

Abstract
Snowflake vitreoretinal degeneration (SVD, MIM 193230) is a developmental and progressive hereditary eye disorder that affects multiple tissues within the eye. Diagnostic features of SVD include fibrillar degeneration of the vitreous humor, early-onset cataract, minute crystalline deposits in the neurosensory retina, and retinal detachment. A genome-wide scan previously localized the genetic locus for SVD to a 20 Mb region flanked by D2S2158 and D2S2202. This region contains 59 genes, of which 20 were sequenced, disclosing a heterozygous mutation (484C > T, R162W) in KCNJ13, member 13 of subfamily J of the potassium inwardly rectifying channel family in all affected individuals. The mutation in KCNJ13, the gene encoding Kir7.1, was not present in unaffected family members and 210 control individuals. Kir7.1 localized to human retina and retinal pigment epithelium and was especially prevalent in the internal limiting membrane adjacent to the vitreous body. Molecular modeling of this mutation predicted disruption of the structure of the potassium channel in the closed state located immediately adjacent to the cell-membrane inner boundary. Functionally, unlike wild-type Kir7.1 whose overexpression in CHO-K1 cells line produces highly selective potassium current, overexpression of R162W mutant Kir7.1 produces a nonselective cation current that depolarizes transfected cells and increases their fragility. These results indicate that the KCNJ13 R162W mutation can cause SVD and further show that vitreoretinal degeneration can arise through mutations in genes whose products are not structural components of the vitreous.
Main Text
Snowflake vitreoretinal degeneration (SVD) is one of the vitreoretinal degenerations, a group of hereditary retinal dystrophies characterized by early-onset cataract, congenital liquefaction of the vitreous humor, and abnormalities of the interface between the vitreous and retina leading to increased traction on the retina and retinal detachment.1 SVD shows autosomal-dominant inheritance. It exhibits both congenital abnormalities of the eye and acquired degeneration of multiple ocular tissues that are clinically similar to those seen in the common complex-traits cataract and Fuchs corneal endothelial dystrophy.2 The congenital abnormalities include optic-nerve-head dysmorphism with fibrillar degeneration of the vitreous. Progressive ocular features include peripheral retinal degeneration within which minute crystalline deposits referred to as snowflakes might be seen and include corneal guttae.2,3 These characteristics distinguish SVD from other vitreoretinal degenerations.
The most common vitreoretinal degeneration is Stickler syndrome. Mutations leading to haploinsufficiency of the collagen 2A1(COL2A1) gene cause Stickler syndrome type I (STL1, MIM 108300).4 These patients have a vitreous degeneration characterized by vestigial vitreous gel occupying the immediate retrolental space and no discernible gel in the central vitreous cavity.5 The expression of systemic features exhibits variability both between and within families.
Mutations leading to haploinsufficiency of the collagen 11A1 (COL11A1) gene cause Stickler syndrome type II (STL2, MIM 604841). Unlike COL2A1 disease, such COL11A1 mutations lead to a fibrillar vitreous degeneration with limited and random fibrils throughout the vitreous cavity.5 Mutations altering intron-exon splicing of the COL11A1 gene lead to Marshall syndrome (MIM 154780), distinguished from Stickler syndrome by more pronounced facial dysmorphism and less frequent retinal detachment. Mutations in the related collagen 11A2 gene lead to the systemic features of Stickler syndrome but without ocular features because this gene (COL11A2) is not expressed in the eye. There is evidence that mutations in other, as-yet-unknown genes can lead to Stickler syndrome.
Wagner disease (MIM 143200) is caused by noncoding mutations that are thought to affect the splicing of chondroitin sulfate proteoglycan-2 (CSPG2), the gene encoding versican.6,7 These mutations might lead to disease through abnormal ratios of the isoforms of versican. The distinguishing features of Wagner disease are pseudostrabismus, thickened and partially detached posterior hyalodi with an empty vitreous cavity, variable degeneration of the retina and choroid, and the absence of systemic manifestations.8
The originally described SVD family, an American family of European extraction containing 31 individuals (13 affected individuals, 14 unaffected individuals, and four unaffected spouses) were enrolled in this study.2,9 The 13 subjects diagnosed with SVD ranged from 12 to 85 years of age at the time of diagnosis. The inheritance pattern was autosomal dominant, and no obligatory carriers of the Snowflake trait were found to be normal. The relevant institutional review boards approved this study, and informed consent was obtained from all participating family members, consistent with the Declaration of Helsinki. The examination and diagnostic criteria and enrollment of subjects have been described previously.2 In brief, a comprehensive eye and focused physical examination were performed by a vitreoretinal specialist and internist prospectively for 13 subjects. Blood was obtained from each participant for isolation of genomic DNA.
Because of the demonstrated ability of mutations in genes encoding vitreous proteins to cause vitreoretinal degeneration, particular attention was paid to COL4A3, part of which lies in the linked region bounded by D2S2158 and D2S2202. Exons 2–52 of COL4A3 were amplified and subjected to direct DNA sequencing along with flanking DNA sequences as previously described.9 In addition, COL4A3 mRNA from two affected individuals was amplified in overlapping segments from total human RNA by reverse transcriptase-PCR and directly sequenced. No mutations were identified through either approach, and identification of heterozygous SNPs excluded deletion of large parts of COL4A3 as a cause of disease as well as indicated that the coding sequences of COL4A3 lie outside the linked region (data not shown).
Exons of candidate genes in the linked region were amplified by PCR and sequenced directly in the order of their suitability as judged by expression in the retina, identified or predicted function in the retina, and any known effects of mutations in other systems. For mutation screening, coding exons and adjacent intronic sequences were amplified in accordance with standard techniques from genomic DNA of two affected patients and two unaffected individuals. The PCR amplified sequences were sequenced with ABI dye Terminator version 3.1, and the products of the sequencing reactions were analyzed on a 3100 ABI genetic analyzer. Sequences were aligned with the Seqman program of the DNASTAR program package (DNASTAR, Madison, WI, USA). COL4A3, SLC19A3, DNER, SLC17, 16A14, LOC646839, CAB39, PSMD1, ARMC9, NCL, PTMA, PDE6D, COPS7B, ECEL1, EFHD1, TNRC15, ATG16L1, SAG, USP40, and CENTG2 were sequenced and did not show sequence changes predicted to cause dysfunction.
However, sequencing of KCNJ13 (genomic accession CH471063, mRNA-NM_002242) showed a heterozygous c.484C > T transition, changing a CGG codon to TGG and resulting in an R162W change in the amino acid sequence (Figure 1A). This sequence change is present in all affected individuals in the family and not present in unaffected family members or 210 unrelated ethnically matched control individuals (420 chromosomes, providing a power of more than 95% to detect a polymorphism with a frequency of 0.01). The R-to-W change has a −5 score on the blosum 80 matrix and represents a change from a positively charged hydrophilic amino acid to an aromatic and hydrophobic amino acid. KCNJ13 R162 is conserved in a broad group of species including Macaca mulatta, Cavia porcellus, Rattus Norvegicus, and Danio rerio (Table 1). Among the human Kcnj (Kir) family, this residue is conserved among the evolutionary close sequences Kcnj15, Kcnj10, and Kcnj110 but not in the more distant family members except for the distant cluster including Kcnj12, Kcnj4, and Kcnj2 has a conservative arginine-to-lysine change. All of the remaining members of this branch (Kcnj or Kir) of the potassium channel family show conservative changes to lysine or glutamine, except the distantly related Kcnj16, which has a threonine at this position.
Figure 1.
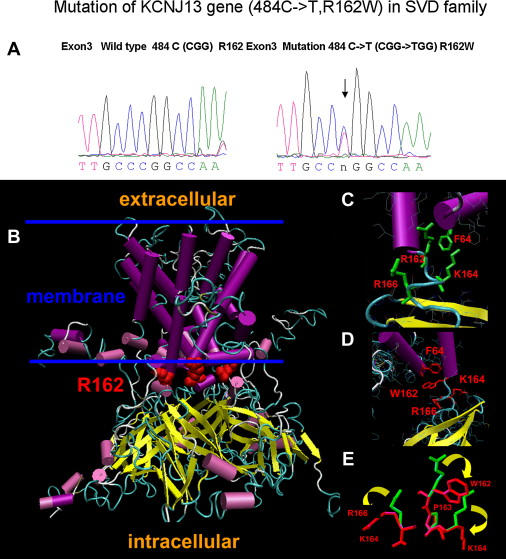
Sequence Tracings and Structural Changes of the Normal and R162W Kir7.1 Proteins
(A) Sequence tracings of normal and predicted structure of the normal and 484C > T KCNJ13 gene resulting in the arginine-to-tryptophan (R162W) amino acid change.
(B) An overview of the normal human Kir7.1 tetramer built by homology modeling. Magenta cylinders and yellow arrows indicate α helices and β strands, respectively. R162 residues in each monomer are shown as red spheres. The estimated location of the membrane is shown by two blue lines.
(C) Detail of R162 and its interactions.
(D) Detail of W162 interface area of the Kir7.1 monomer. Residues predicted to undergo a significant change of conformation because of the R162W mutation are shown in red.
(E) Predicted structural changes for residues K164, P163, and R166 due to the R162W mutation. Wild-type and mutant side chains are shown in green and red, respectively. Yellow arrows show the direction of the change.
Table 1.
Alignment of Kir7.1 Protein Sequences in Region of R162W Mutation
| 155 |
A |
F |
V |
A |
K |
I |
A |
R |
P |
K |
N |
R |
A |
F |
Consensus |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| W | R162W Mutation | ||||||||||||||
| 155 | A | F | V | A | K | I | A | R | P | K | N | R | A | F | Homo sapiens |
| 155 | A | F | V | A | K | I | A | R | P | K | N | R | A | F | Macaca mulatta |
| 155 | A | F | V | A | K | I | A | R | P | K | N | R | A | F | Rattus norvegicus |
| 155 | A | F | V | A | K | I | A | R | P | K | N | R | A | F | Cavia porcellus |
| 167 | A | F | V | A | K | F | S | R | P | Q | K | R | C | D | Danio rerio |
Alignment was carried out by the ClustalW method. Nonidentical amino acids are shown in italics.
In order to assess the structural implications of the R162W mutation more precisely, we carried out molecular modeling of Kir7.1. The K+-selective inward-rectified channel Kir7.1 is a membrane protein and a member of the Kir1-ATP-regulated inward rectifier K+ [ROMK] family.11 Protein fold recognition with 1D and 3D sequence profiles coupled with secondary structure and solvation potential information (3D-PSSM, Kelley 2000) showed the closest match (E value 5.26 × 10−6) with the structure of the prokaryotic potassium-selective inward rectifier channel KirBac 1.1. Protein sequence alignment of these two proteins performed with ClustalW shows a sequence identity (similarity) of 28% (55%). Unfortunately, the structure of the KirBac 1.1 channel is available for the closed state only,12 so that the structure of the closed state for the Kir7.1 channel was built by homology modeling based on crystal coordinates for KirBac 1.1 (Brookhaven protein database [PDB] file: 1p7b),13 as the structural template. The primary sequences of Kir7.1 and 1p7b were aligned by the method of Needleman and Wunsch14 and incorporated in the program Look, version 3.5.15 Structures forming the Kir7.1 tetramer were built with the automatic segment matching method in the program Look,16 and this was followed by 500 cycles of energy minimization. We used the same program to generate the conformation near the R162W mutation and to refine it by self-consistent ensemble optimization,15 which applies the statistical mechanical mean-force approximation iteratively to achieve the global-energy-minimum structure. The geometry of the predicted structures was tested with the program Procheck.17
Structural modeling of normal and mutant Kir7.1 are demonstrated in Figure 1. Figure 1B shows the structural model of the wild-type Kir7.1 tetramer. The part of the structure incorporated into the membrane is shown between two blue lines as approximated by analogy with the template protein.12 The Kir7.1 structure consists of an α-helical integral membrane domain and an intracellular domain formed mostly of β sheets similar to those in KirBac 1.1. Amino acid R162 is located in the polypeptide chain connecting these domains (Figure 1B). Normally, R162 forms part of the central layer of positively charged arginines, similar to that observed in the KirBac 1.1 structure. This interaction is predicted to be supported by hydrogen bonding between the donor-acceptor pairs of the polypeptide chain (Figures 1B and 1C). Analysis of the predicted structure of the R162W mutant channel suggests that this mutation causes major structural changes in the vicinity of residue 162 including residues R166 and K164, as well as residue 162 itself (Figures 1C, 1D, and 1E). These changes are expected to break several hydrogen bonds present in the wild-type channel (interactions between residues R162 and F64 or K159, A62 and K159, R165 and R195) and to form two new hydrogen bonds (interactions between A62 and K159 and between K164 and R284) in the mutant channel (not shown). This modeling suggests that this kind of change might affect both the stability and normal function of the closed state of the K+-selective inward-rectified channel Kir7.1.
In order to investigate the functional consequences of the R162W mutation, we transfected expression constructs with normal and R162W mutant rat Kir7.1 fused to GFP and driven by a CMV promoter into Chinese hamster ovary (CHO-K1) cells, similar to the approach used by Krapivinsky et al.11 These experiments were performed with a rat cDNA sequence similar to NP_446060 (NCBI), except that there is an A139S background in both the control and R162W mutant sequences. Results with the control Kir7.1 were similar to those seen previously,11 at physiological conditions (2.5 mM [K+]o, 150 mM [Na+]o in the external solution and 150 mM [K+]i in the patch pipette) showing a typical bell-shape voltage-current relationship (I-V) of Kir7.1 with reversal potential −81.2 ± 6.7 mV (n = 10) that indicates a high selectivity of this channel for potassium versus sodium ions (Figure 2A). At symmetrical K+ (150 mM [K+] in both external and pipette solutions), I-V shows a nonlinear increase of current at negative potentials typical of inwardly rectifying channels (Figure 2A).
Figure 2.
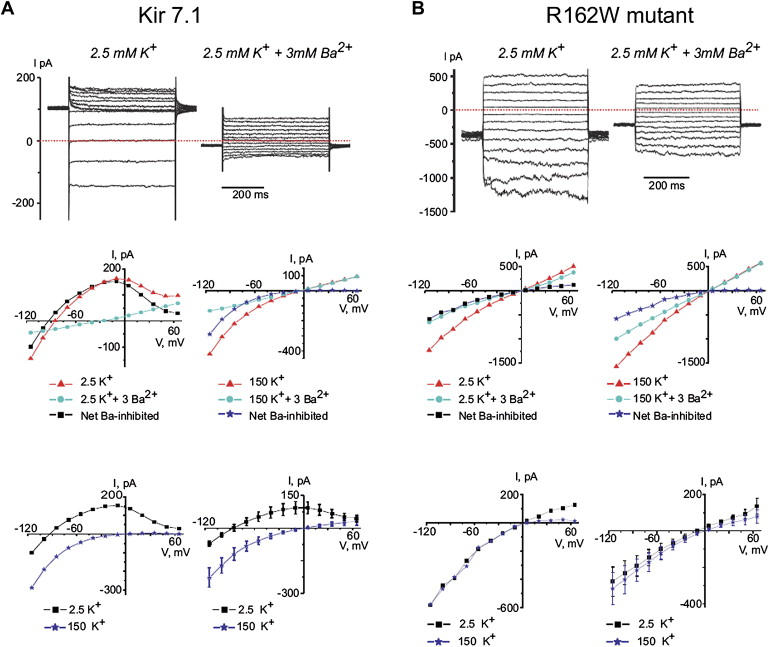
Electrophysiological Properties of Normal and Mutant Rat Kir7.1 Expressed in CHO Cells
The top line show examples of currents measured in Kir7.1 (A) or R162W mutant Kir7.1 (B) transfected CHO cells in response to voltage-clamp steps from the holding potential −50 mV to voltages between −115 mV and 65 mV. The middle-left (2.5 K) and -right (150K) panels show single-experiment examples of I-V relations measured in cells expressing Kir 7.1 (A) and R162W mutant Kir7.1 (B). The mean current measured isochronally between 250 ms and 450 ms of the 500 ms pulse is shown. The net Ba2+ inhibited current differs between the normal and mutant Kir7.1 both in the shape of the curve and its reversal potential (the point at which it crosses the abscissa). The bottom-left panels show superimposed traces from the middle panels, and the bottom-right panels show mean data comparing normal ([A], n = 10 cells from three experiments) and R162W mutant ([B], n = 13 cells per three experiments) Kir7.1. In the R162W mutant, the 2.5K current has lost its potassium selectivity (Er = −9 mV), has a less pronounced inwardly rectifying quality, and appears qualitatively similar to the 150K current in both normal and mutant molecules. Transfected cells showing increased leak current (I > 800 pA at 50 mV; n = 1 and n = 136 for cells transfected with Kir 7.1 and R162W mutant, respectively) were omitted from analysis.
In contrast, expression of the mutant Kir7.1 results in a dramatic shift of the I-V curve toward positive potentials at physiological conditions. The reversal potentials of the mutant current become −9.3 ± 2.5 mV (n = 13) at 2.5 mM [K+]o, and the I-V curves measured with 2.5 mM and 150 mM [K+]o are identical. The resting membrane potential (Er) of CHO-K1 cells expressing mutant Kir7.1 is more depolarized (−17.8 ± 2.3 mV, n = 25) as compared to the Er of cells expressing wild-type Kir 7.1 (−57.8 ± 6.7 mV, n = 8, p < 0.001) and cells transfected with only GFP (−29.9 ± 5.2 mV, n = 6, p < 0.05). In addition, CHO-K1 cells expressing the mutant Kir7.1 are characterized by decreased survival compared to cells expressing GFP alone or wild-type Kir7.1 (not shown).
The fact that the reversal potential (−9 mV) of current induced by expression of mutant R162W and measured in asymmetrical K+ conditions (150 mM [K+]i/2.5 mM [K+]o + 150 mM [Na+]o) is close to zero indicates that this current is equally conducted by both potassium and sodium ions. Activation of such a current depolarizes CHO-K1 cells and increases their fragility. Additional specialized studies are necessary to evaluate the exact origin of the differences in current properties induced by expression of the mutant Kir7.1. One possibility is that the R162W mutation modifies channel selectivity and renders it permeable to Na+ ions.
Kir7.1 has been shown to be present in the inner nuclear layer and especially the retinal pigment epithelial (RPE) cells of the bovine retina,18 in which it seems likely to allow precise regulation of resting membrane potential and sustain cellular ionic composition.19,20 A Kir7.1 having identical electrophysiological properties also has been detected in lower amounts in porcine iris pigment epithelium.21 Immunoreactivity against Kir7.1 in frozen sections of human cadaveric retina embedded in OCT with antibodies described by Krapivinsky et al.11 localizes the protein to the RPE and inner nuclear layer, as in the cow, but also shows diffuse staining throughout the nerve fiber layer and inner plexiform layer and intense reactivity in the internal limiting membrane (ILM) (Figures 3A and 3B). This is confirmed by immunofluorescence confocal microscopy, which shows Kir7.1 primarily in the internal limiting membrane and RPE, in which it colocalizes with the autofluorescence of lipofuschin. Weak staining is observed in the ganglion cell layer, inner plexiform layer, inner nuclear layer, and the outer plexiform layer (Figures 3C–3F). In addition, in some sections there is mild reactivity around retinal vessels (Figure 3B).
Figure 3.
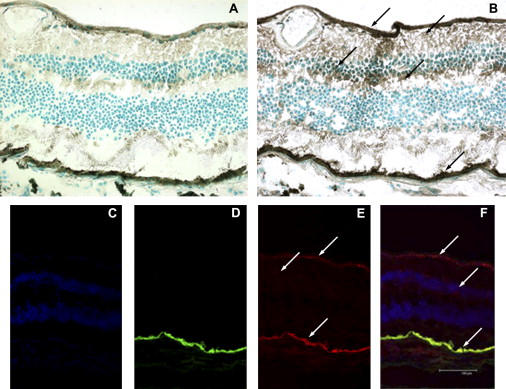
Localization of Kir7.1 in the Retina and RPE
(A) Control section without anti-Kir7.1 antibody.
(B) Retinal section stained with antibody to Kir7.1 showing localization to the internal limiting membrane (ILM), nerve fiber layer (NFL), inner nuclear layer (INL), inner plexiform layer (IPL), and retinal pigment epithelium (RPE). These retinal layers are indicated by the arrows from top to bottom of (B).
(C) DAPI-stained retinal section showing the INL and ONL.
(D) Autofluorescence showing the RPE.
(E) Immunofluorescent staining showing Kir7.1, especially in the ILM and RPE.
(F) Merged images of DAPI, autofluorescence, and Kir7.1 images showing colocalization of Kir7.1 within the RPE, INL, and ONL.
There are two major families of potassium channels, Kv (voltage-gated delayed rectifying channels) and Kir (inward rectifying channels), each having numerous subfamilies. Kir channels are able to pass K+ ions much more readily into the cell than out of it. The highly conserved pore structure of Kir channels has a simple ball-valve design that seems likely to reduce the work cells' need to depolarize their membrane potential and likely to allow modulation by various internal blockers.22 Kir7.1 is characterized by a low single-channel conductance and low sensitivity to external Ba2+ blockade, both of which probably result from the specific amino acids forming its pore region, especially M125,11 with outward conductance increasing as extracellular K+ concentration decreases. Besides the retina, Kir7.1 is expressed in the brain, kidney, intestines, testis, liver, and prostate.23 The net flux of K+ across the RPE and into or out of the subretinal space is determined by the balance between K+ efflux mediated by Kir channels, in large part Kir7.1,18,24,25 and K+ influx mediated by the Na+/K+ pump26 and Na+–K+–2Cl− cotransporter.27
Although two other families have been reported to have SVD, their clinical features are not consistent with our current understanding of SVD.1 At this time, no other families with SVD are available to confirm the association between the Kir7.1 mutation and SVD. In additional to the modeling studies presented above, the mutation in Kir7.1 is an excellent candidate for SVD for several clinical reasons. First, Kir7.1 was found in both the internal limiting membrane, in which the vitreous is generated during development, and the RPE, which is observed to undergo degenerative changes in SVD. Second, the location of Kir7.1 in the internal limiting membrane is most likely to be explained by the presence of Muller cells on its outer surface. The reduced b wave of the electroretinogram seen in Snowflake subjects28 could be explained by disruption of the flow current from the inner to outer retina as a consequence of the R162W mutation. Because Kir7.1 has not previously been localized in the human retina or studied in the ILM, it is not possible to determine at the present time whether the various features of SVD are mediated by different functions of Kir7.1 in different layers of the retina or they are mediated by abnormal function of one retinal layer. Kir7.1 mutations might also have complicated indirect results, as is seen in zebrafish, in which mutant Kir7.1 causes changes in the stripe patterns, probably as a result of an α2-adrenergic signaling defect.29 It seems likely that the change in Kir7.1 function causes a change in K+ and Na+ conductance, leading to cell depolarization. This would both change signaling and induce damage of the cells, thereby initiating a cascade of events that result in the various features of SVD. Further detailed functional studies of the R162W mutant Kir7.1 will help to elucidate the mechanism through which this mutation causes SVD.
Acknowledgments
We thank Dr. Grigory and Dr. Luba Krapivinsky (Children's Hospital, Harvard Medical School, Boston, MA) for the generous gift of antibodies to Kir7.1. We also thank the members of the Snowflake family (family 55001, HED Family 2) for their participation in this study. This work was supported by grants from the National Eye Institute (EY014467), the Foundation Fighting Blindness, Owing Mills, MD, and Research to Prevent Blindness, New York, NY. Additional support from anonymous donors and the Mayo Clinic is gratefully acknowledged.
Web Resources
The URLs for data presented herein are as follows:
GenBank, http://www.ncbi.nlm.nih.gov/genbank/
Genethon, http://www.genethon.fr
National Center for Biotechnology Information (NCIB), http://www.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim/
References
- 1.Edwards A.O., Robertson J.E. Hereditary vitreoretinal degenerations. In: Ryan S.J., Hinton D.R., Schachat A.P., Wilkenson P., editors. Retina. Elsevier Inc.; Philadelphia: 2006. pp. 519–538. [Google Scholar]
- 2.Lee M.M., Ritter R., III, Hirose T., Vu C.D., Edwards A.O. Snowflake vitreoretinal degeneration: Follow-up of the original family. Ophthalmology. 2003;110:2418–2426. doi: 10.1016/S0161-6420(03)00828-5. [DOI] [PubMed] [Google Scholar]
- 3.Hirose T., Lee K.Y., Schepens C.L. Snowflake degeneration in hereditary vitreoretinal degeneration. Am. J. Ophthalmol. 1974;77:143–153. doi: 10.1016/0002-9394(74)90665-5. [DOI] [PubMed] [Google Scholar]
- 4.Ahmad N.N., Ala-Kokko L., Knowlton R.G., Jimenez S.A., Weaver E.J., Maguire J.I., Tasman W., Prockop D.J. Stop codon in the procollagen II gene (COL2A1) in a family with the Stickler syndrome (arthro-ophthalmopathy) Proc. Natl. Acad. Sci. USA. 1991;88:6624–6627. doi: 10.1073/pnas.88.15.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Snead M.P., Yates J.R. Clinical and molecular genetics of Stickler syndrome. J. Med. Genet. 1999;36:353–359. [PMC free article] [PubMed] [Google Scholar]
- 6.Kloeckener-Gruissem B., Bartholdi D., Abdou M.T., Zimmermann D.R., Berger W. Identification of the genetic defect in the original Wagner syndrome family. Mol. Vis. 2006;12:350–355. [PubMed] [Google Scholar]
- 7.Mukhopadhyay A., Nikopoulos K., Maugeri A., de Brouwer A.P., van Nouhuys C.E., Boon C.J., Perveen R., Zegers H.A., Wittebol-Post D., van den Biesen P.R. Erosive vitreoretinopathy and wagner disease are caused by intronic mutations in CSPG2/Versican that result in an imbalance of splice variants. Invest. Ophthalmol. Vis. Sci. 2006;47:3565–3572. doi: 10.1167/iovs.06-0141. [DOI] [PubMed] [Google Scholar]
- 8.Meredith S.P., Richards A.J., Flanagan D.W., Scott J.D., Poulson A.V., Snead M.P. Clinical characterisation and molecular analysis of Wagner syndrome. Br. J. Ophthalmol. 2007;91:655–659. doi: 10.1136/bjo.2006.104406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiao X., Ritter R., III, Hejtmancik J.F., Edwards A.O. Genetic linkage of snowflake vitreoretinal degeneration to chromosome 2q36. Invest. Ophthalmol. Vis. Sci. 2004;45:4498–4503. doi: 10.1167/iovs.04-0722. [DOI] [PubMed] [Google Scholar]
- 10.Friedland D.R., Eernisse R., Popper P. Potassium channel gene expression in the rat cochlear nucleus. Hear. Res. 2007;228:31–43. doi: 10.1016/j.heares.2007.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krapivinsky G., Medina I., Eng L., Krapivinsky L., Yang Y., Clapham D.E. A novel inward rectifier K+ channel with unique pore properties. Neuron. 1998;20:995–1005. doi: 10.1016/s0896-6273(00)80480-8. [DOI] [PubMed] [Google Scholar]
- 12.Kuo A., Gulbis J.M., Antcliff J.F., Rahman T., Lowe E.D., Zimmer J., Cuthbertson J., Ashcroft F.M., Ezaki T., Doyle D.A. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science. 2003;300:1922–1926. doi: 10.1126/science.1085028. [DOI] [PubMed] [Google Scholar]
- 13.Abola E., Bernstein F.C., Bryant S.H., Koetzle T.F., Weng J. Protein data bank. In: Allen F.H., Bergerhoff G., Sievers R., editors. Crystallographic Databases-Information Contene, Software Systems, Scientific Applications. Data Comission of the International Union of Crystallography; Cambridge: 1987. pp. 107–132. [Google Scholar]
- 14.Needleman S.B., Wunsch C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- 15.Lee C. Predicting protein mutant energetics by self-consistent ensemble optimization. J. Mol. Biol. 1994;236:918–939. doi: 10.1006/jmbi.1994.1198. [DOI] [PubMed] [Google Scholar]
- 16.Levitt M. Accurate modeling of protein conformation by automatic segment matching. J. Mol. Biol. 1992;226:507–533. doi: 10.1016/0022-2836(92)90964-l. [DOI] [PubMed] [Google Scholar]
- 17.Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:283–291. [Google Scholar]
- 18.Yang D., Pan A., Swaminathan A., Kumar G., Hughes B.A. Expression and localization of the inwardly rectifying potassium channel Kir7.1 in native bovine retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci. 2003;44:3178–3185. doi: 10.1167/iovs.02-1189. [DOI] [PubMed] [Google Scholar]
- 19.Reimann F., Ashcroft F.M. Inwardly rectifying potassium channels. Curr. Opin. Cell Biol. 1999;11:503–508. doi: 10.1016/S0955-0674(99)80073-8. [DOI] [PubMed] [Google Scholar]
- 20.Kofuji P., Newman E.A. Potassium buffering in the central nervous system. Neuroscience. 2004;129:1045–1056. doi: 10.1016/j.neuroscience.2004.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yasuda K., Shimura M., Nakazawa T., Sato H., Tomita H., Sugano E., Tamai M. Expression and functional properties of unique inward rectifier K+ channel Kir7.1 in the porcine iris and retinal pigment epithelium. Curr. Eye Res. 2003;27:279–287. doi: 10.1076/ceyr.27.5.279.17226. [DOI] [PubMed] [Google Scholar]
- 22.Nichols C.G., Lopatin A.N. Inward rectifier potassium channels. Annu. Rev. Physiol. 1997;59:171–191. doi: 10.1146/annurev.physiol.59.1.171. [DOI] [PubMed] [Google Scholar]
- 23.Partiseti M., Collura V., Agnel M., Culouscou J.M., Graham D. Cloning and characterization of a novel human inwardly rectifying potassium channel predominantly expressed in small intestine. FEBS Lett. 1998;434:171–176. doi: 10.1016/s0014-5793(98)00972-7. [DOI] [PubMed] [Google Scholar]
- 24.Quinn R.H., Miller S.S. Ion transport mechanisms in native human retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci. 1992;33:3513–3527. [PubMed] [Google Scholar]
- 25.Shimura M., Yuan Y., Chang J.T., Zhang S., Campochiaro P.A., Zack D.J., Hughes B.A. Expression and permeation properties of the K(+) channel Kir7.1 in the retinal pigment epithelium. J. Physiol. 2001;531:329–346. doi: 10.1111/j.1469-7793.2001.0329i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller S.S., Steinberg R.H., Oakley B. The electrogenic sodium pump of the frog retinal pigment epithelium. J. Membr. Biol. 1978;44:259–279. doi: 10.1007/BF01944224. [DOI] [PubMed] [Google Scholar]
- 27.Miller S.S., Edelman J.L. Active ion transport pathways in the bovine retinal pigment epithelium. J. Physiol. 1990;424:283–300. doi: 10.1113/jphysiol.1990.sp018067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirose T., Wolf E., Schepens C.L. Retinal functions in snowflake degeneration. Ann. Ophthalmol. 1980;12:1135–1146. [PubMed] [Google Scholar]
- 29.Iwashita M., Watanabe M., Ishii M., Chen T., Johnson S.L., Kurachi Y., Okada N., Kondo S. Pigment pattern in jaguar/obelix zebrafish is caused by a Kir7.1 mutation: Implications for the regulation of melanosome movement. PLoS Genet. 2006;2:e197. doi: 10.1371/journal.pgen.0020197. [DOI] [PMC free article] [PubMed] [Google Scholar]