

Abstract
In response to oxidative stress, the nuclear factor E2-related factor 2 (Nrf2) transcription factor translocates from the cytoplasm into the nucleus and transactivates expression of genes with antioxidant activity. Despite this cellular mechanism, oxidative damage is abundant in Alzheimer and Parkinson disease (AD and PD). To investigate mechanisms by which Nrf2 activity may be aberrant or insufficient in neurodegenerative conditions, we assessed Nrf2 localization in affected brain regions of AD, Lewy body variant of AD (LBVAD), and PD. By immunohistochemistry, Nrf2 is expressed in both the nucleus and the cytoplasm of neurons in normal hippocampi with predominant expression in the nucleus. In AD and LBVAD, Nrf2 was predominantly cytoplasmic in hippocampal neurons and was not a major component of beta amyloid plaques or neurofibrillary tangles. By immunoblotting, we observed a significant decrease in nuclear Nrf2 levels in AD cases. In contrast, Nrf2 was strongly nuclear in PD nigral neurons but cytoplasmic in substantia nigra of normal, AD, and LBVAD cases. These findings suggest that Nrf2-mediated transcription is not induced in neurons in AD despite the presence of oxidative stress. In PD, nuclear localization of Nrf2 is strongly induced, but this response may be insufficient to protect neurons from degeneration.
Keywords: Antioxidant, Brain, Neurodegeneration, Neuron, Oxidative stress, Transcription
INTRODUCTION
Although Alzheimer disease (AD) and Parkinson disease (PD) have distinct pathologic features, there is considerable evidence to support oxidative stress as a common pathogenetic mechanism in both disorders. Evidence of lipid peroxidation, protein nitration and nucleic acid oxidation is abundant in affected brain regions of both AD and PD (1–6). Oxidative damage occurs early in disease (5, 7), suggesting that oxidative stress plays a role in disease progression. Increased antioxidant activity confers protection in mouse and culture models (8, 9) and has been reported to lower the risk of AD (10, 11). However, how disease mechanisms affect endogenous antioxidant defenses is still not completely understood.
Reducing cellular oxidative stress occurs through an endogenous mechanism regulated at the transcriptional level. Genes whose products participate in reducing oxidative stress, inflammation, and accumulation of toxic metabolites contain a common promoter element called the antioxidant response element (ARE) or electrophile response element. ARE-containing gene promoters include glutathione-S-transferase (GST), coenzyme Q10 (Q10), NAD(P)H:quinone oxidoreductase (QR), and superoxide dismutase 1. The ARE promoter element is bound by several transcription factors; however, the nuclear factor E2-related factor 2 (Nrf2) is responsible for activating transcription in response to oxidative stress (12). Nrf2 transcriptional activity is known to be regulated by several mechanisms, including protein interaction, protein stability, nuclear cytoplasmic shuttling, and phosphorylation (13–28). Several reports suggest that interaction with the kelch-like ECH-associated protein (Keap1) sequesters Nrf2 in the cytoplasm, leading to ubiquitination and subsequent degradation by the proteasome (13–15). Either oxidation of sulfhydryl groups on specific cysteines in Keap1 (29) or phosphorylation of Keap1 and/or Nrf2 induces Keap1 to release Nrf2 (19–28). Free of Keap1, Nrf2 is stabilized and translocates from the cytoplasm to the nucleus through a bipartite nuclear localization signal where it transactivates expression of detoxification enzymes, antioxidant enzymes, reducing molecules, and Nrf2 itself (13, 14, 16, 30–33). These gene products go on to protect the cell from oxidative damage. Nrf2 also contains a nuclear export sequence near its nuclear localization signal, presumably to remove Nrf2 from the nucleus when the antioxidant response is no longer needed (16–18).
In primary murine cortical cultures, neurons lacking Nrf2 are more susceptible to oxidative stress through H2O2 and nonexcitotoxic glutamate (34) and are rescued by overexpression of Nrf2 (30). Overexpression of Nrf2 can rescue neurons from mitochondrial complex II inhibition and ischemic insult in animal models of Huntington disease and stroke, respectively (35, 36). Thus, neurons and astrocytes depend on Nrf2 activation of ARE-containing genes for protection from oxidative death.
In AD, expression of 2 Nrf2 target genes, GST and QR, exhibit altered activity and expression, respectively, in AD brain (37, 38). Whereas the increase in QR protein levels suggests activation of the antioxidant response, the reduction in GST activity suggests that the response is incomplete, aberrant, and/or insufficient. In PD, 2 other Nrf2 targets, GSH and Q10, display reduced levels suggesting a loss of Nrf2 response (39, 40). Providing GSH and Q10 exogenously lessens neuronal loss in animal and culture models of PD (41–43).
Given the extensive oxidative damage in AD and PD, we would expect an upregulation of Nrf2 activity in the nuclei of neurons and astrocytes during disease progression; however, in AD and PD, levels of some ARE-containing gene products are reduced, suggesting disruption of the pathway. Because subcellular trafficking is critical to activity of this pathway, we examined expression and localization of Nrf2 in susceptible neuron populations in AD and PD brain tissues. Nrf2 expression patterns and protein levels in AD and PD suggest aberrant regulation of Nrf2 in AD, whereas disrupted or insufficient ARE responses likely occur downstream of Nrf2 nuclear localization in PD.
MATERIALS AND METHODS
Human Subjects
Autopsy brain tissues were obtained from patients with PD, AD, and age-matched controls from the Alzheimer Disease Core Center tissue bank at the University of Pennsylvania and the Alzheimer Disease Research Center at the University of Pittsburgh Medical Center. Both AD and PD were defined by clinical history, neurologic examination, and neuropathologic assessment of Braak neurofibrillary tangle (NFT) stage (44) for all cases and the Braak PD stage for PD cases only (45), because this staging criteria was developed for nondemented cases (Table 1). Using these criteria, 12 cases were diagnosed as AD, 5 as normal age-matched controls, 5 as PD, and 3 as Lewy body variant of AD (LBVAD). All cases have postmortem intervals of less than 12 hours before autopsy with a single exception. Age characteristics are shown in Table 1. From these cases, paraffin-embedded, formalin-fixed tissue sections from the hippocampus and substantia nigra were used. In the hippocampus, special attention was paid to the CA1 region, because these neurons are particularly vulnerable to injury. Frozen tissue was obtained from the Alzheimer's Disease Core Center at the University of Pennsylvania. Midfrontal cortex was dissected from fresh-frozen tissue of 9 AD cases and 9 control cases with similar characteristics as our fixed tissue cohort. Average ages between AD and control cases were not significantly different (79.1 ± 10.6 and 79.9 ± 9.6, respectively). Postmortem interval for AD cases ranged from 5 to 12 hours, whereas control cases ranged from 4 to 24 hours. As for the paraffin-embedded, formalin-fixed tissue, AD cases were Braak stages 4, 5, or 6 and control cases were Braak stage 1.
TABLE 1.
Neuropathologic and Clinical Case Information
| Diagnosis | Average Age | Age Range | PMI Average | PMI Range | Braak Stage |
|---|---|---|---|---|---|
| Normal | 78.6 ± 8.5 | 70–92 | 12.45 | 6–24 | 1 |
| Alzheimer disease | 77.9 ± 9.7 | 56–89 | 8.5 | 5.5–12 | 4–6 |
| Lewy body variant of Alzheimer disease | 82 ± 5.6 | 76–87 | 5.2 | 2.5–10 | 4 |
| Parkinson disease | 79 ± 7.6 | 67–87 | 7.3 | 4–8 | 1–2 |
Clinical data for patient groups used for this study. Average age in years with standard deviation and range are shown. Postmortem interval (PMI) is represented in hours. Braak stage diagnostics are given for each group.
Immunohistochemistry and Immunofluorescent Confocal Microscopy
Paraffin-embedded sections were heated to 50°C overnight and deparaffinized in Histoclear (3 × 15 minutes; National Diagnostics, Atlanta, GA). Sections were rehydrated as follows: 100% alcohol twice for 10 minutes, 95% alcohol for 10 minutes, 90% alcohol for 10 minutes, 70% alcohol for 10 minutes, and water for 5 minutes. Endogenous peroxidase activity was inactivated by immersing in 3% hydrogen peroxide for 30 minutes. Antigen unmasking was performed by placing slides in target retrieval solution (DAKO, Carpinteria, CA) at 95°C for 1 hour. After gradual cooling to room temperature, tissue sections were blocked with 10% normal goat serum (Chemicon International, Temecula, CA) in phosphate-buffered saline (PBS). Antibodies for Nrf2 (Table 2) were characterized for immunohistochemistry and used at empirically determined concentrations using a dilution series with tyramide amplification (NEN-Perkin Elmer, Wellesley, MA). Similar results were seen with both Nrf2 antibodies indicated. Immunostaining was visualized by Vector Red (Vector Laboratories, Inc., Burlingame, CA). Slides were counterstained with Gill's hematoxylin (Vector Laboratories, Inc.) for 30 seconds, dehydrated, and mounted in Permount (Fisher, Fair Lawn, NJ).
TABLE 2.
Antibody Information
| Antigen | Clone or Catalog | Dilution | TSA | Species | Application | Manufacturer |
|---|---|---|---|---|---|---|
| Nrf2 | H300 | 1:200 | Yes | Rb | IHC, IF | Santa Cruz Biotechnology, Santa Cruz, CA |
| Nrf2 | C20 | 1:2,000 | Yes | Rb | IHC, IF | Santa Cruz Biotechnology |
| Nrf2 | KAP-TF125 | 1:200 | Yes | Rb | IHC | Stressgen, Victoria, BC, Canada |
| Nrf2 | H300 | 1:1000 | No | Rb | IB | Santa Cruz Biotechnology |
| GFAP | Z0334 | 1:80 | No | Rb | IF | DAKO, Carpinteria, CA |
| MAP2 | SMI-52 | 1:500 | No | Ms | IF | Covance, Berkeley, CA |
| HNE | HNE11-S | 1:50 | No | Rb | IF | Alpha Diagnostics International, San Antonio, TX |
| GAPDH | MAB374 | 1:100 | No | Ms | IB | Chemicon International, Temecula, CA |
| APP, amyloid fragment | 4G8 | 1:20,000 | No | Ms | IF | Gift from Drs. Lee and Trojanowski, University of Pennsylvania |
| NFT | AT8 | 1:300 | No | Ms | IF | Pierce, Rockford, IL, |
Summary of antibodies used in this study are shown. If antibody staining was amplified with tyramide amplification system (TSA) for immunofluorescent or immunostaining, it is indicated (yes). The species of animal in which the primary antibody was raised is indicated (species; mouse—Ms, rabbit—Rb). Applications using antibodies include immunohistochemistry (IHC), immunofluorescence (IF), and immunoblotting (IB).
Nrf2, nuclear factor E2-related factor 2; GFAP, glial fibrillary acid protein; MAP2, microtubule associate protein 2; HNE, hydroxylnonenal; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; APP, amyloid precursor protein; NFT, neurofibrillary tangle.
For immunofluorescent staining, the Nrf2 was detected as previously stated except the tyramide was directly conjugated to fluorescent isothiocyanate conjugated. At the concentration of Nrf2 antibody used, no Nrf2 staining can be detected with a secondary directly conjugated to a fluor; therefore, a second label with a polyclonal antibody will not crossreact with Nrf2 antibody complexes. This is similar to methods we have described previously (46). Nucleic acids were labeled with DAPI (10 μM for 30 minutes; Invitrogen; Molecular Probes, Carlsbad, CA) or propidium iodide (PI, 10 μg/mL for 30 minutes; Sigma, St. Louis, MO) after RNase A treatment (10 μg/mL for 30 minutes) to remove RNA. The following antibodies were each used in combination with Nrf2 in double-label immunofluorescent microscopy (see Table 2 for experimental details and commercial source): MAP2 antibody to detect neurons; glial fibrillary acid protein antibody to detect astrocytes; beta amyloid antibody to determine amyloid plaques (4G8; gift from Dr. Virginia M.Y. Lee, University of Pennsylvania); AT8 to detect tau; and hydroxylnonenal (HNE) as a marker of oxidative damage. These antibodies were detected using appropriate secondary antibodies (Table 1, goat anti-mouse or goat anti rabbit; Jackson Immunologicals, West Grove, PA) conjugated to Cy5 at 1:200. Slides were mounted in gelvetol and analyzed by immunofluorescent confocal microscopy.
Confocal microscopy was performed on a Bio-Rad Radiance 2100 (Bio-Rad, Hercules, CA) with the 488-nm line of an argon laser to excite fluorescein isothiocyanate conjugated (FITC), a red diode laser (637 nm) to excite Cy5, and a blue diode laser (405 nm) to excite DAPI. The following filters were used for capture respective fluor emissions (FITC—515 df30; Cy5—650 LP; DAPI—476 df 48). Each fluor was excited and captured sequentially to eliminate bleed through. Furthermore, FITC and Cy5 fluors were chosen because there is minimal overlap from the emission spectra.
Protein Extraction and Immunoblotting
Protein extracts were prepared from the midfrontal cortex of 9 frozen normal human brains and 9 brains from patients with a diagnosis of definite AD. Tissues were homogenized on ice with 4 strokes in a manual dounce homogenizer in 2 tissue volumes of Buffer A (10 mM Hepes pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, and 1:1000 protease inhibitor cocktail [Sigma-Aldrich, St. Louis, MO]) and spun at 3000 rpm for 5 minutes at 4°C. The supernatant was removed and the pellet was resuspended in 4 pellet volumes of NP40 lysis buffer (0.1% NP–40, 10 mM Tris [pH 8.0], 10 mM MgCl2, 15 mM NaCl) with 1:1000 protease inhibitor cocktail (Sigma-Aldrich) and incubated on ice for 15 minutes. The tissue pellet was then homogenized on ice with 30 manual strokes in the dounce homogenizer and centrifuged for 30 minutes at 10,000 rpm at 4°C. The supernatant was collected and labeled as the “cytoplasmic” fraction for immunoblotting. The pellet was resuspended in 2 pellet volumes of buffer B (20 mM Hepes pH 7.9, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 25% glycerol, and 1:1000 protease inhibitor cocktail [Sigma-Aldrich]), incubated on ice for 15 minutes, and centrifuged for 30 minutes at 10,000 rpm at 4°C. The supernatant (the “nuclear” fraction) was collected and used in immunoblots. Cytoplasmic extraction was verified by the presence of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Nuclear fractions lacked appreciable GAPDH by immunoblot.
Before immunoblotting, protein concentrations were determined in the nuclear and cytoplasmic fraction using the Bio-Rad protein assay (Bio-Rad). Equal amounts of nuclear extracts or cytoplasmic extracts from 9 AD and 9 normal midfrontal cortices were electrophoresed through a discontinuous SDS-polyacrylamide gel (8% resolving gel) with prestained protein standards (Bio-Rad) and 10 μg of protein extracts from NIH3T3 cells transfected to overexpress Nrf2 as a control.
The proteins were transferred from the 8% SDS-polyacrylamide gel to Immun-Blot PVDF membrane (Bio-Rad) and blocked in 2% bovine serum albumin in PBST (PBS + 0.1% Tween 20). Details of the antibodies used are contained in Table 2. All antibodies were used at 1:1000 in PBST overnight at 4°C for 10 minutes. Goat anti-rabbit HRP (for Nrf2) (1:10,000; Southern Biotechnologies, Inc., Birmingham, AL) was used to detect the appropriate primary antibodies. The secondary antibody was washed in PBST 4 times for 10 minutes. The antibody was then visualized using enhanced chemiluminescence (Amersham Biosciences, Buckinghamshire, UK). Enhanced chemiluminescence was captured on the Kodak Image Station 440 CF and by autoradiography. Blots were stained with Colloidal Gold Total Protein Stain (Bio-Rad) to verify equal loading and protein integrity.
Quantification of appropriate bands on Western images was done using the Kodak 1D 3.5 software (Rochester, NY) on scanned autoradiographs. All defined regions of interest were the same size. Nrf2 intensity for each case was divided by the Nrf2 intensity of AD1 for each blot to view relative Nrf2 levels. To normalize Nrf2 levels, Nrf2 intensity was divided by GAPDH intensity (which also serves as a cytoplasmic marker) for cytoplasmic blots and the intensity of a band near Nrf2 in size stained by colloidal gold for cytoplasmic and nuclear blots. Average values of Nrf2 intensity normalized to colloidal gold loading control and standard deviation for AD cases or normal cases were determined.
Quantification of Nuclear and Cytoplasmic Nuclear Factor E2-Related Factor 2 Staining
Quantification of nuclear and cytoplasmic Nrf2 staining was determined from 3 contiguous 87.5 μm × 87.5 μm fields of CA1 hippocampus for each case in AD, PD, LBVAD, and control. Presence of Nrf2 in nuclear and cytoplasmic compartments was scored by an individual blind to the disease state. The total number of neurons inspected (ranging from 20–100 per case) was used to calculate the percentage of neurons exhibiting staining in either compartment. An average and standard deviation was determined for each category (AD, PD, LBVAD, and control).
Statistical Analysis
Significant differences between disease states and controls for quantification of the percentage of nuclear and cytoplasmic Nrf2 were determined using a one-way analysis of variance with Student-Newman-Keuls using Graphpad Prism 3.02 for Windows (San Diego, CA). To determine if our observed changes in Nrf2 compartment staining were significant for disease sets, we used a Fisher exact text using SPSS Version 11.5 (SPSS, Inc., Chicago, IL). Quantification of western images was performed using digital images captured on the Kodak Image Station 440 CF using the Kodak 1D 3.5 software. Two independent Western blots were quantified. The average and standard deviations for each data set is shown. A 2-tailed Student t-test (Microsoft Office Excel 2003) was used to determine significant differences between Nrf2 levels in nuclear and cytoplasmic fractions from AD and normal tissue extracts.
RESULTS
Nuclear Factor E2-Related Factor 2 Expression Patterns Are Altered in Hippocampi of Alzheimer Disease and Lewy Body Variant of Alzheimer Disease
The Nrf2 transcription factor controls expression of the genes necessary to reduce oxidative stress (30). Because evidence of increased oxidative damage is observed in AD, PD, and LBVAD (2, 5–7), we sought to determine if Nrf2 was localized appropriately for activation of the endogenous antioxidant response in affected brain regions of these diseases. Furthermore, because LBVAD has pathologic features of both AD and PD, we were interested in determining whether it more closely resembled AD or PD with regard to Nrf2 localization. In AD, damage initiates in the entorhinal cortex and hippocampus but progresses to the temporal and frontal cortex resulting in memory deficits and cognitive decline (47). In PD, a preponderance of damage occurs in the substantia nigra resulting in loss of neuromelanin-positive dopaminergic neurons. Therefore, we compared Nrf2 localization by immunohistochemistry in hippocampi and substantia nigra neurons from 12 AD, 3 LBVAD, 5 PD, and 5 age-matched control cases (Tables 1, 3). In normal age-matched control hippocampi, we observed Nrf2 staining in both the nucleus and cytoplasm of cells with the morphology of neurons and astrocytes (Fig. 1A, column 1, normal). In contrast, Nrf2 immunostaining was predominantly cytoplasmic in hippocampal neurons from individuals with AD and LBVAD (Fig. 1A, columns 2 and 3). Consistent with minimal pathologic findings in hippocampi of patients with PD, Nrf2 staining in patients with PD more closely resembled age-matched controls, although occasional neurons exhibited exclusively cytoplasmic Nrf2 staining similar to AD (Fig. 1A, column 4). In the 5 normal cases, 99% of neurons in the CA1 region of the hippocampus had nuclear staining for Nrf2 (Fig. 1C). However, only 12.5% or 1.0% of neurons in CA1 region of the hippocampus exhibited nuclear Nrf2 staining in pure AD cases or LBVAD cases, respectively (Fig. 1C). Interestingly, there is statistically less nuclear Nrf2 in hippocampal neurons of PD cases as compared with normal age-matched controls (86.7%, p < 0.001, Fig. 1C). In normal, AD, LBVAD, and PD cases, a similar percentage of neurons contained Nrf2 staining in the cytoplasm (Fig. 1C). Therefore, in AD and LBVAD, we see a dramatic and significant reduction in nuclear Nrf2 in hippocampal neurons, which is different from the expected localization in neurons responding to oxidative stress.
TABLE 3.
The Number of Cases Exhibiting Nuclear or Cytoplasmic Nuclear Factor E2-Related Factor 2 Staining in Neurons of Parkinson Disease, Two Diagnostic Categories of Alzheimer Disease, or Age-Matched Controls
| Hippocampus
|
Substantia Nigra
|
|||
|---|---|---|---|---|
| Diagnosis | Nuclear | Cytoplasmic | Nuclear | Cytoplasmic |
| Control | 5/5 | 5/5 | 0/5 | 5/5 |
| AD | 1/12* | 12/12 | 0/8 | 8/8 |
| LBVAD | 0/3† | 3/3 | 0/3 | 3/3 |
| PD | 5/5 | 5/5 | 5/5* | 5/5 |
The fraction of cases exhibiting nuclear or cytoplasmic nuclear factor e2-related factor 2 staining in hippocampus and substantia nigra among 3 diagnostic groups: age-matched control (control), Alzheimer disease (AD), Lewy body variant of Alzheimer disease (LBVAD), and Parkinson disease (PD).
Indicates significantly different from control (p < 0.01) using the Fisher exact test.
Indicates significantly different from control (p < 0.05).
FIGURE 1.
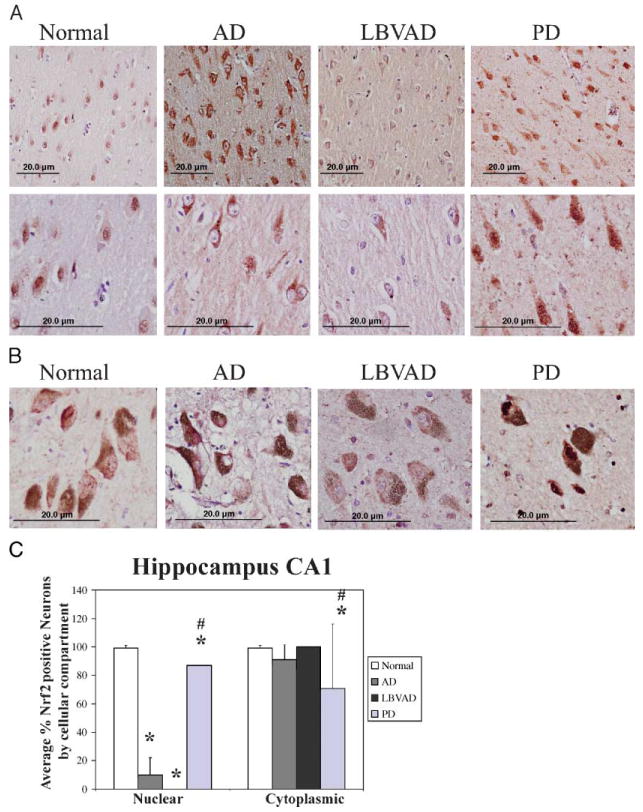
Nrf2 expression patterns are altered in hippocampal neurons of Alzheimer disease and Lewy body variant of Alzheimer disease and neurons of the substantia nigra in Parkinson disease. (A) Immunohistochemistry for nuclear factor e2-related factor (Nrf2) 2 (red) is shown from the CA1 region of hippocampus from control (column 1), Alzheimer disease (column 2), Lewy body variant of Alzheimer disease (column 3), and Parkinson disease (column 4). Similar Nrf2 localization was observed in CA2, CA3, and CA4 of the hippocampus (data not shown). These slides stained with the C20-Nrf2 antibody are representative of our findings with the H300 antibody and KAP-125 antibody (Table 2). Nuclei are counterstained with hematoxylin (purple). Row 1 shows images captured with a 20× objective and row 2 shows an image captured with a 40× objective. Scale bar = 20 μm. Images in row 1 and row 2 are from 2 different cases for each disease group. (B) In control substantia nigra (column 1), Alzheimer disease (column 2), and Lewy body variant of Alzheimer disease (column 3), Nrf2 (red) was weakly cytoplasmic in dopaminergic neurons. In Parkinson disease, Nrf2 immunostaining (red) was stronger and exhibited more nuclear staining in remaining dopaminergic neurons. All images also show counterstaining with hematoxylin (purple). Scale bar = 20 μm. Original magnification: 40× objective was used. (C) The percentage of cells with nuclear Nrf2 (left) and cytoplasmic Nrf2 (right) were counted in the CA1 region of the hippocampus of 12 Alzheimer disease cases, 3 Lewy body variant of Alzheimer disease cases, 5 Parkinson disease cases, and 5 control cases. Shown are the average percentages of Nrf2-positive nuclei (left) and Nrf2-positive cytoplasm (right). Error bars indicate standard deviation. *, Significantly different from control (p < 0.001). #, Significantly different from AD (p < 0.001) as determined by one-way analysis of variance, Student-Newman-Keuls.
Nuclear Factor E2-Related Factor Exhibits Increased Nuclear Staining in Substantia Nigra Neurons From Patients With Parkinson Disease
In PD, the neuronal population of the substantia nigra is selectively lost during disease progression. Loss of dopaminergic neurons manifests as movement abnormalities such as slow voluntary movements, shuffling gait, rigidity, and stooped posture (48). It is also the substantia nigra that prominently accumulates oxidative damage (49). Therefore, we assessed Nrf2 expression patterns by immunohistochemistry in the substantia nigra from 5 PD cases, 8 AD, 3 LBVAD, and 5 normal age-matched control cases to determine if Nrf2 is translocating to the nucleus in PD. In substantia nigra of all 5 control cases, 8 AD cases, and 3 LBVAD cases, weak Nrf2 immunostaining was observed primarily in the cytoplasm (Table 2; Fig. 1B, control, AD, LBVAD, columns 1–3). Neuromelanin-containing neurons (brown), however, exhibited strong nuclear Nrf2 staining and cytoplasmic staining in substantia nigra of all 5 PD cases (Fig. 1C, PD, column 4). Thus, changes in Nrf2 localization in affected brain regions of PD are consistent with activation of the endogenous antioxidant response in neurons, whereas nigral neurons in AD and LBVAD resemble control neurons. These findings suggest that deficits in Nrf2 target gene expression observed in the PD literature (39, 40) either occur downstream of Nrf2 nuclear translocation or may simply reflect nigral neuron loss.
Altered Expression Patterns of Nuclear Factor E2-Related Factor Are Observed in Neurons and Astrocytes of Alzheimer Disease Hippocampus
Using triple label laser confocal microscopy, we assessed localization of Nrf2 staining in specific cell types within the hippocampus from normal and AD cases. In normal cases, the most intense Nrf2 staining (green) was observed in the nucleus (blue) of hippocampal neurons MAP2—red; Fig. 2A, row 1). This confirms our immunohistochemical observations that Nrf2 is seen in neurons. In AD cases, we observed cytoplasmic Nrf2 staining (green) in neurons (red) of the CA1 region of the hippocampus (Fig. 2B, row 2). Nrf2 staining (green) overlapped considerably with immunostaining for the neuronal marker MAP2 (red) as shown in Figure 2B, whereas Nrf2 staining did not overlap with a nuclear stain (PI, blue; Fig. 2B). Astrocytes labeled with glial fibrillary acidic protein (GFAP, red) exhibited minimal Nrf2 staining, but when observable, it was predominantly nuclear (green) in the CA1 region of AD hippocampus (Fig. 2C, row 3). These findings demonstrate that a majority of hippocampal neurons lack nuclear Nrf2 staining in AD, whereas nuclear Nrf2 staining is predominant in normal hippocampal neuronal nuclei.
FIGURE 2.
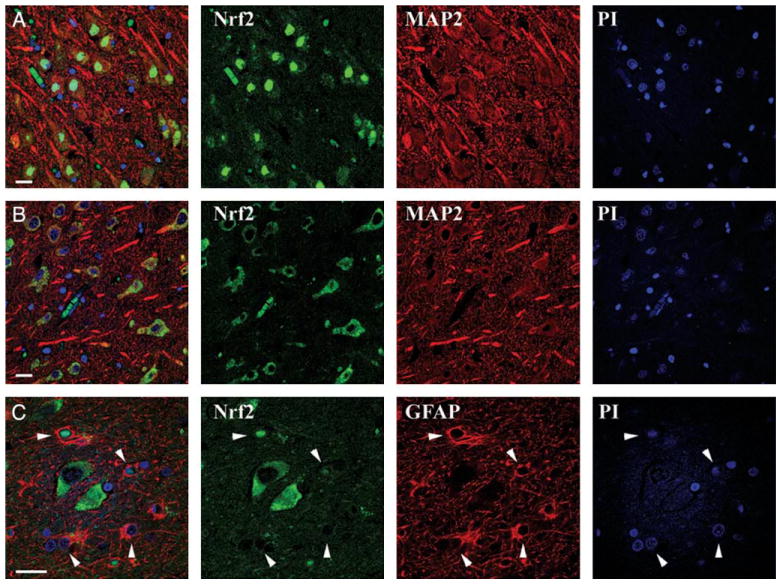
Altered expression patterns of nuclear factor e2-related factor (Nrf2) are observed in neurons and astrocytes of Alzheimer disease hippocampus. (A) Triple label immunofluorescent confocal microscopy revealed Nrf2 (green) is predominantly expressed in the nucleus (blue) of neurons (MAP2, red) present in the CA1 region of the control hippocampus. (B) In contrast, Nrf2 (green) is predominantly cytoplasmic in neurons (MAP2, red) in the CA1 region of the Alzheimer disease hippocampus. (C) In the CA1 region of the Alzheimer disease hippocampus, Nrf2 (green) does not stain astrocyte cytoplasm intensely (glial fibrillary acidic protein, red) but is occasionally observed in the nuclei (blue, arrows). Merged images are shown in the first column. Scale bar = 20 μm.
Altered Nuclear Factor E2-Related Factor Localization Occurs in Proximity to Pathologic Features of Alzheimer Disease Hippocampus, But Is Not a Major Component of Beta Amyloid Plaques or Neurofibrillary Tangles
To determine if Nrf2 staining occurred in association with pathologic features of AD, we used triple label confocal microscopy for Nrf2 (green), nuclei (blue), and either beta amyloid plaques (Aβ, red) or NFTs (AT8, red) in AD hippocampal tissue. Nrf2 immunostaining was observed in proximity to beta amyloid plaques (Fig. 3A); however, the majority of Nrf2 was seen in the cytoplasm of neurons in the absence of plaque structures. Similarly, we did not observe colocalization between Nrf2 staining (green) and tau containing NFTs using the AT8 antibody (red; Fig. 3B, C). When present in AT8-positive neurons of the hippocampus (red), Nrf2 was still cytoplasmic but did not colocalize with tau (Fig. 3B, C, arrows). A subset of AT8-positive neurons contained little or no observable Nrf2 (arrowheads). However, neurons without NFTs (AT8-negative) exhibited predominantly cytoplasmic Nrf2 (green; asterisks). These findings indicate that Nrf2 does not colocalize with beta amyloid or tau, the major components of the hallmark pathologic features of AD. However, the altered localization of Nrf2 is observed in neurons in the vicinity of Aβ plaques and neurofibrillary tangles.
FIGURE 3.
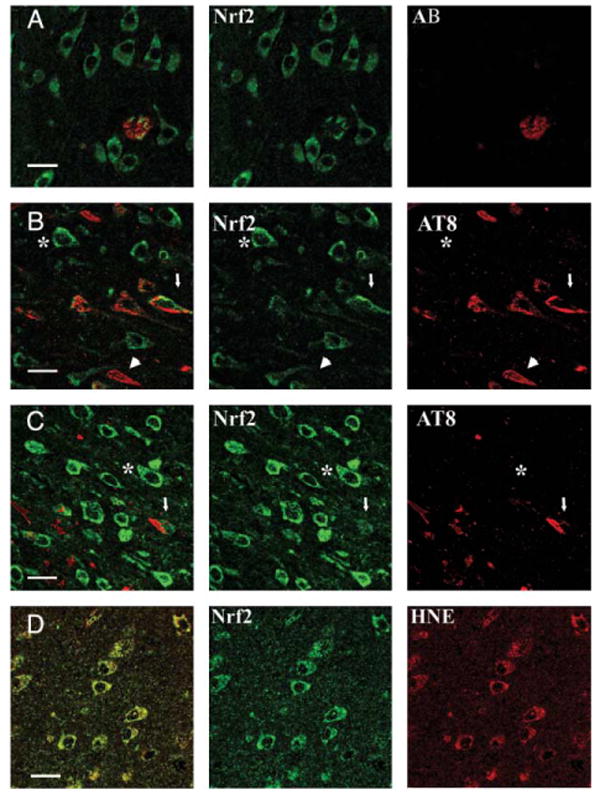
Altered nuclear factor e2-related factor (Nrf2) localization occurs in proximity to pathologic features of Alzheimer disease but does not colocalize with beta amyloid plaques or tau. (A) In CA1 of the hippocampus of Alzheimer disease cases, Nrf2 (green) was observed within a beta amyloid plaque (AB-red), however, did not significantly colocalize with beta amyloid (colocalization appears yellow in the merged field). Instead, the majority of Nrf2 was cytoplasmic in neurons adjacent and distal to the beta amyloid plaque. (B, C) Tau as labeled by AT8 (red) was present in a subset of cells staining for Nrf2 (green) in CA1 of AD hippocampi, but tau and Nrf2 did not colocalize in these neurons (arrows). When observed in AT8-positive neurons, Nrf2 (green) staining was predominantly cytoplasmic (arrow). A subset of AT8-positive neurons contained little or no observable Nrf2 (arrowhead). Neurons without neurofibrillary tangles (AT8-negative) also exhibited predominantly cytoplasmic Nrf2 staining (green; asterisk). (D) In CA1 of Alzheimer disease hippocampi, oxidative damage of lipids was determined by labeling with an antibody to 7-hydroxynonenal (7-hydroxynonenol [HNE]—red). In neurons with evidence of oxidative processes as indicated by hydroxynonenal staining (red), Nrf2 is cytoplasmic (green). For all rows, merged images are shown in the first column, colocalization appears yellow when present. Scale bar = 20 μm.
As Nrf2 changes localization in response to oxidative stress, we wanted to determine if there was evidence of oxidative stress in neurons positive for Nrf2. Using an antibody to 7-hydroxynonenol (HNE), a marker of lipid peroxidation, we determined if neurons positive for Nrf2 (Fig. 3D, green) exhibited evidence of oxidative damage (Fig. 3D, red) in AD hippocampi. We observed cytoplasmic Nrf2 in neurons with abundant evidence of oxidative damage (Fig. 3D) in the CA1 region of the hippocampus. These findings suggest that Nrf2 is not localizing to the nucleus despite the presence of oxidative stimuli in neurons.
Reduced Nuclear Factor E2-Related Factor Protein Levels Were Observed in Nuclear Fractions From Alzheimer Disease Frontal Cortex as Compared With Normal Control Frontal Cortex
It has been reported that cytoplasmic Nrf2 interacts with the Keap1:Cul3 complex, an ubiquitin E3 ligase complex that leads to Nrf2 degradation by the proteasome (15). However, dissociation of Nrf2 from the complex in response to oxidative stress stabilizes Nrf2 and allows its translocation to the nucleus. To determine if the alterations in Nrf2 localization in AD reflects increased Nrf2 protein stability, we assessed Nrf2 protein levels by immunoblotting protein extracts from the midfrontal cortex of 9 AD and 9 normal cases. Subcellular fractionation resulted in cytoplasmic and nuclear fractions, which were immunoblotted for Nrf2 and appropriate subcellular compartment markers. In cytoplasmic extracts, we observed Nrf2 in both normal and AD cases (Fig. 4A). Quantification of cytoplasmic Nrf2 did not reveal a trend for more or less Nrf2 in AD cases as compared with control cases whether using the raw values (which were controlled for by protein quantity) normalized to the GAPDH or normalized to a band stained for colloidal gold (Fig. 4C, E). Average Nrf2 levels in AD cases were not significantly different from Nrf2 levels in normal cases using either raw or normalized values (Fig. 4G). However, in the GAPDH-negative nuclear fractions less Nrf2 was observed in AD cases as compared with normal cases by immunoblotting (Fig. 4B; data not shown). Quantification of Nrf2 in the nuclear fractions demonstrated less Nrf2 in AD cases using both raw Nrf2 levels (controlled for protein quantity at loading) and Nrf2 levels normalized to a band stained by colloidal gold (Fig. 4D, F). Average nuclear Nrf2 levels in AD cases were significantly less than Nrf2 levels in normal cases using either raw or normalized values (p < 0.001, Fig. 4H). These findings indicate that there is less Nrf2 in nuclear fractions from cortex in AD cases consistent with our immunohistochemical observations in the hippocampus.
FIGURE 4.
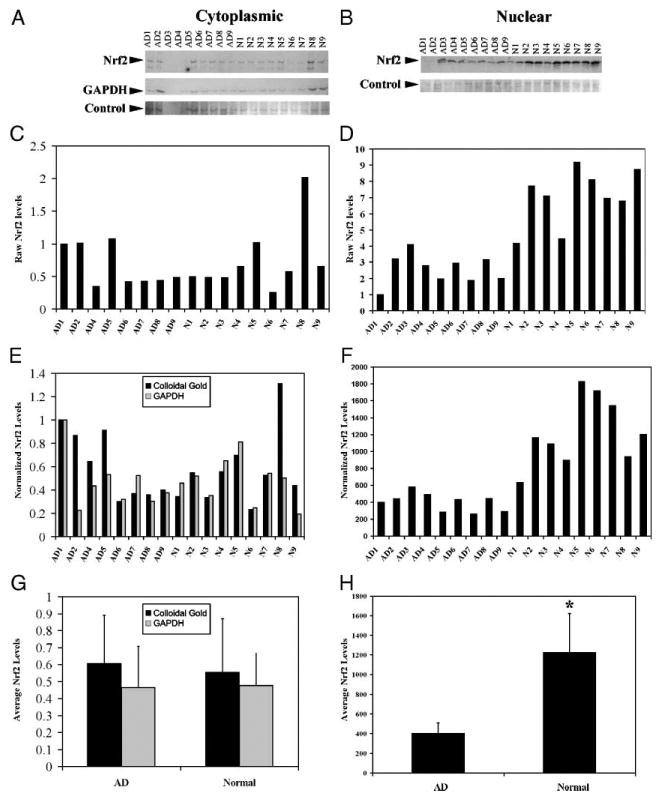
Reduced nuclear factor e2-related factor (Nrf2) proteins levels were observed in nuclear fractions from Alzheimer disease frontal cortex as compared to age-matched control frontal cortex. Cytoplasmic and nuclear protein extracts were obtained from the midfrontal cortex from 9 Alzheimer disease and 9 control cases. Equal quantities (21 μg of protein for cytoplasmic and nuclear) were fractionated by size on an 8% discontinuous SDS polyacrylamide gel and immunoblotted for Nrf2. (A) By immunoblot, Nrf2 levels in cytoplasmic extracts (21 μg) from 9 Alzheimer disease (AD) cases (AD1–9) and 9 normal control cases (N1–9) do not exhibit a difference. Ten micrograms of protein extract from NIH3T3 cells transfected with pcDNA3.1-Nrf2 was included as a control. In addition to Nrf2 (top row), we immunoblotted for glyceraldehyde-3-phosphate dehydrogenase (GAPDH; bottom row) to demonstrate cytoplasmic extraction. To control for protein degradation, we stained the membrane with colloidal gold and quantified the intensity of a band near Nrf2 in size (bottom row; control). (B) By immunoblot, Nrf2 levels in nuclear extracts from 9 AD (AD1–9) cases are lower than Nrf2 levels in 9 normal age-matched control cases (N1–9). As a control, 10 μg of protein extract from NIH3T3 cells transfected with pcDNA3.1-Nrf2 was included as a control. To control for protein degradation, we stained the membrane with colloidal gold and quantified the intensity of a band near Nrf2 in size (bottom row). Quantification of the Nrf2 band from the cytoplasmic immunoblot (C) did not show any statistical differences for control or AD cases; however, quantification of Nrf2 levels from the nuclear immunoblot (D) demonstrated lower Nrf2 levels in AD. Cytoplasmic sample AD3 was dropped from further consideration as the protein from this sample was degraded. (E) Comparison of Nrf2 levels normalized to GAPDH from cytoplasmic extracts showed no evident trend or statistical difference. (F) Normalizing nuclear Nrf2 to the selected colloidal gold band by dividing Nrf2 band intensity by the colloidal gold band intensity demonstrated decreased levels in AD as compared with age-matched control. (G) Average Nrf2 levels in normal cytoplasmic extracts were not statistically different from average Nrf2 levels in AD cytoplasmic extracts. Error bars indicate standard deviation. (H) Average Nrf2 levels in normal nuclear extracts were significantly higher (*, p < 0.001) than average Nrf2 levels in AD nuclear extracts. Error bars indicate standard deviation. *, Indicates statistically significant difference from normal (p < 0.001), Student t-test.
DISCUSSION
Oxidative stress is a common pathogenic mechanism implicated in AD, PD, and other diseases associated with aging (1–6). The cellular mechanism for eradicating oxidative stress involves release, stabilization, and nuclear translocation of the Nrf2 transcription factor (13, 14). By activating expression of genes whose products reduce oxidative stress and inactivate toxic chemicals, Nrf2 acts as the “on” switch for the endogenous antioxidant response. It would be expected that Nrf2 would be activated in AD and PD based on the presence of oxidative damage in these diseases. Our observations indicate that Nrf2 is not translocating to the nucleus in hippocampal neurons in AD cases despite evidence of oxidative stress in these cells and abundant nuclear Nrf2 in normal age-matched controls. In contrast, in PD, nuclear Nrf2 is more abundant in the remaining dopaminergic neurons within the substantia nigra. These observations demonstrate a difference in neuronal response to oxidative stress associated with AD as compared with PD.
Our observation that Nrf2 is predominantly cytoplasmic in hippocampal neurons in AD indicates that Nrf2 is not being translocated to the nucleus or is less antigenically available in AD neuronal nuclei. However, the biochemical extracts from frontal cortex confirm decreased levels of nuclear Nrf2 in AD cases. One explanation for these findings is that Nrf2 is not responding properly to oxidative stress in AD neurons. Neither neurons nor astrocytes exhibited abundant Nrf2 staining in their nuclei in AD cases. These findings suggest several scenarios with regard to Nrf2 regulation in the central nervous system. Based on known functions of Nrf2 in nonneuronal cells, the abundant nuclear Nrf2 observed in age-matched controls suggests that there is enough oxidative stress or some other inducer (i.e. toxins, misfolded proteins, and inflammation) in neurons of control patients to activate the endogenous antioxidant response. Alternatively, Nrf2 may have an additional role that requires nuclear localization in age-matched control hippocampal neurons. The lack of Nrf2 in the nuclei of AD hippocampal neurons suggests that Nrf2 is not performing the activities seen in age-matched control individuals. Whether Nrf2 is responding to oxidative stress in control cases or has a novel role in neuronal nuclei, AD hippocampal neurons are deficient in this activity. This suggests that in AD, some process is blocking Nrf2 nuclear activity, potentially contributing to neuronal dysfunction and/or loss. Our immunoblots indicate that there is a loss of nuclear Nrf2 in AD but that the level of cytoplasmic Nrf2 is not different between age-matched control cases and AD. Thus, the nuclear deficit is not the result of generalized loss of total Nrf2 protein, but may reflect impaired nuclear trafficking, as has been observed with other transcription factors (50). In either case, our observations suggest that the Nrf2 pathway is likely dysfunctional in hippocampal neurons of AD.
In PD, our observations are different. The localization of Nrf2 immunostaining in the hippocampal neurons in PD is somewhere between the predominantly cytoplasmic Nrf2 seen in AD and LBVAD and the strong nuclear staining in observed in age-matched control cases. However, in the most severely affected brain region, the substantia nigra, dopaminergic neurons exhibit a Nrf2 expression pattern consistent with a neuronal response to increased oxidative stress: weakly cytoplasmic in control cases, but more abundant with increased nuclear staining in PD neurons. The different Nrf2 localizations observed in hippocampal neurons as compared with nigral neurons in age-matched controls suggest distinct regulation of Nrf2 in these neuronal subpopulations and potentially divergent functions. It is interesting to speculate that altered Nrf2 regulation may be one of the functional differences between these 2 neuronal types. It may also indicate a difference in susceptibility to oxidative stress between these neuronal populations. Nrf2 localization in PD nigral neurons is consistent with the expected Nrf2 response to oxidative stress. The neurons we observe in PD are the surviving neurons, which may indicate that these neurons were able to delay death, but protection by Nrf2 was insufficient in neurons that have been lost. Alternatively, additional downstream mechanisms may interfere with ARE transactivation despite nuclear stabilization of Nrf2. Further investigation is needed to understand why Nrf2 translocation is not sufficient to protect oxidatively stressed nigral neurons in PD.
In addition to interaction with keap1, Nrf2 activity is also regulated by phosphorylation via several kinases including phosphoinositol-3 kinase (PI3K), extracellular signal-regulated protein kinase (ERK), protein kinase C (PKC), and pancreas enriched kinase (PERK) (19–25, 27). Interestingly, several of these kinases localize to mitochondria, a major source of intracellular oxidative stress (51), and the subcellular distribution of ERK is also altered in degenerating AD and PD neurons (52–54). Although PI3K and ERK activity are required for Nrf2 activation in certain cell types or in response to specific stimuli, direct phosphorylation of Nrf2 has been demonstrated for PKC and PERK (19, 28). Phosphorylation by PKC can disrupt interaction with Keap1 but is not required for stabilization and translocation of Nrf2 to the nucleus. Disruption of Nrf2:Keap1 interaction by PKC further suggests this complex can be regulated by extracellular signaling cascades as well as oxidative insult. Because there is a pronounced inflammatory component to amyloid plaques (55), one possibility is that Nrf2 translocation is inhibited by inflammatory signaling molecules such as cytokines, chemokines, and neurotrophic factors. Phosphorylation of Nrf2 by PERK also disrupts the interaction between Nrf2 and Keap1 and leads to nuclear translocation (15, 19). PERK is one of several kinases activated in response to endoplasmic reticulum (ER) stress. It has been shown that cell survival after ER stress is mediated by the increase in Nrf2-induced glutathione (19). A major cause of ER stress is accumulation of unfolded proteins (56). Because 2 of the major pathologic features of AD (amyloid plaques and NFTs) are composed of misfolded proteins, it is likely that ER stress is activated in neurons and astrocytes at some point during AD progression. Together, these findings suggest that several pathways act to regulate Nrf2 function. Understanding how these pathways intersect to regulate Nrf2 will be important for uncovering why Nrf2 is not responding to oxidative stress, misfolded proteins, or signaling proteins present in the neuronal environment in AD brain tissue.
Previous studies have shown that astrocytic Nrf2 is sufficient to rescue neurons from oxidative stress (33, 34). Our observations suggest that astrocytes have also lost nuclear Nrf2 in AD. By triple label immunofluorescent confocal microscopy, we only identified a few astrocytes with weak nuclear staining in the hippocampus. Similarly, our immunoblotting studies indicate minimal nuclear Nrf2 from all cell populations in the midfrontal cortex. Kraft et al have shown that Nrf2 induction activates expression of different gene products in cultured astrocytes as compared with cultured cortical neurons (34). Although some genes, including glutathione-S-transferase m1, were upregulated in both neurons and astrocytes treated with an inducer of Nrf2 activity (tert butylhydroquinone [tBHQ]), the genes upregulated in astrocytes included detoxification enzymes, glutathione synthesis, and other protective molecules. In contrast, the genes upregulated in neurons treated with tBHQ included those that regulated synaptosome formation and function and neurotransmitter homeostasis. The ability of Nrf2 to regulate distinct subsets of genes in neurons and astrocytes supports a distinct role for Nrf2 in neurons in addition to protection from oxidative stress. Our data suggest that Nrf2 is not nuclear in AD hippocampal neurons and therefore is not transactivating expression of proteins that regulate synapse function or antioxidant activity. The absence of this activity is likely detrimental to neuronal function and implicates Nrf2 dysfunction in AD.
Our findings indicate that Nrf2 is not localizing to the nucleus in neurons or astrocytes in AD despite the presence of oxidative stress and misfolded proteins, 2 stimuli that can induce nuclear Nrf2. However, Nrf2 does translocate to the nucleus of substantia nigra neurons in PD. In light of the findings by Kraft et al showing Nrf2 regulates unique subsets of genes in neurons versus astrocytes, it is also possible that Nrf2 may regulate different gene products in hippocampal neurons versus nigral neurons (34). Future studies are needed to determine how Nrf2 localization is regulated, why it is aberrant in AD, and how it differs in various neuronal subpopulations. By understanding this pathway and how it has been altered by AD progression, we will gain insight into how neurons lose their endogenous protection, leading to potential strategies to restore this function before neuronal loss.
Acknowledgments
The authors thank Kathleen L. Morgan, Daniel Martinez, and Molly Ainsman for technical support and Dr. Michael G. White and Dr. Mark J. Sciutto for statistical support. The authors also appreciate helpful discussion, advice, support, and reagents from Drs. Nigel Cairns, Virginia M.-Y. Lee, and John Q. Trojanowski.
This work was supported by the following grants: NS41202 (KLJ-S), NS40817 (CTC), and a pilot grant to KLJ-S from the University of Pennsylvania Alzheimer Disease Center Grant (AG10124, John Q. Trojanowski, PI). RLH was supported in part by NIH grant AG05133 (Pittsburgh ADRC, Steve DeKosky, PI).
References
- 1.Beall MF. Mitochondria, oxidative damage, and inflammation in Parkinson's disease. Ann N Y Acad Sci. 2003;991:120–31. doi: 10.1111/j.1749-6632.2003.tb07470.x. [DOI] [PubMed] [Google Scholar]
- 2.Butterfield DA, Drake J, Pocernich C, et al. Evidence of oxidative damage in Alzheimer's disease brain: Central role for amyloid beta-peptide. Trends Mol Med. 2001;7:548–54. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- 3.Butterfield DA, Griffin S, Munch G, et al. Amyloid beta-peptide and amyloid pathology are central to the oxidative stress and inflammatory cascades under which Alzheimer's disease brain exists. J Alzheimers Dis. 2002;4:193–201. doi: 10.3233/jad-2002-4309. [DOI] [PubMed] [Google Scholar]
- 4.Maccioni RB, Munoz JP, Barbeito L. The molecular bases of Alzheimer's disease and other neurodegenerative disorders. Arch Med Res. 2001;32:367–81. doi: 10.1016/s0188-4409(01)00316-2. [DOI] [PubMed] [Google Scholar]
- 5.Pratico D, Clark CM, Liun F, et al. Increase of brain oxidative stress in mild cognitive impairment: A possible predictor of Alzheimer disease. Arch Neurol. 2002;59:972–76. doi: 10.1001/archneur.59.6.972. [DOI] [PubMed] [Google Scholar]; Erratum appears in Arch Neurol. 2002;59:1475. [Google Scholar]
- 6.Sayre LM, Smith MA, Perry G. Chemistry and biochemistry of oxidative stress in neurodegenerative disease. Curr Med Chem. 2001;8:721–38. doi: 10.2174/0929867013372922. [DOI] [PubMed] [Google Scholar]
- 7.Nunomura A, Perry G, Aliev G, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–67. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 8.Callio J, Oury TD, Chu CT. Manganese superoxide dismutase protects against 6-hydroxydopamine injury in mouse brains. J Biol Chem. 2005;280:18536–42. doi: 10.1074/jbc.M413224200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kulich SM, Chu CT. Role of reactive oxygen species in extracellular signal-regulated protein kinase phosphorylation and 6-hydroxydopamine cytotoxicity. J Biosci. 2003;28:83–89. doi: 10.1007/BF02970136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engelhart M, Geerlings M, Ruitenberg A, et al. Dietary intake of antioxidants and risk of Alzheimer disease. JAMA. 2002;287:3223–29. doi: 10.1001/jama.287.24.3223. [DOI] [PubMed] [Google Scholar]
- 11.Tuppo E, Forman L. Free radical oxidative damage and Alzheimer's disease. J Am Osteopathic Assoc. 2001;101:S11–15. [PubMed] [Google Scholar]
- 12.Itoh K, Chiba T, Takahashi S, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–22. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 13.Itoh K, Wakabayashi N, Katoh Y, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McMahon M, Itoh K, Yamamoto M, et al. The Cap'n'Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 2001;61:3299–307. [PubMed] [Google Scholar]
- 15.Cullinan SB, Gordan JD, Jin J, et al. The Keap1-BTB protein is an adaptor that bridges Nrf2 to the Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-Keap1 ligase. Mol Cell Biol. 2004;24:8477–86. doi: 10.1128/MCB.24.19.8477-8486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jain AK, Bloom DA, Jaiswal AK. Nuclear import and export signals in control of Nrf2. J Biol Chem. 2005;280:29158–68. doi: 10.1074/jbc.M502083200. [DOI] [PubMed] [Google Scholar]
- 17.Li W, Jain MR, Chen C, et al. Nrf2 Possesses a redox-insensitive nuclear export signal overlapping with the leucine zipper motif. J Biol Chem. 2005;280:28430–38. doi: 10.1074/jbc.M410601200. [DOI] [PubMed] [Google Scholar]
- 18.Velichkova M, Hasson T. Keap1 regulates the oxidation-sensitive shuttling of Nrf2 into and out of the nucleus via a Crm1-dependent nuclear export mechanism. Mol Cell Biol. 2005;25:4501–13. doi: 10.1128/MCB.25.11.4501-4513.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cullinan SB, Zhang D, Hannink M, et al. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23:7198–209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang HC, Nguyen T, Pickett CB. Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc Natl Acad Sci U S A. 2000;97:12475–80. doi: 10.1073/pnas.220418997. [DOI] [PMC free article] [PubMed] [Google Scholar]; erratum appears in Proc Natl Acad Sci U S A. 2001;98:379. [Google Scholar]
- 21.Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem. 2002;277:42769–74. doi: 10.1074/jbc.M206911200. [DOI] [PubMed] [Google Scholar]
- 22.Kang KW, Lee SJ, Park JW, et al. Phosphatidylinositol 3-kinase regulates nuclear translocation of NF-E2-related factor 2 through actin rearrangement in response to oxidative stress. Mol Pharmacol. 2002;62:1001–10. doi: 10.1124/mol.62.5.1001. [DOI] [PubMed] [Google Scholar]
- 23.Lee JM, Hanson JM, Chu WA, et al. Phosphatidylinositol 3-kinase, not extracellular signal-regulated kinase, regulates activation of the antioxidant-responsive element in IMR-32 human neuroblastoma cells. J Biol Chem. 2001;276:20011–16. doi: 10.1074/jbc.M100734200. [DOI] [PubMed] [Google Scholar]
- 24.Numazawa S, Ishikawa M, Yoshida A, et al. Atypical protein kinase C mediates activation of NF-E2-related factor 2 in response to oxidative stress. Am J Physiol. 2003;285:C334–42. doi: 10.1152/ajpcell.00043.2003. [DOI] [PubMed] [Google Scholar]
- 25.Zipper LM, Mulcahy RT. Inhibition of ERK and p38 MAP kinases inhibits binding of Nrf2 and induction of GCS genes. Biochem Biophys Res Commun. 2000;278:484–92. doi: 10.1006/bbrc.2000.3830. [DOI] [PubMed] [Google Scholar]
- 26.Zipper LM, Mulcahy RT. The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J Biol Chem. 2002;277:36544–52. doi: 10.1074/jbc.M206530200. [DOI] [PubMed] [Google Scholar]
- 27.Zipper LM, Mulcahy RT. Erk activation is required for Nrf2 nuclear localization during pyrrolidine dithiocarbamate induction of glutamate cysteine ligase modulatory gene expression in HepG2 cells. Toxicol Sci. 2003;73:124–34. doi: 10.1093/toxsci/kfg083. [DOI] [PubMed] [Google Scholar]
- 28.Bloom DA, Jaiswal AK. Phosphorylation of Nrf2 at Ser40 by protein kinase C in response to antioxidants leads to the release of Nrf2 from INrf2, but is not required for Nrf2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response element-mediated NAD(P)H: Quinone oxidoreductase-1 gene expression. J Biol Chem. 2003;278:44675–82. doi: 10.1074/jbc.M307633200. [DOI] [PubMed] [Google Scholar]
- 29.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, et al. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A. 2002;99:11908–13. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee J, Johnson JA. An important role of Nrf2-ARE pathway in cellular defense mechanism. Biochem Mol Bio. 2004;37:139–43. doi: 10.5483/bmbrep.2004.37.2.139. [DOI] [PubMed] [Google Scholar]
- 31.Kwak MK, Itoh K, Yamamoto M, et al. Role of transcription factor Nrf2 in the induction of hepatic phase 2 and antioxidative enzymes in vivo by the cancer chemoprotective agent, 3H-1, 2-dimethiole-3-thione. Mol Med. 2001;7:135–45. [PMC free article] [PubMed] [Google Scholar]
- 32.Lee JM, Calkins MJ, Chan K, et al. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J Biol Chem. 2003;278:12029–38. doi: 10.1074/jbc.M211558200. [DOI] [PubMed] [Google Scholar]
- 33.Shih AY, Johnson DA, Wong G, et al. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci. 2003;23:3394–406. doi: 10.1523/JNEUROSCI.23-08-03394.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kraft AD, Johnson DA, Johnson JA. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J Neurosci. 2004;24:1101–12. doi: 10.1523/JNEUROSCI.3817-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Calkins MJ, Jakel RJ, Johnson DA, et al. Protection from mitochondrial complex II inhibition in vitro and in vivo by Nrf2-mediated transcription. Proc Natl Acad Sci U S A. 2005;102:244–49. doi: 10.1073/pnas.0408487101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shih AY, Imbeault S, Barakauskas V, et al. Induction of the Nrf2-driven antioxidant response confers neuroprotection during mitochondrial stress in vivo. J Biol Chem. 2005;280:22925–36. doi: 10.1074/jbc.M414635200. [DOI] [PubMed] [Google Scholar]
- 37.Lovell MA, Xie C, Markesbery WR. Decreased glutathione transferase activity in brain and ventricular fluid in Alzheimer's disease. Neurology. 1998;51:1562–66. doi: 10.1212/wnl.51.6.1562. [DOI] [PubMed] [Google Scholar]
- 38.Wang Y, Santa-Cruz K, DeCarli C, et al. NAD(P)H:quinone oxidoreductase activity is increased in hippocampal pyramidal neurons of patients with Alzheimer's disease. Neurobiol Aging. 2000;21:525–31. doi: 10.1016/s0197-4580(00)00114-7. [DOI] [PubMed] [Google Scholar]
- 39.Nakamura K, Wang W, Kang UJ. The role of glutathione in dopaminergic neuronal survival. J Neurochem. 1997;69:1850–58. doi: 10.1046/j.1471-4159.1997.69051850.x. [DOI] [PubMed] [Google Scholar]
- 40.Shults CW, Haas RH, Passov D, et al. Coenzyme Q10 levels correlate with the activities of complexes I and II/III in mitochondria from parkinsonian and nonparkinsonian subjects. Ann Neurol. 1997;42:261–64. doi: 10.1002/ana.410420221. [DOI] [PubMed] [Google Scholar]
- 41.Klivenyi P, Andreassen OA, Ferrante RJ, et al. Mice deficient in cellular glutathione peroxidase show increased vulnerability to malonate, 3-nitropropionic acid, and 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. J Neurosci. 2000;20:1–7. doi: 10.1523/JNEUROSCI.20-01-00001.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Przedborski S, Kostic V, Jackson-Lewis V, et al. Transgenic mice with increased Cu/Zn-superoxide dismutase activity are resistant to N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity. J Neurosci. 1992;12:1658–67. doi: 10.1523/JNEUROSCI.12-05-01658.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang J, Graham DG, Montine TJ, et al. Enhanced N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity in mice deficient in CuZn-superoxide dismutase or glutathione peroxidase. J Neuropathol Exp Neurol. 2000;59:53–61. doi: 10.1093/jnen/59.1.53. [DOI] [PubMed] [Google Scholar]
- 44.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 45.Braak H, Del Tredici H, Rub U, et al. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 46.Jordan-Sciutto KL, Wang G, Murphey-Corb M, et al. Cell cycle proteins exhibit altered expression patterns in lentiviral-associated encephalitis. J Neurosci. 2002;22:2185–95. doi: 10.1523/JNEUROSCI.22-06-02185.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mirra SS, Hyman BT. Ageing and dementia. In: Graham DI, Lantos PL, editors. Greenfield's Neuropathology. London: Oxford University Press, Inc; 2002. pp. 195–226. [Google Scholar]
- 48.Cote L, Crutcher M. The basal ganglia. In: Kandel E, Schwartz J, Jessell T, editors. Principles of Neural Science. New York: Elsevier Publishing; 1991. pp. 653–56. [Google Scholar]
- 49.Castellani RJ, Perry G, Siedlak SL, et al. Hydroxynonenal adducts indicate a role for lipid peroxidation in neocortical and brainstem Lewy bodies in humans. Neurosci Lett. 2002;319:25–28. doi: 10.1016/s0304-3940(01)02514-9. [DOI] [PubMed] [Google Scholar]
- 50.Chalovich E, Zhu J, Caltagarone J, et al. Functional repression of cAMP response element in 6-hydroxydopamine-treated neuronal cells. J Biol Chem. 2006;281:17870–81. doi: 10.1074/jbc.M602632200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Horbinski C, Chu CT. Kinase signaling cascades in the mitochondrion: A matter of life or death. Free Radic Biol Med. 2005;38:2–11. doi: 10.1016/j.freeradbiomed.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 52.Chu CT, Levinthal DJ, Kulich SM, et al. Oxidative neuronal injury. The dark side of ERK1/2. Eur J Biochem. 2004;271:2060–66. doi: 10.1111/j.1432-1033.2004.04132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ferrer I, Blanco R, Carmona M, et al. Phosphorylated map kinase (ERK1, ERK2) expression is associated with early tau deposition in neurones and glial cells, but not with increased nuclear DNA vulnerability and cell death, in Alzheimer disease, Pick's disease, progressive supranuclear palsy and corticobasal degeneration. Brain Pathol. 2001;11:144–58. doi: 10.1111/j.1750-3639.2001.tb00387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhu JH, Kulich SM, Oury TD, et al. Cytoplasmic aggregates of phosphorylated extracellular signal-regulated protein kinases in Lewy body diseases. Am J Pathol. 2002;161:2087–98. doi: 10.1016/S0002-9440(10)64487-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tuppo EE, Arias HR. The role of inflammation in Alzheimer's disease. Int J Biochem Cell Biol. 2005;37:289–305. doi: 10.1016/j.biocel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 56.Forman M, Lee VMY, Trojanowski J. ‘Unfolding’ pathways in neurodegenerative disease. Trends Neurosci. 2003;27:407–10. doi: 10.1016/S0166-2236(03)00197-8. [DOI] [PubMed] [Google Scholar]