

Summary
In a large consanguineous family of Turkish origin, genome-wide homozygosity mapping revealed a locus for recessive nonsyndromic hearing impairment on chromosome 14q24.3–q34.12. Fine mapping with microsatellite markers defined the critical linkage interval to a 18.7 cM region flanked by markers D14S53 and D14S1015. This region partially overlapped with the DFNB35 locus. Mutation analysis of ESRRB, a candidate gene in the overlapping region, revealed a homozygous 7 bp duplication in exon 8 in all affected individuals. This duplication results in a frame shift and premature stop codon. Sequence analysis of the ESRRB gene in the affected individuals of the original DFNB35 family and in three other DFNB35-linked consanguineous families from Pakistan revealed four missense mutations. ESRRB encodes the estrogen-related receptor beta protein, and one of the substitutions (p.A110V) is located in the DNA-binding domain of ESRRB, whereas the other three are substitutions (p.L320P, p.V342L, and p.L347P) located within the ligand-binding domain. Molecular modeling of this nuclear receptor showed that the missense mutations are likely to affect the structure and stability of these domains. RNA in situ hybridization in mice revealed that Esrrb is expressed during inner-ear development, whereas immunohistochemical analysis showed that ESRRB is present postnatally in the cochlea. Our data indicate that ESRRB is essential for inner-ear development and function. To our knowledge, this is the first report of pathogenic mutations of an estrogen-related receptor gene.
Introduction
Autosomal-recessive nonsyndromic hearing impairment (ARNSHI) is a genetically heterogeneous disorder. To date, 67 loci for ARNSHI, referred to as DFNB loci, have been mapped, and 24 of the causative genes have been identified.1 The proteins encoded by DFNB genes vary greatly with respect to their functions and their temporal and spatial patterns of expression in the inner ear.
The majority of DFNB loci have been identified in families with consanguineous marriages, and some of these DFNB loci span large genomic regions containing many genes. Linkage analysis in additional families segregating hearing loss might result in the identification of linkage intervals that overlap known DFNB loci, thereby refining the critical regions and reducing the number of candidate genes that would need to be screened for mutations. Expression profiling of genes preferentially or predominantly expressed in the inner ear2–5 has also helped to prioritize candidate genes for mutation screening. In addition, large-scale microarray analysis of regenerating inner hair cells from several avian species has recently revealed indications of a number of distinct pathways being important for inner-ear development.6 Among these were known pathways such as those involving NOTCH, TGFβ, and WNT, of which the latter is essential for planar cell polarity and thus for stereocilia formation in inner-ear hair cells.7 Furthermore, components of other pathways, for instance estrogen-receptor signaling, were found to be involved in hair cell regeneration.6
In the present study, we describe the mapping of ARNSHI segregating in a large consanguineous family (TR-21) of Turkish origin to chromosome 14q24.3–q34.12. The critical region had an overlap of 1.1 cM with DFNB35 (MIM %608565).8 One of the genes in this overlapping region was the estrogen-related receptor beta gene ESRRB (MIM #602167) that is a member of the estrogen-receptor family. Sequence analysis of this gene in the affected individuals of family TR-21, in the original DFNB35 family, and in three additional DFNB35-linked families from Pakistan indicate that mutations of ESRRB are causative for early-onset hearing impairment.
Material and Methods
Subjects and Clinical Evaluations
Consanguineous family TR-21 of Turkish origin has eight affected individuals (Figure 1). Other than hearing loss, general examinations did not reveal any abnormalities in the participating individuals of family TR-21. Several participating family members underwent otoscopic examination and pure-tone audiometry. Both air conduction (frequencies 250–8000 Hz) and bone conduction (500–4000 Hz) were evaluated in a sound-treated room with an Interacoustics AC5 audiometer (Interacoustics). In addition, we further characterized the hearing loss in two of the affected individuals of this family (IV:5 and V:2, Figure 1C) by recording the auditory brainstem response (ABR) and otoacoustic emission (OAE) spectra. Click-evoked ABRs were measured with the ICS Chartr MCU-90 system (GN Otometrics). Transiently evoked OAE spectra were recorded with a Madsen Capella Cochlear Emissions Analyzer (GN Otometrics). Emissions of 6 dB or greater for at least three frequency bands were accepted as a positive result indicating functionally intact outer hair cells.
Figure 1.
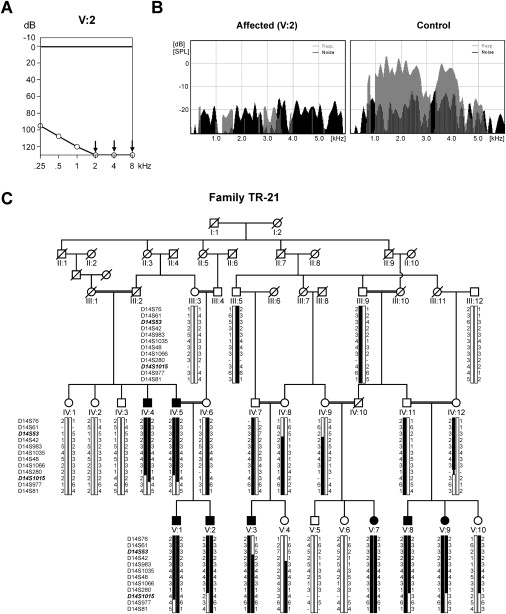
Clinical Characterization and Linkage Analysis of Family TR-21
(A) Binaural mean hearing-level thresholds of affected individual V:2 (TR-21) in decibels (dB) for each frequency in kilohertz (kHz).
(B) Representative examples of transiently evoked OAE recordings for individual V:2 (TR-21, left panel) and one control individual (right panel). The x axis shows the frequencies at which OAE responses were measured, and the y axis shows the emissions in dB. Noise floor levels are shown in black, and the responses to the stimuli are shown in gray.
(C) Pedigree and haplotypes of family TR-21 for STR markers within the linkage interval. Flanking markers D14S53 and D14S1015 are indicated in bold and italic. The disease haplotype is indicated in black.
We addressed vestibular function and vision by performing Romberg tests and questionnaires including questions on childhood motor development, insecure feelings during walking in darkness or on an uneven surface, sport activities, motion sickness, reading during walking and visual problems, and night blindness. For individuals IV:5 and V:2, computerized tomography of the temporal bone, caloric testing, and funduscopy were performed. In addition, complete urinalysis, urine-concentration and -acidification tests, tubular reabsorption of phosphate, and measurements of plasma urea, creatinine levels, and blood gases were carried out so that renal functions could be assessed. Furthermore, 131 index patients from Turkish families that have prelingual sensorineural hearing loss with indications of a recessive mode of inheritance participated in this study. For all affected individuals in this study, we excluded the GJB2 gene as the causative gene by sequencing both exons and intron-exon boundaries. In addition, for all Turkish patients from consanguineous parents, the known ARNSHI genes were excluded by homozygosity mapping with flanking microsatellite markers. Control DNAs were used from 120 unrelated Turkish individuals. The clinical features of affected subjects in the original DFNB35 family have been described previously.8
Families PKDF191, PKDF159, and PKDF327 were ascertained from the Punjab in Pakistan. All participating members of these families were evaluated by a physician so that obvious extra-auditory phenotypes associated with common syndromic forms of deafness were ruled out. Air-conduction pure-tone audiometry tests were performed on selected individuals under quiet ambient conditions at octave frequencies ranging from 250 to 8000 Hz. Vestibular function was evaluated by tandem gait and Romberg testing. This study was approved by human-subject ethics committees in Pakistan, United States, Turkey, and the Netherlands. All participating subjects were ascertained after obtaining written informed consent.
Genetic-Linkage Studies
Genomic DNA was extracted from peripheral blood lymphocytes with either salting out or nonorganic procedures.9,10 Genomic DNA of five affected members of family TR-21 (V:2, V:3, V:7, V:8, and V:9) was subjected to a genome-wide scan with a total of 382 short tandem repeat (STR) polymorphic markers (ABI Prism linkage mapping set MD10.A) located on the 22 autosomal chromosomes. The average distance and heterozygosity of the genotyped markers were 10 cM and 0.76, respectively. The STR markers were amplified by polymerase chain reaction (PCR) with a RoboAmp 4,200 (Isogen Life Science) and were analyzed on an ABI prism 3700 genetic analyzer. The alleles were assigned with the GeneMapper software (Applied Biosystems) and linkage designer software (Microsoft EXCEL macro by Guy van Camp, University of Antwerpen). In three Pakistani families, STR polymorphic markers were typed for all of the reported DFNB loci including DFNB35.8 Markers were amplified by the polymerase chain reaction (PCR) on a Gene Amp PCR system 9700 (Applied Biosystems) and were analyzed on an ABI Prism 3100 Genetic Analyzer. For refining the DFNB35 locus in the original family, individuals were genotyped at the Center for Inherited Disease Research (CIDR) with the Illumina fine-mapping panel.
The calculation of the two-point log of odds (LOD) scores was carried out with the easyLINKAGE program, version 3.03.11 An autosomal-recessive mode of inheritance with complete penetrance was assumed, and the disease allele frequency was set at 0.01. For fine mapping, all TR-21 family members were genotyped for the following STR markers: D14S76, D14S61, D14S53, D14S42, D14S983, D14S1035, D14S48, D14S1066, D14S280, D14S1015, D14S977, and D14S81. The genomic localization of the markers was derived from the Marshfield map and the UCSC human genome database (build hg18, March 2006). For Pakistani families, LOD scores were calculated with LINKMAP as described previously.12
Mutation Analysis
All exons and intron-exon boundaries of ESRRB were amplified under standard PCR conditions. Primer sequences are presented in Table 1. Sequence analysis was performed with the ABI PRISM Big Dye Terminator Cycle Sequencing V2.0 Ready Reaction kit and the ABI PRISM 3730 DNA analyzer (Applied Biosystems). To determine the presence of the p.Val342GlyfsX44 mutation in healthy control individuals, we performed an amplification refractory mutation system (ARMS)-like approach by using forward primer 5′-ttacgctacacagggaaagc-3′, wild-type reverse primer 5′-cttgagcgtcacaaactcctcc-3′, and mutant reverse primer 5′-ccttgagcgtcacaaactccaaac-3′. To determine the presence of the various missense mutations in Pakistani control individuals, we amplified the corresponding exons under standard PCR conditions and sequenced them as described above.
Table 1.
Primer Sequences Used to Amplify ESRRB
| Exon | Forward Primer | Reverse Primer |
|---|---|---|
| 1 | 5′-atgtttccgcagcatttatc-3′ | 5′-gccacatgctctctaaatcc-3′ |
| 2 | 5′-agcaaccagtactcaccagg-3′ | 5′-ggagaaaaggaggcagaaac-3′ |
| 3 | 5′-atgcaatgtgaccctagagc-3′ | 5′-aagacagcatggtctgcatc-3′ |
| 4 | 5′-taatgccagaaacttgctcc-3′ | 5′-cacagaagtaccgctccaac-3′ |
| 5 | 5′-tgctttgagaacactagggg-3′ | 5′-aagaaattccaattcccacc-3′ |
| 6 | 5′-ggatgcgccattactgttag-3′ | 5′-cccaagatccacattgtctc-3′ |
| 7 | 5′-ggagctcttaggaacccaac-3′ | 5′-tcctctccaatgctacaagg-3′ |
| 8 | 5′-ttacgctacacagggaaagc-3′ | 5′-cttaggaaatgctcagccag-3′ |
| 9 | 5′-acctcttgagaaatgtcccc-3′ | 5′-catgatacaggggttgaagg-3′ |
| 10 | 5′-atgcccacctacagacagac-3′ | 5′-aagctgagtggaaactgtgg-3′ |
| 11 | 5′-tcccaggaaactcctctacc-3′ | 5′-agctgctctgcagtttgtg-3′ |
| 12 | 5′-ctcactgtgctgtgtccttg-3′ | 5′-atggaccccttcagtaccag-3′ |
Molecular Modeling
The structure of the DNA-binding domain of the estrogen receptor beta ERB (PDBid = 11o1) has been solved experimentally13 and is 99% identical to that of the ESRRB protein. The ligand-binding domain shows high sequence similarity with the corresponding domain of ESRRG (PDBid = 1kv6).14 The crystal structure of ESRRG was used as a template for building a model of the ligand-binding domain of the human ESRRB protein. Modeling was performed with the WHAT IF software15 with standard parameter settings and protocols as described previously.16,17 This modeling is straightforward because the percentage sequence identity is high (79%) and all key residues are conserved between the template and the model. The effects of the four missense mutations were analyzed with the WHAT IF/YASARA TwinSet.18
Tissue Distribution of ESRRB Isoforms
Total RNA from human placenta, adult brain, testis, kidney, and retina and from fetal heart, skeletal muscle, liver, and lung was obtained from Clontech Laboratories. Total RNA from cochlea of 16- to 22-week-old human fetuses was isolated with RNAzol B (Campro Scientific) as described previously.3 For cDNA synthesis, 2 μg of total RNA was incubated with 5 ng/μl of random hexamers [pd(N)6; Pharmacia] and 0.3 mM dNTPs (Invitrogen Life Sciences). Subsequently, cDNA was synthesized with the M-MLV Reverse Transcriptase kit (Invitrogen Life Sciences) with a final concentration of 10 mM DTT, 11 U Reverse Transcriptase, and 0.33 U RNAguard (American Biosciences) per reaction.
For detection of the distribution patterns of the ESRRBlong, ESRRB-Δ10, and ESRRBshort isoforms19 in various human tissues, RT-PCR was carried out with the GC-rich PCR system (Roche) with forward primer 5′-ctatagcgtcaaactgcagggcaaagtg-3′ and reverse primer 5′-ctgctcttggccaacctgccctct-3′ for the ESRRBlong isoform, forward primer 5′-tgacgacaagctggtgtacg-3′ and reverse primer 5′-atccctgcttgtgaaggcag-3′ for the ERRSB-Δ10 isoform, and forward primer 5′-tgacgacaagctggtgtacg-3′ and reverse primer 5′-tctgtaggtgggcattggtc-3′ for the ESRRBshort isoform. As a control, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was amplified with forward primer 5′-accacagtccatgccatcac-3′ and reverse primer 5′-tccaccaccctgttgctgta-3′. PCR products were analyzed via agarose gel electrophoresis and sequenced as described above.
Digoxigenin Labeling of cRNA In Situ Hybridization Probes
To obtain probes for RNA in situ hybridization, we generated PCR products that correspond to the 3′ end of murine Esrrb (GenBank ID AK052256). PCR reactions were carried out with forward primer 5′-catcgtgtaccgctcgctc-3′ and reverse primer 5′-caggagcatccactgagaac-3′. Amplimers were cloned in the pCR4-TOPO vector with the TOPO TA cloning kit (Invitrogen) and sequenced with T7 and T3 sequencing primers. Subsequently, PCR reactions were performed with T7 and T3 polymerase-specific primers with the pCR4-TOPO construct as a template. We generated digoxigenin (DIG)-cRNA probes by incubating 400 ng of these PCR products with 2 μl 10× DIG RNA labeling mix (Roche), 2 μl T3 (sense) or T7 (antisense)-dependent RNA polymerase (Roche), 2 μl 10× Transcription buffer (Roche), and RNase-free H2O added to a final volume of 20 μl, at 37°C for 2 hr. To terminate the reaction, we added 2 μl of 0.2 M EDTA (pH 8.0). The cRNA probe was ethanol precipitated and dissolved in RNase-free H2O. DIG concentrations were determined by dot-blot analysis on Hybond N+ nylon membranes (Amersham Biosciences), compared with a control DIG-cRNA probe (Roche), and stored at −80°C.
RNA In Situ Hybridization
Mouse embryos collected at various embryonic stages (E12.5–E18.5) as well as eyes of adult mice (P90) were frozen on crushed dry ice and stored at −80°C. The tissues were embedded in Tissue-Tek O.C.T. compound (Sakura Finetek) and sagitally cut into 16 μm sections at temperatures of −16°C to −20°C. Serial sections were collected on SuperFrost Plus microscope slides (Menzel-Gläser). After quick drying, these sections were stored at −80°C. The DIG-labeled cRNA probes were hybridized to the mouse-tissue sections as follows: Samples were rapidly defrosted and fixed in 4% paraformaldehyde in PBS (pH 7.4) for 10 min. Slides were washed three times in PBS (pH 7.4) and acetylated for 10 min in a solution containing 3.3 ml triethanolamine, 0.438 ml 37% HCl, and 0.625 ml acetic anhydride in a final volume of 250 ml H2O. Slides were washed three times for 5 min in PBS and prehybridized for 2 hr at room temperature (RT) with hybridization mix [50% deionized formamide, 5× SSC, 5× Denhardt's solution, 250 μg/ml brewer's yeast tRNA (Roche), and 500 μg/ml sonicated salmon sperm DNA (Sigma)]. Subsequently, hybridization was performed overnight at 72°C with 150 μl hybridization mix containing 400 ng/ml DIG-labeled probe, washed briefly at 72°C in 2× SSC, and subsequently washed for 2 hr at 72°C in 0.2× SSC. Slides were allowed to cool, transferred to 0.2× SSC at RT for 5 min, and subsequently transferred to buffer 1 (100 mM Tris-HCl [pH 7.4], and 150 mM NaCl). Slides were incubated for 1 hr at RT with 10% heat inactivated fetal-calf serum (FCS) in buffer 1 and then incubated overnight at 4°C with 0.7 ml 1% heat inactivated FCS in buffer 1 with 1:5000 diluted anti-DIG-AP, fab fragment from sheep (Roche) per slide. Slides were washed three times in buffer 1 and once in buffer 2 (100 mM Tris-HCl [pH 9.5], 100 mM NaCl, and 50 mM MgCl2) at RT. A total of 1 ml of staining solution [200 μl of NBT-BCIP stock solution (Roche), 1 ml Levamisole (Sigma, 2.4 mg/ml), and 8.8 ml buffer 2 per 10 ml] was applied to the slides, and staining was allowed to take place overnight in a dark environment. Subsequently, the slides were washed once in T10E5 (10 mM Tris [pH 8.0] and 5 mM EDTA) for the reaction termination. To increase structural detail, we incubated some slides in Nuclear Fast Red (Sigma) for 10 s. Slides were dehydrated and sealed with Entellan rapid mounting media (ProSciTech). Images were recorded on a Zeiss Axioskop2 plus microscope with a Sony power HAD DXC-950P 3CCD color video camera.
Antibodies
A primary polyclonal antibody directed against mouse Esrrb (Ab19331: aa 376–388, peptide DYELSQRHEEPRR) was purchased from Abcam. Specificity of the ESRRB antiserum was confirmed by preincubation of the antiserum with 10 μg of the peptide (Princeton BioMolecules) used as antigen prior to immunohistochemistry (data not shown). Mouse monoclonal antibody M3F7 (DSHB) directed against collagen type IV and mouse monoclonal antibody RNF402 (Abcam) directed against the 200 kDa neurofilament heavy chain were used as markers. Secondary antibodies were acquired from Molecular Probes (Invitrogen).
Immunohistochemistry
Cochleae from P4 rats were isolated and fixed as described before.20 After fixation, cryosections were made at a thickness of 10 μm and treated with 0.01% Tween-20 in PBS; this was followed by a blocking step with blocking solution (0.1% ovalbumin and 0.5% fish gelatin in PBS). Antibodies diluted in blocking solution were incubated overnight at 4°C. Secondary antibodies were also diluted in blocking solution and incubated for 1 hr at room temperature. Sections were embedded with Prolong Gold Antifade (Molecular Probes). Pictures were made with an Axioskop2 Mot plus fluorescence microscope (Zeiss) equipped with an AxioCam MRC5 camera (Zeiss). Images were processed with Axiovision 4.3 (Zeiss).
Results
Clinical Characterization
Affected individuals of family TR-21 have bilateral, severe-to-profound sensorineural hearing loss (Figure 1A), and acoustic brainstem responses were absent in affected individuals (data not shown). Transient otoacoustic emission spectra (TEOAEs) were absent in two individuals (IV:5 and V:2, measured at the age of 48 and 23 years, respectively) that were tested (Figure 1B), indicating a deficiency of outer hair cell function in these patients. For these two individuals, computerized tomography of the temporal bone and caloric testing for evaluation of the vestibular function were performed, and no abnormalities were found. In the affected family members, there were no indications of a visual problem, kidney failure, or morphological features that suggested a syndromic form of hearing loss.
Linkage and Mutation Analyses
Genome-wide homozygosity mapping revealed only one interval located on chromosome 14q24–31. Fine mapping with microsatellite markers in the complete family refined the linkage interval to 18.7 cM between markers D14S53 and D14S1015 (Figure 1C) with a maximum two-point LOD score of 5.45 for marker D14S48. The linkage interval partially overlapped with the DFNB35 locus for which the causative gene had not yet been identified.8 Within the original DFNB35 family, the linkage interval was reduced, and rs1035159 was determined to be the distal flanking marker (Figure 2A). Therefore, only a 1.1 cM-region, flanked by markers D14S53 and rs1035159, is shared between the DFNB35 locus and the linkage interval described here (Figure 2B). This shared interval contains seven RefSeq genes and includes only exons 5–12 of the estrogen-related receptor beta gene ESRRB. This gene was represented in a cochlear cDNA library that was generated via a suppression subtractive hybridization technique.3
Figure 2.

Linkage Interval and Mutation Analysis of ESRRB in Family TR-21 and the Original DFNB35 Family
(A) Haplotype analysis for part of the original DFNB35 family reduced the DFNB35 locus. The recombination event in V:1 was determined to have occurred between rs8020517 and rs1035159.
(B) Schematic representation of the reduced DFNB35 locus and the critical region identified in family TR-21. Genetic positions of the markers, according to the Marshfield map, are shown in centimorgan (cM). The intervals share a 1.1 cM (0.7 Mb) region flanked by markers D14S53 and rs1035159, encompassing part of ESRRB.
(C) Partial sequence of ESRRB from an affected individual of family TR-21 homozygous for the 7 bp duplication (c.1018_1024 dupGAGTTTG; depicted in red), a family member with two wild-type alleles and a heterozygous carrier. The effect of the mutation at the amino acid level is indicated above the sequence.
(D) Partial sequence of ESRRB from an affected member of the original DFNB35 family, carrying the nucleotide substitution c.1024G→T homozygously, from a family member with two wild-type alleles and from a heterozygous carrier. The predicted amino acid substitution (p.V342L) and the surrounding amino acids are indicated above the sequence.
Sequence analysis of ESRRB in an affected individual of family TR-21 revealed a 7 bp duplication in exon 8 (c.1018_1024 dupGAGTTTG; Figure 2C), and such a duplication is predicted to change the reading frame and cause premature termination of the protein (p.V342GfsX44). Subsequently, mutation analysis was performed in the original DFNB35 family8 as well as in three other families of Pakistani origin (PKDF159, PKDF191, and PKDF327) segregating severe-to-profound hearing loss linked to the DFNB35 interval. In the affected individuals of the original DFNB35 family, a homozygous missense mutation (c.1024G→T; Figure 2D) was identified that is predicted to substitute a leucine residue for a valine residue (p.V342L). Also in the other DFNB35-linked families, missense mutations were identified. In family PKDF191, affected individuals were homozygous for an alanine-to-valine substitution (c.329C→T; p.A110V), whereas in family PKDF159, a leucine-to-proline change (c.959T→C; p.L320P) was detected (Figure 3). Similarly, in family PKDF327, a leucine-to-proline change (c.1040C→T; p.L347P) (Figure 3) was segregating and homozygous in the affected individuals. In all five families, the homozygous mutations cosegregated with hearing impairment and were not present in at least 120 ethnically matched normal-hearing individuals. In addition to the mutations described above, one other variant (c.1156C→T; p. P386S) was present homozygously in both the original DFNB35 family and family PKDF191. This variant, however, was also present in 9 out of 100 Pakistani control individuals, homozygously in two persons, and was, therefore, considered to be a polymorphism. All disease-causing variants detected in the various families and the maximum LOD scores indicating linkage to DFNB35 are presented in Table 2.
Figure 3.
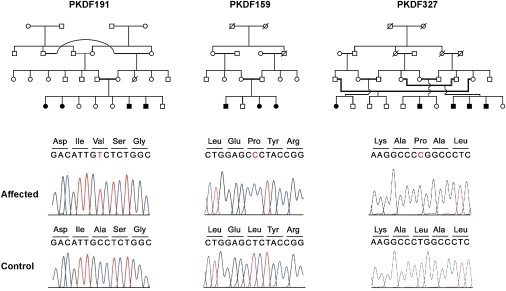
Mutation Analysis of ESRRB in Additional DFNB35 Families
Pedigrees of families PKDF191, PKDF159, and PKDF327 in which linkage to the DFNB35 locus was suggested. Sequence traces of ESRRB carrying the respective nucleotide substitutions c.329C→T, c.959T→C, and c.1040C→T from an affected and normal-hearing individual of these families are shown below the pedigrees. The predicted amino acid substitutions are p.A110V, p.L320P, and p.L347P, respectively.
Table 2.
Mutations in ESRRB that Cause Recessive Hearing Loss
| Family | Max. Two-Point LOD Score | Mutationa | Amino Acid Change | Predicted Effect on ESRRB |
|---|---|---|---|---|
| TR21 | 5.45 | c.1018_1024 dupGAGTTTG | p.V342GfsX44 | Frame-shift; premature truncation |
| Pedigree 2 (DFNB35) | 5.53 | c.1024G→T | p.V342L | Structural defect; ligand-binding domain |
| PKDF159 | 2.06 | c.959T→C | p.L320P | Structural defect; ligand-binding domain |
| PKDF191 | 2.75 | c.329C→T | p.A110V | Structure or function; DNA-binding domain |
| PKDF327 | 5.10 | c.1040C→T | p.L347P | Structural defect; ligand-binding domain |
Overview of the families presented in this study with mutations in ESRRB. Shown are the family name, maximum two-point LOD scores calculated for markers within the DFNB35 locus, the nucleotide change, and the predicted effect of the mutation at the amino acid level and on the ESRRB protein.
Nucleotide positions are numbered relative to the first nucleotide of the translation open reading frame found in RefSeq NM_004452.
For determination of the possible involvement of ESRRB mutations in the Turkish hearing-impaired population, 83 index patients from consanguineous families were genotyped for markers flanking the DFNB35 locus. Seven of these patients were found to be homozygous for these markers. The ESRRB gene was sequenced in these seven patients, as well as in 48 index patients from nonconsanguineous families. In one patient, a nonsynonymous change p.T389M (c.1166C→T) was identified in the heterozygous state and was not present in 120 ethnically matched control individuals. However, in this patient a second mutant allele of ESRRB was not found, and therefore, the disease-causing nature of this missense variant is uncertain.
Structural Analysis of the ESRRB Mutants
ESRRB encodes the estrogen-related receptor protein beta that is a member of the nuclear hormone receptor (NHR) family. One of the NHR subfamilies is composed of the estrogen receptors (ERs), the estrogen-related receptors (ESRRs), and the steroid receptors (SRs).21 In general, these proteins share a zinc finger C4 DNA-binding domain at their N terminus and a ligand-binding domain that is located more C terminally (Figure 4A). The main difference between these three groups is that the ESRRs are considered to be orphan receptors, whereas the ERs and SRs are regulated by molecules such as estrogens and glucocorticoids.22,23 Although no endogenous ligand has yet been identified for the ESRR proteins, the sequence of their ligand-binding domains are well conserved (Figure 4B). The ligand-binding domain of nuclear hormone receptors is a highly organized structure containing 12 α helices (Figures 4B and 4C).
Figure 4.

Molecular Modeling for ESRRB Missense Mutations
(A) Schematic representation of human ESRRB. The predicted zinc finger C4 DNA-binding domain (DBD) and the ligand-binding domain (LBD) are depicted in red and blue, respectively.
(B) Sequence comparison of the ligand-binding domains of five vertebrate ESRRB proteins, the two other human ESRR proteins ESRRA and ESRRG, and the human estrogen receptors ERA and ERB. Residues identical in all sequences are white on a black background, whereas similar amino acids are black on a light-gray background. Residues that are present in at least five of the proteins are indicated in white on a gray background. The missense mutations affecting amino acids in the ligand-binding domain are indicated by arrows. The positions of the various α helices in the ligand-binding domain are marked above the sequence. Species abbreviations are as follows: Hs, Homo sapiens; Mm, Mus musculus; Rn, Rattus norvegicus; Gg, Galus gallus; and Dr, Danio rerio. Accession numbers of the various human protein sequences: Hs ESRRB, NP_004443, Mm ESRRB, NP036064; Rn ESRRB, AAR89824; Gg ESRRB: XP001235147; Dr ESRRB: XP00133980; Hs ESRRA, AAH92470; Hs ESRRG, P62509; Hs ERA, AAI28575; and Hs ERB, NP_001035365.
(C) Molecular modeling of the ligand-binding domain of the human ESRRB protein. The structure was deduced from the known ESRRG structure.14 The various helices are presented by cylinders. The three amino acids that are affected by the missense mutations, marked by arrows in Figure 4B, are indicated in green.
(D) Graphic representation of the predicted effect of the p.L320P mutation. The leucine residue is depicted in green, whereas the side chain of the proline residue is presented in red. The absence of the leucine side chain results in the loss of hydrophic interactions with residues located in helix ten and eleven.
(E) Graphic representation of the predicted effect of the p.V342L mutation. The substitution of a leucine for a valine residue results in a larger side chain (leucine side chain is depicted in red; original valine residue in green) that does not fit into the molecular surface between helix eight and helix one, here presented as a grayish shade.
(F) Graphic representation of the predicted effect of the p.L347P mutation. The leucine residue is depicted in green, whereas the side chain of the proline residue is presented in red. The side chain of leucine 347 interacts with a number of other leucine residues (side chains in gray) located in helix nine. These interactions are lost by substituting proline for leucine. Images as presented in (C)–(F) were made with the YASARA program.18
The p.A110V change found in family PKDF191 is located in the DNA-binding domain of the ESRRB protein and is likely to disturb the structure of this domain (data not shown). The three other missense mutations identified in the Pakistani families substitute amino acids that are located in the ligand-binding domain of ESRRB. Leucine 320, replaced by a proline in family PKDF159 (p.L320P), is located in helix seven of this domain and conserved in vertebrate ESRRB proteins as well as in ESRRA and ESRRG (Figure 4B). In general, the introduction of proline residues within α helices reduces the stability of the helix, and therefore, this mutation will probably disrupt the structure of helix seven and the complete ligand-binding domain. In addition, the loss of the leucine side chain abrogates a number of hydrophobic interactions (Figure 4D). The valine residue that is replaced by a leucine in the original DFNB35 family (p.V342L) is less conserved. In the mouse ESRRB sequence, a methionine residue is present at this position, although in chicken and zebrafish ESRRB, the valine is conserved. In the other ESRR proteins and in ERA, the valine residue that is located within helix eight (Figure 4B) is also conserved. However, in ERB there is a leucine present in that position. The substitution of a leucine for this valine residue results in the occurrence of a somewhat larger side chain that bumps into the molecular surface of helix one (Figure 4E), and this substitution is predicted to reduce the strength of the interaction between helix one and eight. Finally, the leucine-to-proline change (p.L347P) in family PKDF327 again substitutes a proline for a leucine that is conserved in all ESRRB proteins and in human ESRRA and that is located in helix eight (Figure 4B). As mentioned for the p.L220P mutation, this leucine-to-proline change will reduce the stability of the α helix and disrupt the conformation of the ligand-binding domain. In addition, and as shown in Figure 4F, the leucine side chain is facing a number of other leucine residues that together form a tightly packed hydrophobic cluster. The presence of a proline residue at that position would also result in the loss of this cluster of hydrophobic interactions (Figure 4F). In summary, the molecular modeling data predict that mutations of ESRRB will result in conformational changes near the substituted amino acids or decreased helix stability and are therefore likely to affect the stability and function of the complete ligand-binding domain. A detailed overview of the molecular modeling is available (see Web Resources).
Cochlear Expression of the Different ESRRB Isoforms
Alternative splicing of the ESRRB gene gives rise to three major isoforms (Figure 5A) of which the distribution in various tissues has previously been studied.19 To determine the presence of these three isoforms in cochlear tissue, we performed RT-PCR analysis by using primers that specifically amplify the transcripts encoding the various isoforms. The transcript for the small isoform of ESRRB (ESRRBshort) was detected in all tissues examined. The transcript that lacks exon 10 (encoding ESRRB-Δ10) was mainly expressed in testis, kidney, cochlea, and retina, although some weak expression was observed in the other tissues examined. Interestingly, the transcript that contains all exons and encodes the ESRRBlong isoform was detected mainly in testis and cochlea, although a weak band was also observed in retina (Figure 5B). These data show that all isoforms are abundantly expressed in human cochlear tissue and that the ESRRBlong isoform appears to be preferentially expressed in cochlea and testis.
Figure 5.
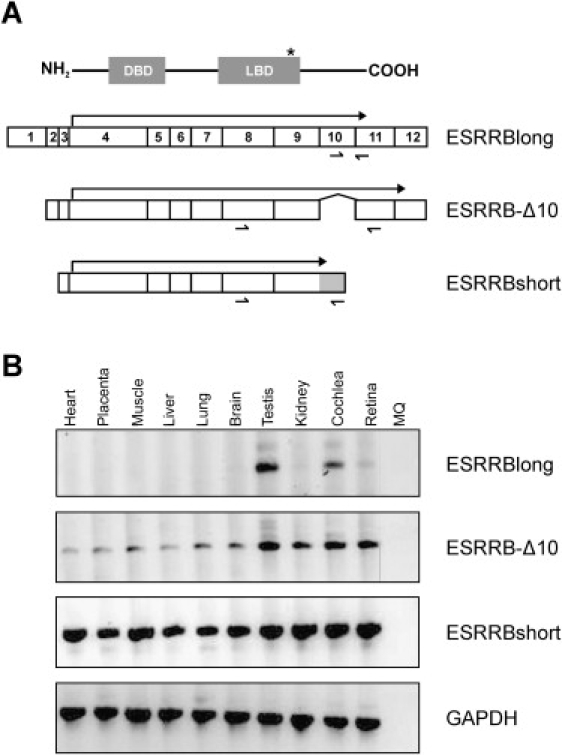
Tissue Distribution of ESRRB Isoforms
(A) Schematic overview of transcripts encoding the three ESRRB isoforms (exons are numbered). The protein corresponding to the long isoform is presented above the isoforms, showing that all isoforms contain both the DNA-binding domain (DBD) and the ligand-binding domain (LBD). Arrows indicate the protein coding regions in the three different transcripts. The gray part at the 3′ end of the ESRRBshort isoform represents an extension of exon 9 that was due to an alternative splicing event. Relative positions of the primers used for amplification of the various transcripts are indicated below the transcripts. The asterisk indicates the relative position of the epitope used for raising the antiserum for immunohistochemical analysis (see Figure 7).
(B) Tissue distribution of transcripts encoding the three different ESRRB isoforms. RT-PCR analysis was performed on total RNA from the various tissues. The expression of GAPDH (lower panel) was used as a control.
Expression of Esrrb during Inner-Ear Development
To characterize the expression of Esrrb during murine embryogenesis, we hybridized Esrrb-specific sense and antisense cRNA probes to sagitally cut embryo sections of various developmental stages. Whereas no signal was detected in any of the sections analyzed with the sense probe (data not shown), a strong signal in the antisense probed sections was visible in a restricted number of developing tissues. At embryonic day 12.5 (E12.5), staining was observed in the midbrain, the spinal cord, the metanephros, and the adrenal primordium (Figures 6A and 6B). In addition, a very specific signal was present in the region of the developing inner ear, both at E12.5 (Figure 6C) and E14.5 (Figure 6D). The observed staining in the midbrain and spinal cord remained throughout embryonic development, although signal was not detected in the developing kidney and adrenal gland after E14.5 (data not shown). At E16.5, Esrrb expression is present in the developing cochlea as well as the developing vestibular system (Figures 6E–6H). In addition, staining was observed in the vestibular ganglion (Figure 6H). Esrrb expression in the inner ear is even more prominent at E18.5 in the cochlear turns, the ampullae of the semicircular canals, the utricle, and the vestibular ganglion (Figures 6I–6L). To obtain more structural detail, a number of sections of an E18.5 embryo were counterstained with Nuclear Fast Red. Detailed images of the inner-ear region show specific staining in the developing stria vascularis in the cochlea (Figure 6M), a subset of nonsensory epithelial cells of the utricle and the ampullae in the vestibulum (Figures 6N and 6O), and part of the cells in the developing vestibular ganglion (Figure 6P).
Figure 6.
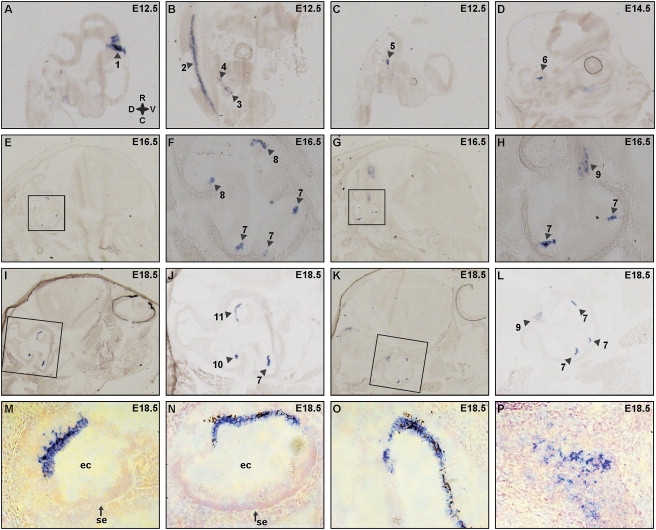
RNA In Situ Hybridization for Esrrb on Mouse Sagittal Sections
(A–C) At embryonic day E12.5, images of the medial side of the head (A), the body (B), and the lateral side of the head (C) are presented. Regions that show specific staining are indicated with arrowheads and are as follows: midbrain (1), mantle layer in the ventral side of the spinal cord (2), metanephros (3), adrenal primordium (4), and developing inner ear (5).
(D) At E14.5, staining in the developing inner ear (6).
(E–H) Serial sections of embryo heads at E16.5 in (E) and (G) with the corresponding inner-ear region enlarged in (F) and (H), respectively. Staining was observed in specific parts of the developing cochlea (7) and vestibulum (8) and the developing vestibular ganglion (9).
(I–L) Serial sections of embryo heads at E18.5 in (I) and (K) with the indicated inner-ear region enlarged in (J) and (L), respectively. Staining was present in the turns of the cochlea (7), in the ampulla (10) and the utricle (11) of the vestibulum, and in the vestibular ganglion (9).
(M–P) Detailed images of the inner ear at E18.5, showing expression in (M) the developing stria vascularis in the cochlea, (N) a subset of the nonsensory epithelial cells in the utricle, and (O) the nonsensory epithelium in the ampulla and in (P) cells of the vestibular ganglion. Sections were counterstained with Nuclear Fast Red for an increase of structural detail. The following abbreviations are used: ec, endolymphatic compartment; and se, sensory epithelia. In (A), the compass shows the orientation of the embryos in all panels; R, rostral; V, ventral; C, caudal; and D, dorsal.
In the eyes of adult mice (P90), a clear and strong signal was detected in the photoreceptor layer of the retina (data not shown), although during development, no signal was detected in the eye. This implies that the expression of Esrrb within a tissue is different when comparing developmental and adult stages. In summary, the in situ hybridization data show that Esrrb is highly expressed during inner-ear development in nonsensory epithelial cells of both the cochlea and the vestibulum. In addition, the gene is expressed in the developing vestibular ganglion.
ESRRB Expression in Inner Ear
For determination of the expression of Esrrb at the protein level, inner-ear sections derived from rats sacrificed at postnatal day four (P4) were incubated with antibodies directed against ESRRB, together with a set of markers. In the cochlea, ESRRB was found to be expressed in cells of mesothelial origin in the spiral limbus and the supporting cells. Moderate staining was also observed in Reissner's membrane, the stria vascularis, and the spiral ligament (Figures 7A and 7D). In addition, ESRRB was highly expressed in the nerve fibers and the spiral ganglion cells (Figure 7D). With an antibody against collagen type IV as a basement membrane marker24 (Figure 7B), it was shown that ESRRB is expressed on the mesothelial side of the basilar membrane (Figure 7C). Because ESRRB was found to be expressed in the nerve fibers and the spiral ganglion cells, an antibody directed against the heavy chain of neurofilaments (NFH) was used to specifically stain these structures25 (Figure 7E). This showed partial colocalization of NFH with ESRRB (Figure 7F). A magnified image of NFH expression in the organ of Corti shows that these nerve fibers protrude from the habenula perforata and contact the inner and outer hair cells at their basolateral side (Figure 7H).
Figure 7.
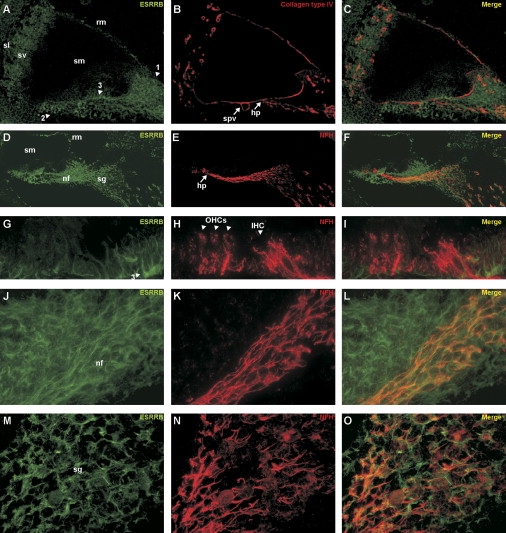
Immunohistochemical Analysis of ESRRB Expression in Inner Ear of P4 Rats
The relative position of the epitope used to raise the antiserum is depicted in Figure 5A.
(A–C) Overview of the cochlea. ESRRB expression is mainly observed in mesothelial cells of the spiral limbus (1) and the basilar membrane (2) and in the supporting cells of the organ of Corti (3). In addition, moderate expression was observed in the stria vascularis (sv), the spiral ligament (sl), and some cells of mesothelial origin in Reissner's membrane (rm). Collagen type IV staining shows the cochlear basement membranes; a merged picture shows that ESRRB is expressed on the mesothelial side of the basement membrane. The following abbreviations are used: sm, scala media; spv, spiral vessel; and hp, habenula perforata.
(D–F) Overview of the cochlea including the organ of Corti, nerve fibers, and the spiral ganglion. In addition to the expression described in (A), ESRRB is abundantly present in the nerve fibers (nf) as well as in the spiral ganglion cells (sg), whose structures are clearly stained by the antibody directed against neurofilament heavy chain NFH. The merged picture shows partial overlap of ESRRB and NFH in the nerve fibers and the spiral ganglion cells.
(G–I) Magnified image of ESRRB and NFH staining in the region of the organ of Corti. NFH is expressed at the base of outer (OHCs) and inner (IHC) hair cells, whereas ESRRB is clearly not expressed in those cells, but in supporting cells (3).
(J–L) Magnified image of ESRRB and NFH staining in the neurites that connect the hair cells to the spiral ganglia. ESRRB is expressed in the nerve fibers as well as in cells surrounding the nerve fibers, whereas NFH is only expressed in the neurons. The merged picture shows colocalization of ESRRB and NFH in the nerve fibers.
(M–O) Magnified image of ESRRB and NFH staining in a spiral ganglion. Both ESRRB and NFH are expressed in the spiral ganglion neurons and fibers, although their localization does not completely overlap.
No ESRRB expression was detected in the sensory cells in the organ of Corti, but only in the supporting cells (Figures 7G and 7I). The localization of ESRRB and NFH in the nerve fibers is depicted in more detail in Figures 7J–7L. ESRRB and NFH also are found to colocalize in the spiral ganglion neurons and fibers (Figures 7M–7O), although their distributions in these cells is not exactly the same (Figure 7O). In the ganglion cells of the vestibulum, a similar pattern of colocalization between ESRRB and NFH was observed (data not shown).
Discussion
In this study, genome-wide homozygosity mapping revealed a locus for nonsyndromic hearing impairment in an 18.7 cM interval on chromosome 14q24.3–q34.12, partly overlapping with the DFNB35 locus.8 One of the genes in the overlapping region was ESRRB. Sequence analysis of this gene revealed a 7 bp duplication in the affected members of family TR-21 (p.V342GfsX44), whereas four missense mutations in ESRRB were identified in the original DFNB35 family and three other families from Pakistan.
ESRRB is a member of the nuclear-hormone-receptor family of transcription factors. All family members share two functional domains, a DNA-binding domain and a ligand-binding domain. The 7 bp duplication in ESRRB of family TR-21 results in a frame shift and premature protein termination. Because the mutation is not in the last exon of the gene, the mRNA molecule might be a target for nonsense-mediated RNA decay.26 Alternatively, an aberrant protein might be present in the cells that would lack an intact ligand-binding domain and thus probably would not function properly. Whereas the substitution of a valine residue for a fully conserved alanine (p.A110V) affects the DNA-binding domain of ESRRB, the other missense mutations affect amino acids in the ligand-binding domain. The p.A110V does not affect a residue directly involved in DNA-contact, but it is likely to disrupt the 3D structure of the DNA-binding domain. Missense mutations in this domain of other nuclear hormone receptors are known to cause specific phenotypes such as androgen insensitivity syndrome27 and maturity-onset diabetes.28
The ligand-binding domain of estrogen-receptor-family members generally consists of twelve α helices, although the second helix is absent in some family members including ESRRB. The α helices fold together with a β sheet into a three-layered helical sandwich. Activation of a receptor depends on the binding of an agonist and one or more cofactors. The ligand-binding pocket is formed by the β sheet and helices three, five, six, and eleven. Upon ligand binding, helix twelve becomes repositioned into a groove between helix three and eleven, thereby forming a new groove for coactivator binding. In contrast, antagonist binding prevents helix twelve from relocating, after which corepressors occupy the available space in the groove between helix three and eleven and prevent activation of the receptor.29 Although the missense variants found in the ligand-binding domain do not affect the helices directly involved in ligand binding, the structural changes that occur near the substituted amino acids probably influence the structure or stability of the complete domain. In addition, leucine-to-proline changes within an α helix are likely to disrupt the helical conformation. The p.V342L substitution in helix eight is predicted to disrupt the interaction between helix eight and helix one and might impair the function of the ligand-binding domain. This is supported by other mutations, either in helix eight of the estrogen receptor30 or in helix one of the androgen receptor,31 that alter the binding affinity of the ligand. In addition, hydrophobic interactions between helix one and helix eight have been reported to be required for maintenance of the conformation of the receptor PPARγ,32 and it is has been suggested that these results can be extrapolated to other members of the nuclear receptor family, including the estrogen-related receptors.
Alternative splicing of ESRRB results in the occurrence of three isoforms,19 two of which were found to be ubiquitously expressed. Interestingly, the ESRRBlong isoform had a narrow tissue distribution and was abundant in cochlea and testis. These results are mainly in accordance with the observed distribution described by Zhou and others.19 Although the mutations found in this study affect all protein isoforms, it is tempting to speculate that the defect in ESRRBlong underlies the nonsyndromic hearing impairment. Because this isoform is also highly expressed in testis, one might expect male individuals to have altered testicular function. One affected male individual of family TR-21 (IV:5), however, has reproduced, indicating that affected males with DFNB35 can be fertile. On the other hand, many other genes that cause nonsyndromic hearing loss are abundantly expressed outside the cochlea, for instance DFNB3133 and PCDH15.34 Therefore, a dysfunction of the other two ESRRB isoforms might contribute to the phenotype.
During mouse embryogenesis, there is a very specific distribution pattern of Esrrb transcripts in the developing inner ear. Previously, RNA in situ hybridizations in zebrafish have shown transcripts of the ESRRB ortholog to be present in the diencephalon, the trigeminal ganglia, and in the anterior and posterior ganglia of the lateral line, which is structurally and functionally related to the mammalian inner ear. No expression was observed in the vestibular organ of the fish.35
Within the developing inner ear of the mouse, we found Esrrb expression in the nonsensory epithelia of the vestibulum, the developing stria vascularis, and lateral wall of the cochlea and in the vestibular ganglion. Very recently, Chen and Nathans36 showed a similar distribution of Esrrb postnatally, namely in the marginal cells of the stria vascularis in the cochlea and in analogous structures in the vestibular system, both by in situ hybridization and by immunohistochemistry. Our immunohistochemical analysis showed a broader expression of ESRRB within the early postnatal cochlea. All of the various cochlear cells that express ESRRB (supporting cells and mesothelial cells) are derived from nonsensory epithelium, and such a finding is consistent with the expression pattern observed during embryonic development. Both the RNA in situ hybridization and the immunohistochemistry show no expression of ESRRB in the developing cochlear hair cells. The more narrow localization of ESRRB in immunohistochemistry as presented by Chen and Nathans might be due to variations in experimental conditions and to the use of different antibodies.
In the affected individuals of family TR-21, TEOAEs were absent; this finding is indicative of compromised outer hair cell function. Because ESRRB is not expressed in these cells, the loss of outer hair cell function is probably a secondary effect. Although ESRRB is abundantly expressed in both the cochlea and the vestibulum during embryonic development in mice, the hearing-impaired individuals of the various families do not show symptoms of vestibular dysfunction. This suggests redundancy for ESRRB in the vestibulum but not in the cochlea.
From a single gene in the ancestor of bilaterian metazoans, two waves of duplication led to the three ESRR genes present in vertebrates, ESRRA, ESRRB, and ESRRG.37 All three paralogues play an important role, both during development and in adulthood. In vertebrates, we have the most knowledge of the function of ESRRA, which has been implicated in estrogen signaling and mammary-tumor biology,38 and the expression levels of both ESRRA and ESRRG are used as prognostic indicators in breast and ovarian cancer.39–41 Furthermore, ESRRA has been implicated in mitochondrial biogenesis and lipid metabolism.42,43 In zebrafish, the esrra receptor has been shown to control morphogenetic movements during gastrulation.44
Little is known about the role of ESRRB in humans. In mice, Esrrb is essential for normal placentation and exhibits a highly specific expression pattern in extra-embryonic tissue from 5.5 until 8.5 days postcoitum (dpc).45 From 11.5 until 16 dpc, Esrrb is expressed in primordial germ cells.46 Esrrb−/− mice die at approximately 10.5 dpc but can be rescued by aggregation with tetraploid wild-type cells.45,46 Rescued Esrrb null mice were fertile but, interestingly, exhibited problems with walking and showed circling and head-tossing behavior.46 The latter are indicative of an inner-ear dysfunction. Whereas the murine Esrrb is essential for placentation, apparently the human ortholog might only be required for proper hearing. Recently, Chen and Nathans confirmed the inner-ear phenotype of rescued Esrrb−/− mice by generating conditional Esrrb−/− mice.36 These studies revealed that ESRRB is essential for the development of the marginal cells and thereby for the development and function of the stria vascularis as a whole. Disturbed endolymph production in these mice results in an aberrant inner-ear fluid homeostasis and as a consequence in defective hearing and balance.
Although defects in ESRRB clearly affect cellular processes within the inner ear, the exact molecular mechanism by which ESRRB acts upon putative target genes remains elusive. ESRRs, including ESRRB, are implicated in mediating the effects of estrogens, thyroid hormone, and glucocorticoid hormone that are all important for inner-ear development and function. Thyroid hormone and its receptors TRHA and TRHB are essential for several aspects of cochlear development and maturation.47–50 Studies in Thra and Thrb knockout mice indicate that both receptors are involved in the development of the endocochlear potential, in which the stria vascularis is known to have a major role.51 Because ESRRB is suggested to influence the thyroid hormone pathway,52,53 the concomitant high expression of Esrrb in the developing stria vascularis suggests that ESRRB might mediate the effect of both thyroid receptors α and β in this region.
ESRRB and other ESRRs bind to both estrogen-responsive elements and steroid-responsive elements, and estrogen has a role in preservation of hearing during aging in human adults. Such a role is consistent with the decline in hearing observed in the estrogen receptor β knockout mouse.54 The early onset, severe-to-profound hearing loss in the DFNB35 families, however, seems not to be in agreement with the proposed role for estrogen in the maintenance of hearing.
Alterations in transcription mediated by the glucocorticoid receptor might also contribute to the phenotype of the present ESRRB mutations because the glucocorticoid receptor is, like ESRRB, widely expressed during and after maturation of the mouse and rat cochlea. In addition, ESRRB might repress transcriptional activity mediated by the glucocorticoid receptor.55
In conclusion, this study describes the identification of ESRRB as the gene involved in autosomal recessive hearing impairment DFNB35. The fact that the frame-shift mutation and the missense mutations result in a comparable clinical phenotype suggests that the molecular mechanism underlying the hearing impairment is similar and probably results from a loss of function. This is corroborated by the phenotype of the conditional Esrrb−/− mice. In these mice, defective hearing and balance coincide with disturbed endolymph production and ion homeostasis, and it is likely that human ESRRB is crucial for these processes as well. Further characterization of the exact molecular mechanisms by which genetic defects in ESRRB cause nonsyndromic hearing loss will contribute to the understanding of the processes that are essential for normal hearing and might therefore be of great importance for therapeutic intervention.
Web Resources
The URLs for data presented herein are as follows:
Marshfield Map, http://research.marshfieldclinic.org/genetics/home/index.asp
Molecular Modeling of ESRRB Mutations, http://www.cmbi.ru.nl/∼hvensela/estrogen
Mouse Genome Informatics (MGI), http://www.informatics.jax.org/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/sites/entrez?db=OMIM
UCSC Human Genome Database Build hg18, March 2006, http://www.genome.ucsc.edu
Accession Numbers
The GenBank accession number for human ESRRB reported in this paper is NM_004452.
Acknowledgments
We express gratitude to all the individuals who participated in this study and thank S. van der Velde-Visser, C. Beumer, O. Kucukoner, and Y. Ofluoglu for technical assistance. Furthermore, we gratefully acknowledge Dr. R. Wansink for providing mouse RNA, and Dr. D. Wu for helpful discussions regarding the in situ hybridizations. This study was financially supported by (1) the European Commission FP6 Integrated Project EUROHEAR, contract number: LSHG-CT-20054-512063; (2) the ARHI KP6 Project; contract number: QLRT-2001-00331; (3) the Heinsius Houbolt foundation; (4) the Karadeniz Technical University Research Fund, contract numbers: 2002.114.001.3 and 2006.114.001.1; (5) intramural funds 1 Z01 DC000035-10 from National Institute on Deafness and Other Communication Disorders/National Institutes of Health (NIDCD/NIH) to T.B.F.; (6) the Higher Education Commission (HEC), EMRO/WHO-COMSTECH and Ministry of Science and Technology (MoST), Islamabad, Pakistan; and (7) NIDCD/NIH grant DC03594. Genotyping services were provided by the Center for Inherited Disease Research (CIDR). CIDR is fully funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, contract number N01-HG-65403.
References
- 1.Morton C.C., Nance W.E. Newborn hearing screening–a silent revolution. N. Engl. J. Med. 2006;354:2151–2164. doi: 10.1056/NEJMra050700. [DOI] [PubMed] [Google Scholar]
- 2.Hildebrand M.S., de Silva M.G., Klockars T., Campbell C.A., Smith R.J., Dahl H.H. Gene expression profiling analysis of the inner ear. Hear. Res. 2007;225:1–10. doi: 10.1016/j.heares.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 3.Luijendijk M.W., van de Pol T.J., van Duijnhoven G., den Hollander A.I., ten Caat J., van Limpt V., Brunner H.G., Kremer H., Cremers F.P. Cloning, characterization, and mRNA expression analysis of novel human fetal cochlear cDNAs. Genomics. 2003;82:480–490. doi: 10.1016/s0888-7543(03)00150-2. [DOI] [PubMed] [Google Scholar]
- 4.Morton C.C. Gene discovery in the auditory system using a tissue specific approach. Am. J. Med. Genet. A. 2004;130:26–28. doi: 10.1002/ajmg.a.30049. [DOI] [PubMed] [Google Scholar]
- 5.Peters L.M., Belyantseva I.A., Lagziel A., Battey J.F., Friedman T.B., Morell R.J. Signatures from tissue-specific MPSS libraries identify transcripts preferentially expressed in the mouse inner ear. Genomics. 2007;89:197–206. doi: 10.1016/j.ygeno.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hawkins R.D., Bashiardes S., Powder K.E., Sajan S.A., Bhonagiri V., Alvarado D.M., Speck J., Warchol M.E., Lovett M. Large scale gene expression profiles of regenerating inner ear sensory epithelia. PLoS ONE. 2007;2:e525. doi: 10.1371/journal.pone.0000525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones C., Chen P. Planar cell polarity signaling in vertebrates. Bioessays. 2007;29:120–132. doi: 10.1002/bies.20526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ansar M., Din M.A., Arshad M., Sohail M., Faiyaz-Ul-Haque M., Haque S., Ahmad W., Leal S.M. A novel autosomal recessive non-syndromic deafness locus (DFNB35) maps to 14q24.1–14q24.3 in large consanguineous kindred from Pakistan. Eur. J. Hum. Genet. 2003;11:77–80. doi: 10.1038/sj.ejhg.5200905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller S.A., Dykes D.D., Polesky H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grimberg J., Nawoschik S., Belluscio L., McKee R., Turck A., Eisenberg A. A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic Acids Res. 1989;17:8390. doi: 10.1093/nar/17.20.8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindner T.H., Hoffmann K. easyLINKAGE: A PERL script for easy and automated two-/multi-point linkage analyses. Bioinformatics. 2005;21:405–407. doi: 10.1093/bioinformatics/bti009. [DOI] [PubMed] [Google Scholar]
- 12.Ahmed Z.M., Riazuddin S., Bernstein S.L., Ahmed Z., Khan S., Griffith A.J., Morell R.J., Friedman T.B., Riazuddin S., Wilcox E.R. Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am. J. Hum. Genet. 2001;69:25–34. doi: 10.1086/321277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwabe J.W., Chapman L., Finch J.T., Rhodes D. The crystal structure of the estrogen receptor DNA-binding domain bound to DNA: How receptors discriminate between their response elements. Cell. 1993;75:567–578. doi: 10.1016/0092-8674(93)90390-c. [DOI] [PubMed] [Google Scholar]
- 14.Greschik H., Wurtz J.M., Sanglier S., Bourguet W., van Dorsselaer A., Moras D., Renaud J.P. Structural and functional evidence for ligand-independent transcriptional activation by the estrogen-related receptor 3. Mol. Cell. 2002;9:303–313. doi: 10.1016/s1097-2765(02)00444-6. [DOI] [PubMed] [Google Scholar]
- 15.Vriend G. WHAT IF: A molecular modeling and drug design program. J. Mol. Graph. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. 29. [DOI] [PubMed] [Google Scholar]
- 16.Chinea G., Padron G., Hooft R.W., Sander C., Vriend G. The use of position-specific rotamers in model building by homology. Proteins. 1995;23:415–421. doi: 10.1002/prot.340230315. [DOI] [PubMed] [Google Scholar]
- 17.De Filippis V., Sander C., Vriend G. Predicting local structural changes that result from point mutations. Protein Eng. 1994;7:1203–1208. doi: 10.1093/protein/7.10.1203. [DOI] [PubMed] [Google Scholar]
- 18.Krieger E., Koraimann G., Vriend G. Increasing the precision of comparative models with YASARA NOVA–a self-parameterizing force field. Proteins. 2002;47:393–402. doi: 10.1002/prot.10104. [DOI] [PubMed] [Google Scholar]
- 19.Zhou W., Liu Z., Wu J., Liu J.H., Hyder S.M., Antoniou E., Lubahn D.B. Identification and characterization of two novel splicing isoforms of human estrogen-related receptor beta. J. Clin. Endocrinol. Metab. 2006;91:569–579. doi: 10.1210/jc.2004-1957. [DOI] [PubMed] [Google Scholar]
- 20.Knipper M., Zinn C., Maier H., Praetorius M., Rohbock K., Kopschall I., Zimmermann U. Thyroid hormone deficiency before the onset of hearing causes irreversible damage to peripheral and central auditory systems. J. Neurophysiol. 2000;83:3101–3112. doi: 10.1152/jn.2000.83.5.3101. [DOI] [PubMed] [Google Scholar]
- 21.Laudet V. Evolution of the nuclear receptor superfamily: Early diversification from an ancestral orphan receptor. J. Mol. Endocrinol. 1997;19:207–226. doi: 10.1677/jme.0.0190207. [DOI] [PubMed] [Google Scholar]
- 22.Ben-Or S. Development of responsiveness to glucocorticoid hormones. J. Steroid Biochem. 1983;19:305–314. [PubMed] [Google Scholar]
- 23.Rochefort H., Borgna J.L. Differences between oestrogen receptor activation by oestrogen and antioestrogen. Nature. 1981;292:257–259. doi: 10.1038/292257a0. [DOI] [PubMed] [Google Scholar]
- 24.Peters T.A., Kuijpers W., Tonnaer E.L., van Muijen G.N., Jap P.H. Distribution and features of melanocytes during inner ear development in pigmented and albino rats. Hear. Res. 1995;85:169–180. doi: 10.1016/0378-5955(95)00043-4. [DOI] [PubMed] [Google Scholar]
- 25.Kuijpers W., Tonnaer E.L., Peters T.A., Ramaekers F.C. Expression of intermediate filament proteins in the mature inner ear of the rat and guinea pig. Hear. Res. 1991;52:133–146. doi: 10.1016/0378-5955(91)90193-d. [DOI] [PubMed] [Google Scholar]
- 26.Frischmeyer P.A., Dietz H.C. Nonsense-mediated mRNA decay in health and disease. Hum. Mol. Genet. 1999;8:1893–1900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]
- 27.Jaaskelainen J., Mongan N.P., Harland S., Hughes I.A. Five novel androgen receptor gene mutations associated with complete androgen insensitivity syndrome. Hum. Mutat. 2006;27:291. doi: 10.1002/humu.9405. [DOI] [PubMed] [Google Scholar]
- 28.Oxombre B., Kouach M., Moerman E., Formstecher P., Laine B. The G115S mutation associated with maturity-onset diabetes of the young impairs hepatocyte nuclear factor 4alpha activities and introduces a PKA phosphorylation site in its DNA-binding domain. Biochem. J. 2004;383:573–580. doi: 10.1042/BJ20040473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shiau A.K., Barstad D., Loria P.M., Cheng L., Kushner P.J., Agard D.A., Greene G.L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 30.Eng F.C., Lee H.S., Ferrara J., Willson T.M., White J.H. Probing the structure and function of the estrogen receptor ligand binding domain by analysis of mutants with altered transactivation characteristics. Mol. Cell. Biol. 1997;17:4644–4653. doi: 10.1128/mcb.17.8.4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Belsham D.D., Pereira F., Greenberg C.R., Liao S., Wrogemann K. Leu-676-Pro mutation of the androgen receptor causes complete androgen insensitivity syndrome in a large Hutterite kindred. Hum. Mutat. 1995;5:28–33. doi: 10.1002/humu.1380050104. [DOI] [PubMed] [Google Scholar]
- 32.Holt J.A., Consler T.G., Williams S.P., Ayscue A.H., Leesnitzer L.M., Wisely G.B., Billin A.N. Helix 1/8 interactions influence the activity of nuclear receptor ligand-binding domains. Mol. Endocrinol. 2003;17:1704–1714. doi: 10.1210/me.2003-0001. [DOI] [PubMed] [Google Scholar]
- 33.Van Wijk E., Van der Zwaag B., Peters T., Zimmermann U., Te Brinke H., Kersten F.F., Marker T., Aller E., Hoefsloot L.H., Cremers C.W. The DFNB31 gene product whirlin connects to the Usher protein network in the cochlea and retina by direct association with USH2A and VLGR1. Hum. Mol. Genet. 2006;15:751–765. doi: 10.1093/hmg/ddi490. [DOI] [PubMed] [Google Scholar]
- 34.Murcia C.L., Woychik R.P. Expression of Pcdh15 in the inner ear, nervous system and various epithelia of the developing embryo. Mech. Dev. 2001;105:163–166. doi: 10.1016/s0925-4773(01)00388-4. [DOI] [PubMed] [Google Scholar]
- 35.Bardet P.L., Schubert M., Horard B., Holland L.Z., Laudet V., Holland N.D., Vanacker J.M. Expression of estrogen-receptor related receptors in amphioxus and zebrafish: Implications for the evolution of posterior brain segmentation at the invertebrate-to-vertebrate transition. Evol. Dev. 2005;7:223–233. doi: 10.1111/j.1525-142X.2005.05025.x. [DOI] [PubMed] [Google Scholar]
- 36.Chen J., Nathans J. Estrogen-related receptor beta/NR3B2 controls epithelial cell fate and endolymph production by the stria vascularis. Dev. Cell. 2007;13:325–337. doi: 10.1016/j.devcel.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 37.Bardet P.L., Laudet V., Vanacker J.M. Studying non-mammalian models? Not a fool's ERRand! Trends Endocrinol. Metab. 2006;17:166–171. doi: 10.1016/j.tem.2006.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giguere V. To ERR in the estrogen pathway. Trends Endocrinol. Metab. 2002;13:220–225. doi: 10.1016/s1043-2760(02)00592-1. [DOI] [PubMed] [Google Scholar]
- 39.Ariazi E.A., Clark G.M., Mertz J.E. Estrogen-related receptor alpha and estrogen-related receptor gamma associate with unfavorable and favorable biomarkers, respectively, in human breast cancer. Cancer Res. 2002;62:6510–6518. [PubMed] [Google Scholar]
- 40.Suzuki T., Miki Y., Moriya T., Shimada N., Ishida T., Hirakawa H., Ohuchi N., Sasano H. Estrogen-related receptor alpha in human breast carcinoma as a potent prognostic factor. Cancer Res. 2004;64:4670–4676. doi: 10.1158/0008-5472.CAN-04-0250. [DOI] [PubMed] [Google Scholar]
- 41.Sun P., Sehouli J., Denkert C., Mustea A., Konsgen D., Koch I., Wei L., Lichtenegger W. Expression of estrogen receptor-related receptors, a subfamily of orphan nuclear receptors, as new tumor biomarkers in ovarian cancer cells. J. Mol. Med. 2005;83:457–467. doi: 10.1007/s00109-005-0639-3. [DOI] [PubMed] [Google Scholar]
- 42.Huss J.M., Torra I.P., Staels B., Giguere V., Kelly D.P. Estrogen-related receptor alpha directs peroxisome proliferator-activated receptor alpha signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol. Cell. Biol. 2004;24:9079–9091. doi: 10.1128/MCB.24.20.9079-9091.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mootha V.K., Handschin C., Arlow D., Xie X., St P.J., Sihag S., Yang W., Altshuler D., Puigserver P., Patterson N. Erralpha and Gabpa/b specify PGC-1alpha-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc. Natl. Acad. Sci. USA. 2004;101:6570–6575. doi: 10.1073/pnas.0401401101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bardet P.L., Horard B., Laudet V., Vanacker J.M. The ERRalpha orphan nuclear receptor controls morphogenetic movements during zebrafish gastrulation. Dev. Biol. 2005;281:102–111. doi: 10.1016/j.ydbio.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 45.Luo J., Sladek R., Bader J.A., Matthyssen A., Rossant J., Giguere V. Placental abnormalities in mouse embryos lacking the orphan nuclear receptor ERR-beta. Nature. 1997;388:778–782. doi: 10.1038/42022. [DOI] [PubMed] [Google Scholar]
- 46.Mitsunaga K., Araki K., Mizusaki H., Morohashi K., Haruna K., Nakagata N., Giguere V., Yamamura K., Abe K. Loss of PGC-specific expression of the orphan nuclear receptor ERR-beta results in reduction of germ cell number in mouse embryos. Mech. Dev. 2004;121:237–246. doi: 10.1016/j.mod.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 47.Bryant J., Goodyear R.J., Richardson G.P. Sensory organ development in the inner ear: Molecular and cellular mechanisms. Br. Med. Bull. 2002;63:39–57. doi: 10.1093/bmb/63.1.39. [DOI] [PubMed] [Google Scholar]
- 48.Forrest D., Reh T.A., Rusch A. Neurodevelopmental control by thyroid hormone receptors. Curr. Opin. Neurobiol. 2002;12:49–56. doi: 10.1016/s0959-4388(02)00289-1. [DOI] [PubMed] [Google Scholar]
- 49.Sendin G., Bulankina A.V., Riedel D., Moser T. Maturation of ribbon synapses in hair cells is driven by thyroid hormone. J. Neurosci. 2007;27:3163–3173. doi: 10.1523/JNEUROSCI.3974-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Winter H., Braig C., Zimmermann U., Engel J., Rohbock K., Knipper M. Thyroid hormone receptor alpha1 is a critical regulator for the expression of ion channels during final differentiation of outer hair cells. Histochem. Cell Biol. 2007;128:65–75. doi: 10.1007/s00418-007-0294-6. [DOI] [PubMed] [Google Scholar]
- 51.Rusch A., Ng L., Goodyear R., Oliver D., Lisoukov I., Vennstrom B., Richardson G., Kelley M.W., Forrest D. Retardation of cochlear maturation and impaired hair cell function caused by deletion of all known thyroid hormone receptors. J. Neurosci. 2001;21:9792–9800. doi: 10.1523/JNEUROSCI.21-24-09792.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Castet A., Herledan A., Bonnet S., Jalaguier S., Vanacker J.M., Cavailles V. Receptor-interacting protein 140 differentially regulates estrogen receptor-related receptor transactivation depending on target genes. Mol. Endocrinol. 2006;20:1035–1047. doi: 10.1210/me.2005-0227. [DOI] [PubMed] [Google Scholar]
- 53.Xie W., Hong H., Yang N.N., Lin R.J., Simon C.M., Stallcup M.R., Evans R.M. Constitutive activation of transcription and binding of coactivator by estrogen-related receptors 1 and 2. Mol. Endocrinol. 1999;13:2151–2162. doi: 10.1210/mend.13.12.0381. [DOI] [PubMed] [Google Scholar]
- 54.Hultcrantz M., Simonoska R., Stenberg A.E. Estrogen and hearing: A summary of recent investigations. Acta Otolaryngol. 2006;126:10–14. doi: 10.1080/00016480510038617. [DOI] [PubMed] [Google Scholar]
- 55.Trapp T., Holsboer F. Heterodimerization between mineralocorticoid and glucocorticoid receptors increases the functional diversity of corticosteroid action. Trends Pharmacol. Sci. 1996;17:145–149. doi: 10.1016/0165-6147(96)81590-2. [DOI] [PubMed] [Google Scholar]