- 75.Tunkel J, Mayo K, Austin C, Hickerson A, Howard P. Practical considerations on the use of predictive models for regulatory purposes. Environ Sci Technol. 2005;39(7):2188–2199. doi: 10.1021/es049220t. [DOI] [PubMed] [Google Scholar]
- 76.Contrera JF, Matthews EJ, Kruhlak NL, Benz RD. Development and application of computational toxicology and informatics resources at the FDA CDER Office of Pharmaceutical Sciences. 2004 http://www.fda.gov/ohrms/dockets/ac/04/slides/2004-4078S1_06_Contrera.ppt#468,10,Slide.
- 77.Contrera JF, Maclaughlin P, Hall LH, Kier LB. QSAR modeling of carcinogenic risk using discriminant analysis and topological molecular descriptors. Curr Drug Discov Technol. 2005;2(2):55–67. doi: 10.2174/1570163054064684. [DOI] [PubMed] [Google Scholar]
- 78.Matthews EJ, Kruhlak NL, Benz RD, Contrera JF. Assessment of the health effects of chemicals in humans: I. QSAR estimation of the maximum recommended therapeutic dose (MRTD) and no effect level (NOEL) of organic chemicals based on clinical trial data. Current drug discovery technologies. 2004;1(1):61–76. doi: 10.2174/1570163043484789. [DOI] [PubMed] [Google Scholar]
- 79.Contrera JF, Matthews EJ, Kruhlak NL, Benz RD. Estimating the safe starting dose in phase I clinical trials and no observed effect level based on QSAR modeling of the human maximum recommended daily dose. Regul Toxicol Pharmacol. 2004;40(3):185–206. doi: 10.1016/j.yrtph.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 80.Geldenhuys WJ, Gaasch KE, Watson M, Allen DD, Van der Schyf CJ. Optimizing the use of open-source software applications in drug discovery. Drug Discovery Today. 2006;11(3–4):127–132. doi: 10.1016/S1359-6446(05)03692-5. [DOI] [PubMed] [Google Scholar]
- 81.Baurin N, Aboul-Ela F, Barril X, Davis B, Drysdale M, Dymock B, Finch H, Fromont C, Richardson C, Simmonite H, Hubbard RE. Design and characterization of libraries of molecular fragments for use in NMR screening against protein targets. J Chem Inf Comput Sci. 2004;44(6):2157–2166. doi: 10.1021/ci049806z. [DOI] [PubMed] [Google Scholar]
- 82.Rees DC, Congreve M, Murray CW, Carr R. Fragment-based lead discovery. Nat Rev Drug Discov. 2004;3(8):660–672. doi: 10.1038/nrd1467. [DOI] [PubMed] [Google Scholar]
- 83.Schneider G, Fechner U. Computer-based de novo design of drug-like molecules. Nat Rev Drug Discov. 2005;4(8):649–663. doi: 10.1038/nrd1799. [DOI] [PubMed] [Google Scholar]
- 84.Rajasethupathy P, Vayttaden SJ, Bhalla US. Systems modeling: a pathway to drug discovery. Curr Opin Chem Biol. 2005;9(4):400–406. doi: 10.1016/j.cbpa.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 85.Butcher EC. Can cell systems biology rescue drug discovery? Nat Rev Drug Discov. 2005;4(6):461–467. doi: 10.1038/nrd1754. [DOI] [PubMed] [Google Scholar]
- 86.Butcher EC, Berg EL, Kunkel EJ. Systems biology in drug discovery. Nat Biotechnol. 2004;22(10):1253–1259. doi: 10.1038/nbt1017. [DOI] [PubMed] [Google Scholar]
- 87.Hood L, Perlmutter RM. The impact of systems approaches on biological problems in drug discovery. Nat Biotechnol. 2004;22(10):1215–1217. doi: 10.1038/nbt1004-1215. [DOI] [PubMed] [Google Scholar]
- 88.Bugrim A, Nikolskaya T, Nikolsky Y. Early prediction of drug metabolism and toxicity: systems biology approach and modeling. Drug Discov Today. 2004;9(3):127–135. doi: 10.1016/S1359-6446(03)02971-4. [DOI] [PubMed] [Google Scholar]
- 89.Ekins S. Systems-ADME/Tox: resources and network approaches. J Pharmacol Toxicol Methods. 2006;53(1):38–66. doi: 10.1016/j.vascn.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 90.Michelson S. Assessing the impact of predictive biosimulation on drug discovery and development. J Bioinform Comput Biol. 2003;1(1):169–177. doi: 10.1142/s0219720003000022. [DOI] [PubMed] [Google Scholar]
- 91.Michelson S. The impact of systems biology and biosimulation on drug discovery and development. Mol Biosyst. 2006;2(6–7):288–291. doi: 10.1039/b602194h. [DOI] [PubMed] [Google Scholar]
- 92.Davidov E, Holland J, Marple E, Naylor S. Advancing drug discovery through systems biology. Drug Discov Today. 2003;8(4):175–183. doi: 10.1016/s1359-6446(03)02600-x. [DOI] [PubMed] [Google Scholar]
- 93.Cho CR, Labow M, Reinhardt M, van Oostrum J, Peitsch MC. The application of systems biology to drug discovery. Curr Opin Chem Biol. 2006;10(4):294–302. doi: 10.1016/j.cbpa.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 94.Kubinyi H. Drug research: myths, hype and reality. Nature Reviews Drug Discovery. 2003;2(8):665–668. doi: 10.1038/nrd1156. [DOI] [PubMed] [Google Scholar]