

Abstract
Autism is a neurodevelopmental disorder of complex etiology in which genetic factors play a major role. We have implicated the neurexin 1 (NRXN1) gene in two independent subjects who display an autism spectrum disorder (ASD) in association with a balanced chromosomal abnormality involving 2p16.3. In the first, with karyotype 46,XX,ins(16;2)(q22.1;p16.1p16.3)pat, NRXN1 is directly disrupted within intron 5. Importantly, the father possesses the same chromosomal abnormality in the absence of ASD, indicating that the interruption of α-NRXN1 is not fully penetrant and must interact with other factors to produce ASD. The breakpoint in the second subject, with 46,XY,t(1;2)(q31.3;p16.3)dn, occurs ∼750 kb 5′ to NRXN1 within a 2.6 Mb genomic segment that harbors no currently annotated genes. A scan of the NRXN1 coding sequence in a cohort of ASD subjects, relative to non-ASD controls, revealed that amino acid alterations in neurexin 1 are not present at high frequency in ASD. However, a number of rare sequence variants in the coding region, including two missense changes in conserved residues of the α-neurexin 1 leader sequence and of an epidermal growth factor (EGF)-like domain, respectively, suggest that even subtle changes in NRXN1 might contribute to susceptibility to ASD.
Main Text
Autism and related developmental disabilities, clinically referred to as autism spectrum disorders (ASDs; MIM 209850), affect up to ∼1 in 150 children1 and include language impairment, difficulties in social interaction and communication, a restricted pattern of interests, unusual behaviors, and/or stereotyped and repetitive motor mannerisms. ASDs are likely to result from a complex interaction of genetic factors, environment, experience, and, possibly, chance. Most readily analyzed are the genes because the fully sequenced human genome presents a finite universe to explore for contributing factors. ASDs are among the most heritable behavioral disorders, on the basis of familial relative risk and twin studies.2 Although several monogenic disorders might involve autism-like symptoms, their relevance to most cases of ASD is not clear. Genetic-linkage studies have suggested a few novel chromosomal locations for ASD genes, but the richest source of candidates might lie in chromosomal abnormalities that target particular genomic segments.3–8 Cytogenetic abnormalities occur in >5% of ASD cases and, although some regions, such as 2q37, 15q11–q13, and 22q13.3, are seen recurrently, the Autism Chromosome Rearrangement Database4 currently reports 391 visible chromosomal breakpoints spread throughout the genome. Recent application of higher-resolution techniques has revealed frequent submicroscopic genomic abnormalities and documented a significant increase in de novo copy-number changes, suggesting that many different genomic disruptions might predispose to ASD.6–8 These genetic analyses are consistent with the view that many different genes contribute, possibly leading by different means to a final common neurodevelopmental pathway that produces the autism phenotype. ASD genes are likely to involve both common variants that contribute broadly to susceptibility and rare variants that contribute in fewer individuals but might have stronger effect. Either could provide fundamental insights into the mechanism(s) leading to ASD.
In the Developmental Genome Anatomy Project (DGAP), we have sought genes of developmental importance disrupted by apparently balanced chromosomal aberrations.9–12 We have focused where possible on “double-hit” cases, in which independent breakpoints in two individuals with related phenotypes occur in the same chromosomal region, maximizing the likelihood that the chromosomal disruption is causative. The independent occurrence among our first 200 DGAP cases, encompassing a wide variety of clinical phenotypes, of two subjects displaying ASD in association with a chromosomal breakpoint in 2p16.3 has revealed NRXN1 (MIM 600565) as an autism-associated gene, consistent with recent reports of both de novo heterozygous deletion of NRXN1 and rare sequence variants in the β-NRXN1 leader sequence in ASD.8,13
Two DGAP subjects with an ASD phenotype, DGAP123 and DGAP200, display chromosomal breakpoints in 2p16.3: DGAP123 is a female subject with a familial rearrangement, 46,XX,ins(16;2)(q22.1;p16.1p16.3)pat, and DGAP200 is a male with a de novo apparently balanced translocation, 46,XY,t(1;2)(q31.3;p16.3)dn (Figure 1). Blood samples were obtained from both and from the parents of DGAP123 for preparation of DNA and EBV-transformed lymphoblastoid cell lines.14 All human studies were performed under informed consent protocols approved by the Partners HealthCare System Human Research Committee (Boston, MA) or the Yale University School of Medicine (New Haven, CT).
Figure 1.
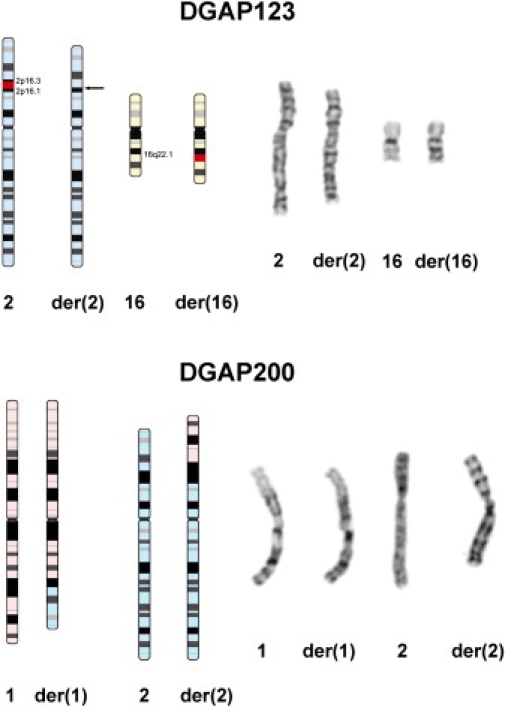
Balanced Chromosome Rearrangements in DGAP123 and DGAP200
The top panel shows an ideogram (excised/inserted region in red) and composite chromosomes for the DGAP123 rearrangement [46,XX,ins(16;2)(q22.1;p16.1p16.3)pat]. The bottom panel shows an ideogram and composite chromosomes for translocation in DGAP200 [46,XY,t(1;2)(q31.3;p16.3)dn].
The detailed phenotypes of DGAP123, her mother (DGAP123-2), and her father (DGAP124) were determined by diagnostic instruments, behavioral questionnaires, and neuropsychological assessment in the Psychiatric and Neurodevelopmental Genetics Unit (PNGU) of the Center for Human Genetic Research, Massachusetts General Hospital. This was not possible for DGAP200, whose phenotypic information and diagnoses were provided by communication with the clinician. DGAP123 meets criteria for autism on both the ADOS and ADI-R, with manifestations that include ritualized behaviors, vocal and motor mannerisms, limited eye contact, minimal verbal output, little change in affect or facial expression, and minimal seeking of interaction.15,16 Her overall level of functioning falls within the range of mental retardation. Neither parent meets formal criteria for autism, although each has minor functional abnormalities. The parents' overall intellectual abilities are in the normal range, although the mother exhibits a relative deficit in working memory. The father, who shares the chromosome rearrangement with his daughter, displayed stuttering as a child, has received speech services in the past, and has some persistent difficulty in articulation. He also exhibits some features of obsessive compulsive disorder (OCD; MIM 143465) and attention deficit disorder (ADHD; MIM 143465), although he has never received a formal diagnosis of either. DGAP200 has diagnoses of PDD-NOS (MIM 209850), along with ADHD, conduct disorder with early onset, and intermittent explosive disorder.
The chromosomal abnormality in DGAP123 involves the excision of ∼8.9 Mb of DNA between 2p16.1 and 2p16.3 and its insertion into 16q22.1 (Figure 1). By array comparative genomic hybridization (array CGH; 244K Human Oligo chip, Agilent Technologies, Palo Alto CA), this anomaly is balanced, showing no significant loss or gain of copy number at either site or elsewhere in the genome. FISH with whole BAC clones, chosen from NCBI and UCSC websites and obtained from CITB-D and RP11 libraries (Invitrogen, San Diego, CA, and the Children's Hospital of Oakland Research Institute, CA), mapped the 2p16 breakpoints within BAC clones RP11-391D19 (Figure 2) and RP11-579K4. Southern-blot analysis in DGAP123 narrowed the breakpoint to a 1368 bp region (Figure 3) in intron 5 of the large NRXN1 gene, which spans 1.1 Mb and encodes α-neurexin 1 from 24 exons and β-neurexin 1 from seven exons (Figure 4). Because each isoform is produced from a different promotor and has a different first exon,17,18 the DGAP123 breakpoint directly disrupts the α-NRXN1 sequence but leaves β-NRXN1 coding sequences intact.
Figure 2.
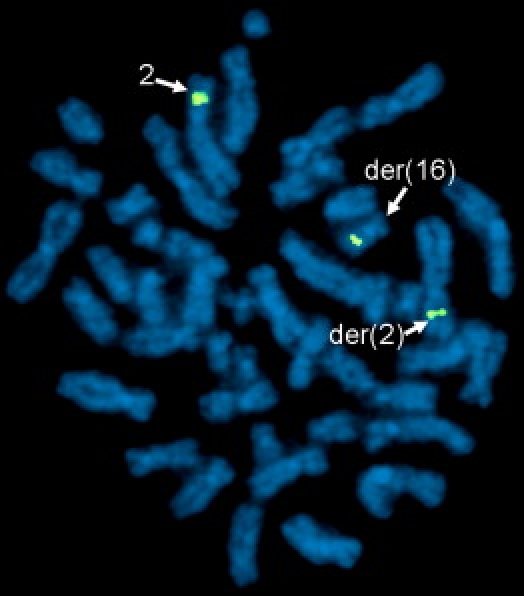
Identification of BAC Clone Crossing the DGAP123 2p16.3 Breakpoint
FISH analysis of DGAP123 with 2p16.3 BAC clone RP11-391D19 (green) shows hybridization to both the der(2) and der(16) chromosomes, indicating that the breakpoint of the insertion is located within the sequence of this genomic clone.
Figure 3.
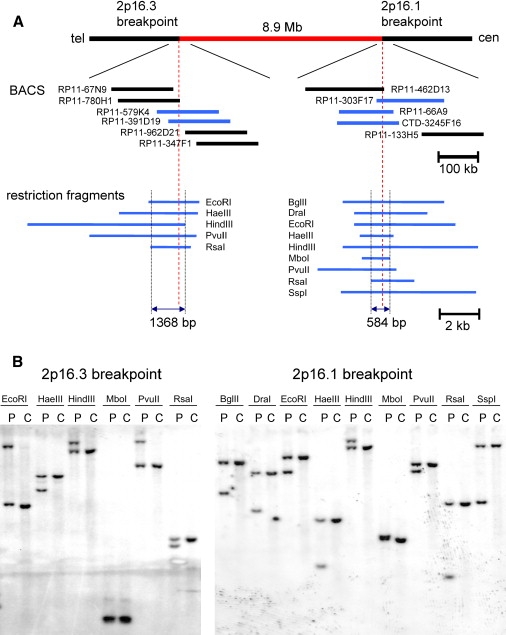
Mapping of the 2p16.3 and 2p16.1 Breakpoints of DGAP123
(A) The mapping of the two chromosome 2 breakpoints (dashed red vertical lines) at the edges of the 8.9 Mb of DNA (shown in red) excised and inserted into chromosome 16. Below the map schematic are BAC clones that in FISH experiments detect both der(2) and der(16) (blue lines) or only one of the derivative chromosomes (black lines). Below the BACs are shown restriction fragments used in DNA-blotting experiments (see [B]) to confine the breakpoints to small segments of 2p16.3 and 2p16.1, respectively.
(B) Genomic DNA blots hybridized with probes from the 2p16.3 breakpoint region (left) and the 2p16.1 breakpoint region (right), respectively. Each lane contains genomic DNA digested with the designated restriction enzyme from either DGAP123 (P) or a normal control (C). Additional bands in the P lanes indicate novel restriction fragments generated by the interchromosomal exchange. The hybridization probes P258C and P328, which detected aberrant bands containing breakpoints at 2p16.3 and 2p16.1, respectively, were amplified using the following primer pairs:
P258C: 5′-ATGTCTGATATTATAAGGTGAAACTCCGGTCTTCC-3′ and
5′-CAAGTCCTGTGTTGCTATATAGCGAATTTGTCTG-3′;
P328: 5′-CTGTTTTCTTCTCTCACTATATGAGTTGAACATATACAAATAGGC-3′ and
5′-GGAAGTGGAAAGCTGCTGTTTCTCAGCCATTGCTCA-3′.
Figure 4.
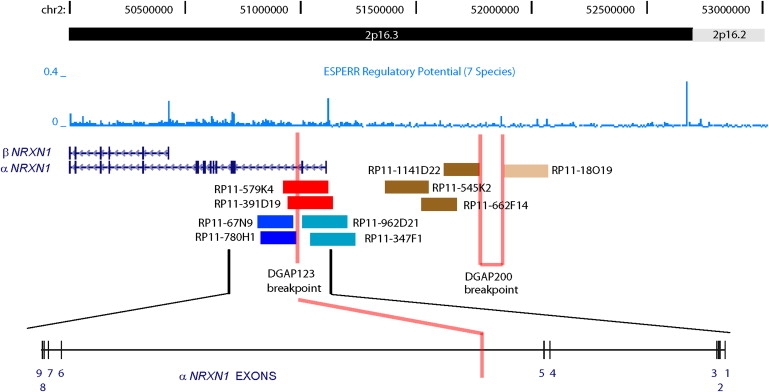
NRXN1 Region of 2p16.3 and DGAP Subject Breakpoints
This schematic diagram, reworked from tracks provided by the UCSC Genome Browser, shows exon locations and transcript orientation for α-NRXN1 and the overlapping β-NRXN1, along with the upstream region devoid of known genes, below a black bar indicating 2p16.3 and base-pair locations from the telomeric side (left) toward the centromere (right). The blue graph between these shows the estimated regulatory potential (0–0.4) calculated by comparing frequencies of short alignment patterns between known regulatory elements and neutral DNA across seven species (human, chimpanzee, macaque, mouse, rat, dog, and cow).19 Regulatory potential is highest at the recognized α-NRXN1 and β-NRXN1 promotors and especially at an anonymous site more than 1 Mb upstream of NRXN1. Red lines indicate the position in α-NRXN1 intron 5 and approximate position upstream of NRXN1 for the DGAP123 and DGAP200 breakpoints, respectively. FISH-mapped BAC clones crossing the former are shown in red, with selected clones mapping to der(2) and der(16) shown in dark blue and teal, respectively. For the latter, FISH-mapped BAC clones mapping to der(1) and der(2) are shown in dark brown and light brown, respectively.
The 2p16.1 breakpoint in DGAP123 was mapped to a 584 bp segment within BAC clones RP11-303F17, RP11-66A9, and CTD-3245F16 (Figure 3), in a gene desert flanked by BCL11A (MIM 606557) and FANCL (MIM 608111), located 589 kb proximal and 1.6 Mb distal, respectively. The chromosome 16 breakpoint was flanked, by RP11-52E21 and RP11-311C24, but not further delineated in a 325 kb segment of 16q22.1 that contains the genes SNTB2 (basic beta 2 syntrophin; MIM 600027), VPS4A (vacuolar-protein-sorting factor 4A; MIM 609982), PDF (peptide deformylase, mitochondrial precursor), COG8 (oligomeric golgi complex component 8; MIM 606979), NIP7 (60S ribosome subunit biogenesis protein NIP7 homolog), TMED6 (transmembrane emp24 domain-containing protein 6 precursor), TERF2 (telomeric repeat binding factor 2; MIM 602027), CYB5B (cytochrome b5 outer mitochondrial membrane isoform; MIM 602027), and NFAT5 (nuclear factor of activated T cells 5 isoform a; MIM 604708).
For DGAP200, FISH mapping placed the 2p16.3 breakpoint between BAC clones RP11-1141D22 and RP11-18O19, ∼750 kb from exon 1 of α-NRXN1, within a 2.6 Mb upstream segment that is devoid of annotated genes (Figure 4). FISH studies localized the chromosome 1 breakpoint between RP11-25C18 and RP11-173E24 in a gene desert with no genes within 1 Mb on either side. Array CGH analysis of DGAP200 genomic DNA revealed a cryptic deletion of ∼534 kb within 2p16.2, 3 Mb proximal to NRXN1, removing the genes ACYP2 (muscle-type acylphosphatase 2; MIM 102595) and TSPYL6 (Testis-specific protein Y-linked-like 6), FLJ40298 (a hypothetical protein), and SPTBN1 (spectrin, beta, nonerythrocytic 1 isoform 1; MIM 182790). It is conceivable that the deletion of one or more of these loci could contribute to the severe behavioral phenotype of DGAP200.
The gene desert upstream of NRXN1 contains sites predicted by ESPERR to have strong regulatory potential on the basis of comparative genomics, in seven species, that used a combination of conservation, composition and short-pattern structure information (Figure 4).19 This suggested that the DGAP200 translocation, although not directly truncating NRXN1, might still disrupt its expression, perhaps by separating a long-range regulatory element from the coding portions of the gene. Because NRXN1 expression has been studied previously mainly in neuronal tissue, we first determined whether the gene is expressed in lymphoblastoid cells, the only tissue readily available from DGAP subjects. We used RT-PCR of RNA from control lymphoblasts to amplify portions of the α-NRXN1 mRNA, as follows: exons 1–2, exons 2–7, exons 7–9, exons 12–14, exons 20–22 (exons 3–5 of β-NRXN1) and exons 22–24 (exons 5–7 of β-NRXN1), as well as exons 1–5 of β-NRXN1. A typical result is shown in Figure 5. All primer pairs produced an appropriately spliced product (albeit at much lower abundance) of expected size and correct DNA sequence compared to brain mRNA, except α-NRXN1 exons 1–2, exons 2–7 and β-NRXN1 exons 1–5, suggesting that while neurexin 1 is expressed, the precise equivalent of neither neuronal α- nor β-neurexin 1 is present. Rather, any neurexin 1 isoform expressed in lymphoblasts is apparently produced from an mRNA that remains to be fully delineated but that does not share a 5′ end with either the brain α or β isoforms.
Figure 5.
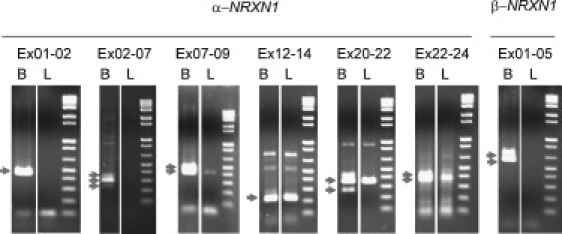
RT-PCR Amplification of NRXN1 mRNA from Lymphoblastoid Cells
Each panel shows the results of amplification of reverse-transcribed mRNA from human brain (B) and control lymphoblastoid cells (L), with primers designed to amplify products of known size and sequence (available upon request), based upon the established brain mRNA sequences of α- and β-NRXN1. The third lane in each panel is a standard marker with band sizes in bp from smallest to largest of 100, 200, 300, 400, 500, 650, 850, and 1,000, in the relevant resolving range. Arrows indicate the expected sizes of PCR products based on known splice variants from brain. Any additional bands not marked by arrows are PCR artifacts, corroborated by DNA sequencing. The lymphoblastoid cell mRNA produced matching NRXN1 products for at least one expected splice product for each of the primer pairs Ex07-09, Ex12-14, Ex20-22 and Ex22-24 (this product is equivalent to β-NRXN1 Ex05-07), but no PCR products for Ex01-02, Ex02-07, or β-NRXN1 Ex01-05. Matching PCR products from the lymphoblastoid cell mRNA were confirmed as the expected NRXN1 products by DNA sequencing.
NRXN1 mRNA was detected in all control lymphoblasts tested, as well as in cell lines from DGAP123 and family members and from DGAP200, but variation in the levels of individual RT-PCR products from different exons and mRNA preparations precluded reproducible quantitation. To test for an effect of the DGAP200 translocation, we capitalized on a polymorphism in the NRXN1 3′ untranslated region in which alleles possess either one or two consecutive copies of a 4 bp TTAC stretch. Control heterozygotes showed expression of both allelic variants in lymphoblastoid cell RNA. By contrast, whereas DGAP200 genomic DNA showed heterozygosity, only one allelic variant was present in the corresponding RNA (Figure 6).
Figure 6.
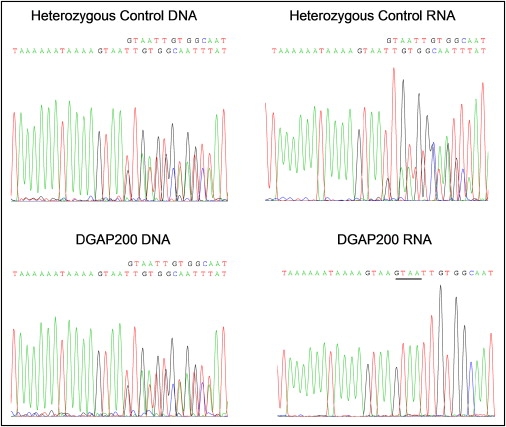
Expression of NRXN1 mRNA from One Allele in DGAP200
Sequence traces for a region of the 3′ UTR of NRXN1 surrounding a four-nucleotide duplication polymorphism are shown for genomic DNA (left) or RNA (right) of lymphoblastoid cells from an unaffected control (top) or DGAP200 (bottom). Both the control and DGAP200 are heterozygous for alleles containing either one or two copies, respectively, of the TTAC polymorphism described in the text, shown here as the reverse strand (GTAA). This introduces mixed sequence after the first copy of the repeat, shown above the traces. The RNA from the control line shows evidence of expression of both alleles, whereas the DGAP200 samples shows expression only of the allele containing two copies of the repeat (second copy underlined). Primers used for genomic DNA were from exon 24, as follows: F: 5′-ATAGCTCTCTGGTATTCAGTG-3′ and R: 5′-TCCAGAAATGTTCATCATG-3′, whereas those mRNA were chosen in exons 23 and 24 to ensure no interference from unspliced RNA or DNA contamination: 4nt-ins F; 5′-AGGACATTGACCCCTGTGAG-3′ and 4nt-ins R; 5′-TGCAACAGAATGAAGGCTGTA-3′. PCR products were isolated by 1% agarose gel and purified with MiniElute Gel Extraction kit (QIAGEN, Hilden, Germany). Extracted DNAs were sequenced with ABI3730XL DNA Analyzer.
We also performed western-blot analyses of protein lysates from cultured lymphoblastoid cell lines of DGAP123, her mother (DGAP123-2), her father (DGAP124), DGAP200, and normal controls with a neurexin-1-specific antibody directed against the carboxyl-terminal region expected to be expressed from our mRNA studies. Figure 7 shows typical results. A specific neurexin 1 band at ∼82 kDa was detected reproducibly in all samples. This band is not seen in brain-tissue extracts, in which α-NRXN1 and β-NRXN11 products migrate at 160–200 kDa and 90–100 kDa, respectively, depending on alternative splicing and glycosylation. The ∼82 kDa band was consistently and significantly reduced in intensity in lysates from DGAP123 and from her father, DGAP124, relative to the proband's mother, DGAP123-2, and to normal controls (Figure 7). Interestingly, DGAP200 also displayed significantly reduced expression of this neurexin 1 band, consistent with a position effect of the upstream breakpoint. The precise structure of this ∼82 kDa neurexin 1 isoform relative to the neuronal isoforms and its extent of glycosylation remain to be determined by more detailed studies.
Figure 7.
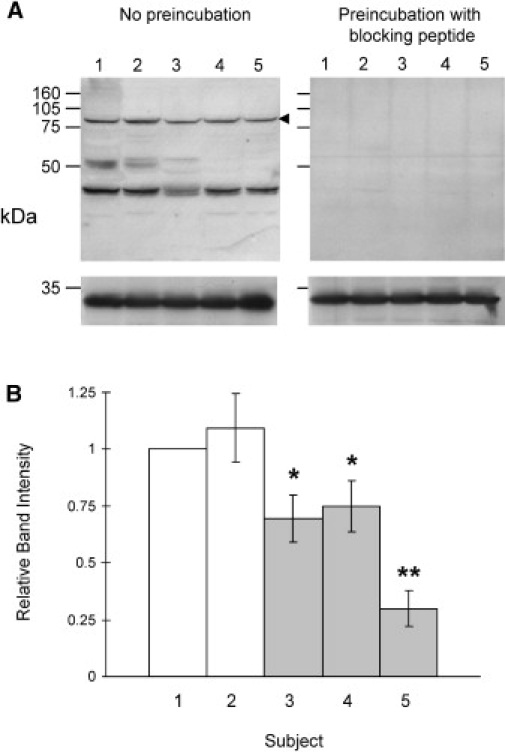
Expression of Neurexin 1 in DGAP Subjects and Control Lymphoblasts
(A) Lymphoblastoid whole-cell protein extracts (prepared by lysis in RIPA buffer containing protease inhibitor mixture [Roche Applied Science, Indianapolis IN], 1 mM PMSF): GUS10928 control (lane 1), DGAP123-2 (lane 2), DGAP124 (lane 3), DGAP200 (lane 4), and DGAP123 (lane 5) were probed on western blots with a C-terminal neurexin 1 antibody (top panel: P-15, Santa Cruz Biotechnology, Santa Cruz, CA, USA), preincubated without or with specific blocking peptide. These same blots were also probed with a glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (bottom panel: FL-335; Santa Cruz Biotechnology) so that the loading amount in each lane could be monitored. Lanes 1 and 2 represent individuals (unrelated control and mother of DGAP123, respectively) who have no cytogenetic abnormality of 2p16.3, whereas lanes 3–5 represent individuals (DGAP124, DGAP200, and DGAP123) with a 2p16.3 chromosomal abnormality. The ∼82 kDa neurexin 1 band quantitated by densitometry in (B) is indicated by an arrow. The smaller bands were not detected reproducibly or in consistent proportion relative to the largest band and could represent degradation products, alternative isoforms or cross-reacting proteins.
(B) The relative band intensity of the ∼82 kDa neurexin 1 band (normalized to glyceraldehyde-3-phosphate dehydrogenase intensity and compared with either one or two different control lymphoblasts in each case) is shown as the mean band intensity (±SD) relative to control from three different experiments. The numbers correspond to the following subjects: 1, Control; 2, DGAP123-2; 3, DGAP124; 4, DGAP200; and 5, DGAP123. Significant reduction compared to control was determined by t test: ∗p < 0.05; ∗∗p < 0.001.
Because the “double-hit” nature of the DGAP cases provided a strong argument for these breakpoints contributing to the phenotypes in the respective subjects, we sought to determine whether NRXN1 coding-sequence alterations, such as nonsense mutations, missense alterations, or frame shifts, might be a frequent predisposing factor in ASD. We performed exon scanning of all NRXN1 coding exons by direct sequencing to identify variants, initially in 57 subjects with ASD (autism, Asperger syndrome, and PDD-NOS; MIM 209850) compared with 57 individuals with OCD (MIM 164230) or Tourette syndrome (TS; MIM 137580) (27 and 30, respectively). In addition to common polymorphisms or previously reported SNPs, we identified seven novel variants that are within the coding sequence (Table 1) and that occurred as heterozygous differences in single individuals, all in the ASD cohort. None was found on the 114 chromosomes of the TS or OCD cohorts or on 354 control chromosomes from 177 unrelated members of the CEPH collection. Two were missense changes, L18Q in the signal peptide of α-neurexin 1 and L748I in an epidermal growth factor (EGF)-like domain, but the other five variants (three in exons specific to α-NRXN1) did not change amino acids. Subsequently, we sequenced the coding exons of 192 individuals (87 affected) from 53 families of the Autism Genetic Resource Exchange (AGRE) cohort but did not identify any rare coding-sequence variants cosegregating with ASD. Of the seven novel variants reported above, only one, L748I, was seen, in two independent ASD families in a total of three of four ASD affected individuals and one of two unaffected individuals, consistent with the possibility that it is an ASD susceptibility allele with incomplete penetrance.
Table 1.
NRXN1 Coding-Sequence Variants in ASD Screening Cohort
| Exon | Alteration | Codon | SNP |
ASD |
non-ASDa |
|---|---|---|---|---|---|
| rs Number | Variant Alleles/Total Chromosomes | Variant Alleles/Total Chromosomes | |||
| 2 | c.53T > A | p.L18Q | – | 1/114 | 0/468 |
| 2 | c.105C > A | p.G35 | – | 1/112 | 0/466 |
| 2 | c.511C > T | p.L171 | rs1045874 | 44/114 | N.D. |
| 5 | c.912C > T | p.G304 | – | 1/114 | 0/464 |
| 7 | c.999C > T | p.P333 | rs2303298 | 3/114 | 3/458 |
| 11 | c.2242C > A | p.L748I | – | 1/114 | 0/476 |
| 16 | c.3165C > T | p.A1055 | – – | 1/114 | 0/462 |
| 22 | c.3975C > T | p.G1325 | – | 1/114 | 0/462 |
| 24 | c.4374A > G | p.P1458 | – | 1/114 | 0/464 |
OCD, TS, and unaffected controls.
The observation of ASD in independent DGAP subjects establishes clearly that disruption of NRXN1 can contribute to autism but further indicates that such disruption, and by extension the consequences of heterozygous deletion, might show incomplete penetrance. DGAP123 and DGAP200 reinforce the recent report from the Autism Genome Project of a family in which two sisters with ASD share a de novo heterozygous deletion of 2p16.3 that directly affects the NRXN1 gene.8 However, the lack of ASD phenotype in DGAP124, the father of DGAP123 who shares her chromosomal abnormality, demonstrates that heterozygous inactivation of NRXN1 is not by itself sufficient to cause the symptoms of autism. This is consistent with the report of a deletion of exons 6–9 of α-NRXN1 in a Japanese participant in the International HapMap Project, an individual expected to be without obvious abnormal phenotype.20 NRXN1 mutation probably contributes by predisposing or sensitizing the individual, but additional factors are also required for ASD phenotypes to be expressed. The phenomenon of incomplete penetrance might well complicate interpretation of other chromosomal abnormalities in ASD. For example, although de novo copy-number changes reported in several studies are likely to target important genes, these changes might be necessary but not sufficient to produce ASD in the research subjects.4–8 Similarly, familial rearrangements inherited from a non-ASD parent or de novo changes also seen in unaffected individuals cannot be excluded as contributing to ASD. The same considerations are likely to apply to the interpretation of chromosomal differences in other multifactorial disorders in which genomic rearrangement is used as a route to the genes underlying the disease mechanism.
Additional investigation will be required for definition of the proportion of autism cases in which NRXN1 is a factor, the nature of NRXN1 genetic variants that confer susceptibility, and the potential functional consequences of these alleles. Our follow-up sequencing analyses revealed several rare variants in autism subjects, suggesting that noninactivating genetic variation in NRXN1 might also predispose to ASD. Consistent with this notion, Feng et al.13 have reported that rare structural variants of the β-neurexin 1 signal peptide are present in several individuals with autism but absent from more than 500 controls. Interestingly, the L18Q missense change in NRXN1 occurs in the signal-peptide region of the α-neurexin 1 isoform and alters a residue that is conserved throughout vertebrate evolution, including zebrafish, chicken, opossum, mouse, rat, dog, and cow. The L748I change in an EGF-like domain changes a residue that is similarly conserved through vertebrate evolution. Still, our findings indicate that the majority of subjects with ASD do not possess dramatic coding-sequence changes in NRXN1 that significantly alter the structure of the protein. However, the notable frequency of rare variants in ASD individuals suggests that even subtle disruptions might contribute to susceptibility. In particular, the seemingly innocuous synonymous variants cannot be dismissed out of hand given evidence that such coding-region changes can affect splicing enhancer or suppressor regulatory sequences, thereby altering the pattern of splicing, and can also alter protein folding, presumably because of effects on the rate of translation.21,22 Additional investigation is clearly required to determine whether any or all of the single-nucleotide variants identified here actually have functional consequences associated with autism and whether screening of larger numbers of individuals will reveal additional alterations. Like the DGAP123/124 alteration, even those rare variants that have dramatic effects on expression of the altered allele or on function of the protein product might not show complete penetrance in ASD. It is also conceivable that in addition to rare variants, common polymorphic variation at NRXN1 might also be associated with ASD, a possibility suggested tentatively by some NRXN1 region SNPs from a low-density scan of the genome.8
The case for NRXN1 in autism is made even more compelling by what is known of the neurobiology of its protein products. Neurexins act as cell-adhesion molecules and receptors in the brain, with over 2000 isoforms produced by use of alternative promotors and extensive alternative splicing.23–26 The α-neurexins contain epidermal EGF-like sequences and laminin G domains and interact with neurexophilins (Ca2+ independent) and dystroglycan (Ca2+ dependent).27–29 The functional consequences of these nteractions remain predominantly to be elucidated.23 The α-neurexins and the β-neurexins, which lack EGF-like sequences and contain one laminin G domain, can both interact variably with a second set of neuronal receptors termed the neuroligins (Ca2+ dependent), dependent on alternative splicing in both the neurexin and neuroligin genes.30 Interestingly, mutations in two of the five neuroligin (NLGN) genes are rare causes of ASD, suggesting a potential role for neurexin-neuroligin interaction in the neurodevelopmental process disrupted in autism.31–36 Notably, NLGN4X mutations might also produce mental retardation, suggesting that the mental retardation seen in DGAP123 might also result from altered neurexin-neuroligin interactions and that NRXN1 mutations might be involved in a broader spectrum of cognitive-impairment phenotypes.35
Neurexins and neuroligins bind to each other across the synaptic cleft, with the neurexin embedded in the presynaptic membrane and the neuroligin embedded in the postsynaptic membrane. This interaction has a dramatic impact on both sides of the synapse because presentation of neuroligins on nonneuronal cells can induce clustering of the neurexins and presynaptic differentiation on glutamatergic axons, whereas presentation of neurexins to dendrites can lead to aggregation of neuroligins and postsynaptic differentiation.37,38 Extensive alternative splicing occurs at conserved sites in both types of genes, controls the selectivity of binding of their products, and appears to underlie differential synaptic function, with differential selectivity, for example, for glutamatergic versus GABAergic synapses. The complexity of isoforms that can be produced and their functional consequences suggest that subtle shifts in the distribution of different isoforms, including reductions in individual forms could affect the balance of excitatory and inhibitory input for any particular neuron.23
α-neurexins also play a role at synapses in normal neurotransmitter release and the function of synaptic calcium channels, defined in part by experiments in knockout mice.39 The effect on presynaptic calcium channels is independent of neuroligin but is required for both glutamate and GABA release in vivo. Individual knockouts of the three α-neurexins are viable, in contrast to the lethality of multiple knockouts, suggesting a degree of functional redundancy in this gene family. Interestingly, although female mutant mice that lack only the α form of neurexin 1 are viable, fertile, and indistinguishable in appearance from wild-type mice, they display maternal indifference that leads to increased death of their pups independent of pup genotype.40 This behavioral abnormality suggests the existence of at least one critical function for Nrxn1 that cannot be compensated by the presence of intact Nrxn2 and Nrxn3.
Overall, the involvement of neurexin 1 in development and function of glutamatergic and GABAergic synapses, its interactions with the neuroligins, and its generation of a behavioral abnormality in knockout mice all make it an attractive candidate for involvement in autism. The complexity of neurexin expression and of its interactions suggests that a shift of the balance in the distribution of neurexin isoforms could have profound consequences, including perhaps altering the relative strengths of excitatory and inhibitory synapses because an increase in the ratio of excitation/inhibition in key neural systems is postulated to be a common pathway for causing autism.41 However, the fact that inactivation of one α-NRXN1 allele by the chromosomal excision is not fully penetrant indicates that other factors must cooperate to produce the ASD phenotype. Delineation of such factors might be essential for full understanding of both the role of NRXN1 and the mechanisms underlying ASD and can be expected to result in part from the discovery of new autism genes with the power of human genetics.
Acknowledgments
We are grateful to the DGAP families and members of the ASD, TS and OCD cohorts for their cooperation and participation in this study. We also gratefully acknowledge the resources provided by the Autism Genetic Resource Exchange (AGRE) Consortium and the participating AGRE families. We thank Heather Ferguson, Chantal Kelly, and Jill Platko for assistance in obtaining subject samples and phenotypic data, Robert Eisenman for technical support and the CHGR Tissue Culture and Genomics Resources for lymphoblast transformation and DNA sequencing services. This work was supported by National Institutes of Health grants P01-GM061354 (to C.C.M.), U19-HD35482 (to F.V., A.K., and D.L.P), P01-HD00300838 (to F.V., A.K., and D.L.P), and R01-NS16648 (D.L.P.) and by a NARSAD Distinguished Investigator Award (J.F.G.). Y.S. received a Young Investigator Award from the Children's Tumor Foundation. AGRE is a program of Cure Autism Now and is supported, in part, by grant MH64547 from the National Institute of Mental Health to Daniel H. Geschwind (PI).
Web Resources
The URLs for data presented herein are as follows:
Autism Chromosome Rearrangement Database, http://projects.tcag.ca/autism/
Autism Genetic Resource Exchange, http://www.agre.org/
CEPH, http://www.cephb.fr/
NCBI and GenBank, http://www.ncbi.nlm.nih.gov/; NRXN1 Accession #EF539882
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Autism and Developmental Disabilities Monitoring Network Surveillance Year 2002 Principal Investigators Prevalence of autism spectrum disorders–autism and developmental disabilities monitoring network, 14 sites, United States, 2002. MMWR Surveillance Summaries. 2007;56:12–28. [PubMed] [Google Scholar]
- 2.Santangelo S.L., Tsatsanis K. What is known about autism: Genes, brain, and behavior. Am. J. Pharmacogenomics. 2005;5:71–92. doi: 10.2165/00129785-200505020-00001. [DOI] [PubMed] [Google Scholar]
- 3.Grice D.E., Buxbaum J.D. The genetics of autism spectrum disorders. Neuromolecular Med. 2006;8:451–460. doi: 10.1385/NMM:8:4:451. [DOI] [PubMed] [Google Scholar]
- 4.Xu J., Zwaigenbaum L., Szatmari P., Scherer S.W. Molecular cytogenetics of autism. Curr. Genomics. 2004;5:347–364. [Google Scholar]
- 5.Vorstman J.A., Staal W.G., van Daalen E., van Engeland H., Hochstenbach P.F., Franke L. Identification of novel autism candidate regions through analysis of reported cytogenetic abnormalities associated with autism. Mol. Psychiatry. 2006;11:18–28. doi: 10.1038/sj.mp.4001781. [DOI] [PubMed] [Google Scholar]
- 6.Sebat J., Lakshmi B., Malhotra D., Troge J., Lese-Martin C., Walsh T., Yamrom B., Yoon S., Krasnitz A., Kendall J. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacquemont M.L., Sanlaville D., Redon R., Raoul O., Cormier-Daire V., Lyonnet S., Amiel J., Le Merrer M., Heron D., de Blois M.C. Array-based comparative genomic hybridisation identifies high frequency of cryptic chromosomal rearrangements in patients with syndromic autism spectrum disorders. J. Med. Genet. 2006;43:843–849. doi: 10.1136/jmg.2006.043166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Szatmari P., Paterson A.D., Zwaigenbaum L., Roberts W., Brian J., Liu X.Q., Vincent J.B., Skaug J.L., Thompson A.P., Senman L. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 2007;39:319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alkuraya F.S., Saadi I., Lund J.J., Turbe-Doan A., Morton C.C., Maas R.L. SUMO1 haploinsufficiency leads to cleft lip and palate. Science. 2006;313:1751. doi: 10.1126/science.1128406. [DOI] [PubMed] [Google Scholar]
- 10.Kim H.G., Herrick S.R., Lemyre E., Kishikawa S., Salisz J.A., Seminara S., MacDonald M.E., Bruns G.A., Morton C.C., Quade B.J., Gusella J.F. Hypogonadotropic hypogonadism and cleft lip and palate caused by a balanced translocation producing haploinsufficiency for FGFR1. J. Med. Genet. 2005;42:666–672. doi: 10.1136/jmg.2004.026989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leach N.T., Sun Y., Michaud S., Zheng Y., Ligon K.L., Ligon A.H., Sander T., Korf B.R., Lu W., Harris D.J. Disruption of diacylglycerol kinase delta (DGKD) associated with seizures in humans and mice. Am. J. Hum. Genet. 2007;80:792–799. doi: 10.1086/513019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu W., van Eerde A.M., Fan X., Quintero-Rivera F., Kulkarni S., Ferguson H., Kim H.G., Fan Y., Xi Q., Li Q.G. Disruption of ROBO2 is associated with urinary tract anomalies and confers risk of vesicoureteral reflux. Am. J. Hum. Genet. 2007;80:616–632. doi: 10.1086/512735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feng J., Schroer R., Yan J., Song W., Yang C., Bockholt A., Cook E.H., Jr., Skinner C., Schwartz C.E., Sommer S.S. High frequency of neurexin 1beta signal peptide structural variants in patients with autism. Neurosci. Lett. 2006;409:10–13. doi: 10.1016/j.neulet.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 14.Anderson M.A., Gusella J.F. Use of cyclosporin A in establishing Epstein-Barr virus-transformed human lymphoblastoid cell lines. In Vitro. 1984;20:856–858. doi: 10.1007/BF02619631. [DOI] [PubMed] [Google Scholar]
- 15.Lord C., Rutter M., Goode S., Heemsbergen J., Jordan H., Mawhood L., Schopler E. Autism diagnostic observation schedule: A standardized observation of communicative and social behavior. J. Autism Dev. Disord. 1989;19:185–212. doi: 10.1007/BF02211841. [DOI] [PubMed] [Google Scholar]
- 16.Lord C., Rutter M., Le Couteur A. Autism diagnostic interview-revised: A revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J. Autism Dev. Disord. 1994;24:659–685. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- 17.Rowen L., Young J., Birditt B., Kaur A., Madan A., Philipps D.L., Qin S., Minx P., Wilson R.K., Hood L., Graveley B.R. Analysis of the human neurexin genes: Alternative splicing and the generation of protein diversity. Genomics. 2002;79:587–597. doi: 10.1006/geno.2002.6734. [DOI] [PubMed] [Google Scholar]
- 18.Tabuchi K., Sudhof T.C. Structure and evolution of neurexin genes: Insight into the mechanism of alternative splicing. Genomics. 2002;79:849–859. doi: 10.1006/geno.2002.6780. [DOI] [PubMed] [Google Scholar]
- 19.Taylor J., Tyekucheva S., King D.C., Hardison R.C., Miller W., Chiaromonte F. ESPERR: Learning strong and weak signals in genomic sequence alignments to identify functional elements. Genome Res. 2006;16:1596–1604. doi: 10.1101/gr.4537706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Redon R., Ishikawa S., Fitch K.R., Feuk L., Perry G.H., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharma S., Black D.L. Maps, codes, and sequence elements: Can we predict the protein output from an alternatively spliced locus? Neuron. 2006;52:574–576. doi: 10.1016/j.neuron.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 22.Kimchi-Sarfaty C., Oh J.M., Kim I.W., Sauna Z.E., Calcagno A.M., Ambudkar S.V., Gottesman M.M. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315:525–528. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
- 23.Craig A.M., Kang Y. Neurexin-neuroligin signaling in synapse development. Curr. Opin. Neurobiol. 2007;17:43–52. doi: 10.1016/j.conb.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dean C., Dresbach T. Neuroligins and neurexins: Linking cell adhesion, synapse formation and cognitive function. Trends Neurosci. 2006;29:21–29. doi: 10.1016/j.tins.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 25.Lise M.F., El-Husseini A. The neuroligin and neurexin families: From structure to function at the synapse. Cell. Mol. Life Sci. 2006;63:1833–1849. doi: 10.1007/s00018-006-6061-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Missler M., Sudhof T.C. Neurexins: Three genes and 1001 products. Trends Genet. 1998;14:20–26. doi: 10.1016/S0168-9525(97)01324-3. [DOI] [PubMed] [Google Scholar]
- 27.Missler M., Hammer R.E., Sudhof T.C. Neurexophilin binding to alpha-neurexins. A single LNS domain functions as an independently folding ligand-binding unit. J. Biol. Chem. 1998;273:34716–34723. doi: 10.1074/jbc.273.52.34716. [DOI] [PubMed] [Google Scholar]
- 28.Missler M., Sudhof T.C. Neurexophilins form a conserved family of neuropeptide-like glycoproteins. J. Neurosci. 1998;18:3630–3638. doi: 10.1523/JNEUROSCI.18-10-03630.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sugita S., Saito F., Tang J., Satz J., Campbell K., Sudhof T.C. A stoichiometric complex of neurexins and dystroglycan in brain. J. Cell Biol. 2001;154:435–445. doi: 10.1083/jcb.200105003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boucard A.A., Chubykin A.A., Comoletti D., Taylor P., Sudhof T.C. A splice code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to alpha- and beta-neurexins. Neuron. 2005;48:229–236. doi: 10.1016/j.neuron.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 31.Chih B., Afridi S.K., Clark L., Scheiffele P. Disorder-associated mutations lead to functional inactivation of neuroligins. Hum. Mol. Genet. 2004;13:1471–1477. doi: 10.1093/hmg/ddh158. [DOI] [PubMed] [Google Scholar]
- 32.Chubykin A.A., Liu X., Comoletti D., Tsigelny I., Taylor P., Sudhof T.C. Dissection of synapse induction by neuroligins: Effect of a neuroligin mutation associated with autism. J. Biol. Chem. 2005;280:22365–22374. doi: 10.1074/jbc.M410723200. [DOI] [PubMed] [Google Scholar]
- 33.Comoletti D., De Jaco A., Jennings L.L., Flynn R.E., Gaietta G., Tsigelny I., Ellisman M.H., Taylor P. The Arg451Cys-neuroligin-3 mutation associated with autism reveals a defect in protein processing. J. Neurosci. 2004;24:4889–4893. doi: 10.1523/JNEUROSCI.0468-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jamain S., Quach H., Betancur C., Rastam M., Colineaux C., Gillberg I.C., Soderstrom H., Giros B., Leboyer M., Gillberg C., Bourgeron T. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laumonnier F., Bonnet-Brilhault F., Gomot M., Blanc R., David A., Moizard M.P., Raynaud M., Ronce N., Lemonnier E., Calvas P. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet. 2004;74:552–557. doi: 10.1086/382137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yan J., Oliveira G., Coutinho A., Yang C., Feng J., Katz C., Sram J., Bockholt A., Jones I.R., Craddock N. Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol. Psychiatry. 2005;10:329–332. doi: 10.1038/sj.mp.4001629. [DOI] [PubMed] [Google Scholar]
- 37.Graf E.R., Zhang X., Jin S.X., Linhoff M.W., Craig A.M. Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell. 2004;119:1013–1026. doi: 10.1016/j.cell.2004.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scheiffele P., Fan J., Choih J., Fetter R., Serafini T. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell. 2000;101:657–669. doi: 10.1016/s0092-8674(00)80877-6. [DOI] [PubMed] [Google Scholar]
- 39.Missler M., Zhang W., Rohlmann A., Kattenstroth G., Hammer R.E., Gottmann K., Sudhof T.C. Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature. 2003;423:939–948. doi: 10.1038/nature01755. [DOI] [PubMed] [Google Scholar]
- 40.Geppert M., Khvotchev M., Krasnoperov V., Goda Y., Missler M., Hammer R.E., Ichtchenko K., Petrenko A.G., Sudhof T.C. Neurexin I alpha is a major alpha-latrotoxin receptor that cooperates in alpha-latrotoxin action. J. Biol. Chem. 1998;273:1705–1710. doi: 10.1074/jbc.273.3.1705. [DOI] [PubMed] [Google Scholar]
- 41.Rubenstein J.L., Merzenich M.M. Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]