

Abstract
Autism is a childhood neuropsychiatric disorder that, despite exhibiting high heritability, has largely eluded efforts to identify specific genetic variants underlying its etiology. We performed a two-stage genetic study in which genome-wide linkage and family-based association mapping was followed up by association and replication studies in an independent sample. We identified a common polymorphism in contactin-associated protein-like 2 (CNTNAP2), a member of the neurexin superfamily, that is significantly associated with autism susceptibility. Importantly, the genetic variant displays a parent-of-origin and gender effect recapitulating the inheritance of autism.
Main Text
Autistic disorder (MIM 290850), first described by Kanner in 1943,1 is a pervasive developmental disorder characterized by a triad of marked features: impaired social interaction, impaired language development, and restricted and repetitive behavior and interests. A diagnosis of autism can typically be made by 4 years of age. The prevalence is approximately 20 per 10,000 for autistic disorder and 60 per 10,000 individuals for all autism spectrum disorders, with males being 4 times as likely, as compared to females, to be affected.2 There is no doubt that autism presents a significant disease burden.
Compelling evidence for a genetic basis for autism has been provided by twin studies, demonstrating a significantly higher concordance rate for monozygous versus dizygous twins, with an overall heritability of 80%–90%.3 Consequently, it is expected that appropriate genomic screens can identify susceptibility genes given the major genetic component to familiality. With the availability of new genotyping technologies that can survey the genome at far higher resolution than before and large family collections with sufficient samples for both discovery and validation, we initiated a two-stage genome-wide study of autism that is not limited by our current understanding of autism pathophysiology.
For stage I, we selected 72 multiplex families (68 with 2 affected children and 4 with 3 affected children) comprising 148 affected offspring and 292 individuals. We attempted to reduce phenotypic heterogeneity and increase the genetic contribution by requiring all affected individuals to be positive for autism on both ADI-R and ADOS instruments4 and to have onset <36 months. This sampling was in contrast to accepting only an ADI-R classification of autism or accepting the broader ADOS classification of autism spectrum disorder. No previously reported genetic study of autism has had similarly strict phenotypic inclusion criteria and equivalent sample size. All samples were obtained from the National Institute of Mental Health (NIMH) Autism Genetics Initiative.
We genotyped all samples by using Affymetrix 500K arrays with genotypes inferred via the Affymetrix BRLMM genotyping algorithm at the default settings. We used relatively stringent quality control cut-offs for including SNPs in our analyses, because even moderate missing data or error rates increase false-positive linkage and family-based association tests such as the transmission disequilibrium test (TDT).5 Specifically, SNPs with >10% missing data, >1% Mendelian error, and lack of fit to Hardy-Weinberg proportions (p < 0.001) were excluded from analysis, leaving 72% (336,121 of 468,411) of the data on autosomal SNPs for further analysis. One family was excluded because of Mendelian errors arising from maternal incompatibility, and one child was excluded because he was incompatible with both parents, resulting in 78 sib-pairs and 145 parent/child trios that we included in the analyses.
Genome-wide association analysis with the TDT was performed for both single-SNP and haplotypes with EATDT,6 and no genome-wide significant SNPs or haplotypes were identified. However, under a scenario in which multiple unlinked variants within a locus contribute to autism susceptibility, as opposed to a single variant of large effect, the incorporation of traditional linkage data can be of great benefit. Indeed, genome-wide linkage analysis by MERLIN7 revealed two loci with LOD scores above 2: one at chromosome 7q35 (maximum LOD score 3.4 at 151.4–154.4 cM; Figure 1A) and the second at chromosome 10p13–14 (maximum LOD score 2.9 at 26.6–34.5 cM). The peak at 7q35 is genome-wide significant and is a novel finding for strictly defined autism, though it is in the same region that has been previously identified as a possible language quantitative trait locus (QTL) in autism families.8 TDT in the 1-LOD genetic interval under the chromosome 7q35 linkage peak revealed a single SNP, rs7794745, with significant association with autism (p < 2.14 × 10−5) (Figure 1B), even after correcting for the number of SNPs tested under the linkage peak by permutation (p < 0.006). rs7794745 had data completeness of 99.7%, no observed Mendelian errors, and was in Hardy-Weinberg equilibrium (p = 0.98). These genotypes were then independently validated by TaqMan assays. The T allele at SNP rs7794745 is overtransmitted with a transmission frequency of τ = 0.68. This SNP is a common polymorphism with minor allele frequency of 0.36 and resides in the intron between exons 2 and 3 of the CNTNAP2 gene (Figure 2). CNTNAP2 (MIM 604569), or contactin-associated protein-like 2, is a large gene spanning 2.5 Mb and encodes a member of the neurexin family9 that are known to mediate cell-cell interactions in the nervous system. CNTNAP2 protein is localized at the juxtaparanodes of myelinated axons and may be involved in axon differentiation.10 Consequently, it is an excellent candidate gene for autism.
Figure 1.
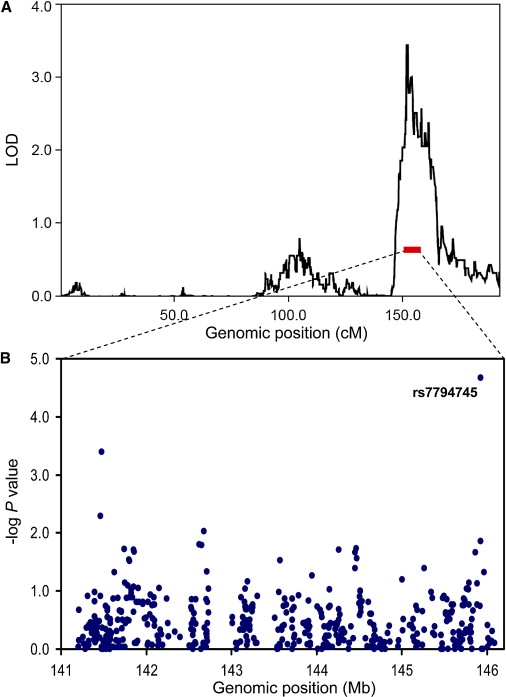
Genome-wide Linkage and Association Studies in Autism
(A) Linkage studies were conducted in 72 multiplex families from the NIMH Repository and yielded a single significant result on chromosome 7q35.
(B) Family-based association test (TDT) results in 145 trios, from the families in (A), for the 1-LOD interval (marked by a red bar) under the linkage peak. Genomic position is on the x axis (cM, centiMorgan; Mb, megabase) and the negative base-10 logarithm of the p value on the y axis.
Figure 2.
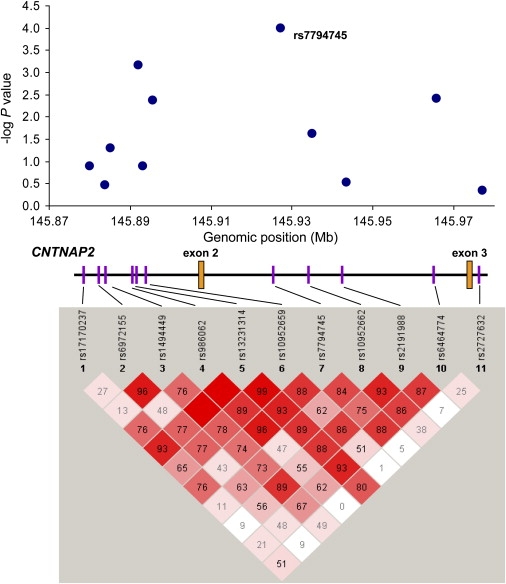
Fine Mapping of CNTNAP2
The top panel shows the results of association tests in 1440 trios with significance for each SNP shown as the negative base-10 logarithm of the p value on the y axis plotted against genomic position in megabase (Mb) on the x axis. The CNTNAP2 exons 2 and 3 are shown in orange. The bottom panel shows all pairwise associations between 11 SNPs at the CNTNAP2 locus. The value within each diamond is the linkage disequilibrium statistic D′. Diamonds without a number correspond to D′ = 1; shading represents the magnitude and significance of pair-wise linkage disequilibrium (LD) with a red-to-white gradient reflecting higher to lower LD values (see Haploview online for further details).
To validate this initial finding, we genotyped an independent sample of 1295 parent-child trios from the NIMH Repository for rs7794745 and again found overtransmission of the T allele (p < 0.005). Genotyping was performed with TaqMan assays and we obtained 98.5% complete data with no observed Mendelian errors or deviation from Hardy-Weinberg equilibrium (p = 0.83). The minor allele frequency was 0.38, similar to that observed in stage I, but the genetic effect was smaller (τ = 0.54). It is important to note that our stage II samples used a broader definition of autism (ADI-R-positive without requiring ADOS classification of either autism or autism spectrum disorder) than in stage I, increasing phenotypic heterogeneity, and this may explain the reduced strength of the effect of rs7794745. However, when we examined 145 multiplex families (303 affected children) from stage II corresponding to the same selection criteria as stage I, the strength of the effect was no different than the remainder of the stage II samples (data not shown), suggesting that the strength of the effect seen in stage I likely reflects a “winner's curse” and is an overestimate of the true effect. Nevertheless, a significant overtransmission of the T allele is observed in two independent family-based samples, confirming that CNTNAP2 is an autism-susceptibility gene. Additional studies incorporating specific domains of autism may shed light on which specific autistic phenotypes are associated with variation in CNTNAP2, because heterogeneity in the genetic effect is observed.
To further characterize the genetic properties of rs7794745, we examined transmission stratified by parental gender and by offspring gender given the marked sex difference in the incidence of autism. As shown in Table 1, the overall transmission frequency (τ = 0.55: p < 7.35 × 10−5) is significantly greater from mothers (τ = 0.61) than from fathers (τ = 0.53) in the combined sample, and this parent-of-origin difference is significant (p < 0.001). Interestingly, this genetic effect and difference is largely observed in affected males than females, although the rarity of affected females implies that the power to detect the observed difference in females is low. To estimate the genetic effect of the T allele, we focused on stage II results, because they are unlikely to reflect any winner's curse and have assumed a normally distributed, but unobservable, liability scale with a threshold determining affectation status.11 Penetrances were then estimated under Morton and Mclean's mixed model of inheritance,12 assuming a prevalence of 0.0032 in males and 0.0008 in females (overall prevalence of 1:500, males 4 times as likely to be affected), and the relative risk stratified by rs7794745 genotype and sex is shown in Figure 3. Our data are compatible with the hypothesis that the common variant we detect is a disease variant only when inherited through the female germline. The cause of this biased transmission is unclear, because a null paternal allele of CNTNAP2 is associated with obsessive-compulsive disorder,13 suggesting that the paternal allele is normally expressed. Our finding of a parent-of-origin bias in the genetic effect is intriguing and needs to be replicated by other studies. Nevertheless, differential genetic effects from the two parents are not unexpected in complex diseases with a sex difference.14
Table 1.
Transmission of the CNTNAP2 Genetic Polymorphism rs7794745 to Affected Children
| n | A Allele | T Allele | τ | τM | τP | p | p∗ | |
|---|---|---|---|---|---|---|---|---|
| Stage I | 137 | 44 | 93 | 0.68 | 0.75 | 0.67 | 2.14 × 10−5 | 2.58 × 10−5 |
| Stage II | 1219 | 561 | 658 | 0.54 | 0.59 | 0.51 | 0.005 | 0.003 |
| Combined | 1356 | 605 | 751 | 0.55 | 0.61 | 0.53 | 7.35 × 10−5 | 0.001 |
| Males | 1077 | 468 | 609 | 0.57 | 0.64 | 0.53 | 1.74 × 10−5 | 3.75 × 10−4 |
| Females | 279 | 137 | 142 | 0.51 | 0.51 | 0.51 | 0.77 | 0.93 |
n is the total number of transmissions; τ is the transmission frequency of the T allele; τM and τP refer to maternal and paternal transmission frequencies, respectively; and p and p∗ refer to the overall significance of τ and the significance of the parent-of-origin effect, respectively.
Figure 3.
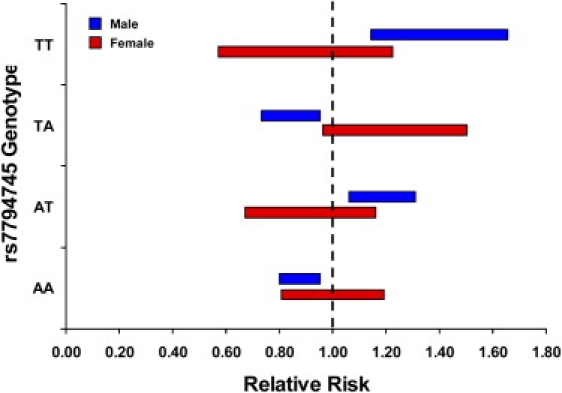
Relative Risk of rs7794745 Genotype Stratified by Sex
The first allele of each genotype represents the paternal allele, and the second allele represents the maternal allele. Relative risk is compared to the sex-specific population risk, assuming overall prevalence of 1:500 and males 4 times more likely to be affected than females. The bars indicate 95% confidence intervals.
In an attempt to fine-map the functional variant in CNTNAP2 contributing to the observed association with autism, we genotyped 10 additional SNPs flanking rs7794745 in the combined stage I and II samples. These SNPs were chosen to tag the LD block containing rs7794745 based on data from the HapMap CEU population. No single SNP showed greater significance than rs7794745 (Figure 2), and no haplotypes showed a marked increased in significance (data not shown), suggesting that either rs7794745, or any other variant highly correlated with it, may be candidates or surrogates for the functional variant.
Our findings are particularly intriguing in light of the recent study by Strauss and colleagues who linked recessive loss-of-function alleles of CNTNAP2 with cortical dysplasia-focal epilepsy (CDFE [MIM 610042]); 67% of the children with CDFE are also diagnosed with autism.15 CNTNAP2 is a very large gene spanning more than 2.5 Mb and maps to a region of chromosomal fragility.16 Consequently, it is possible that additional variants in this gene, including genomic copy number alterations, could also contribute to autism. Indeed, two additional genetic studies in this issue of AJHG, one identifying an association with a rare nonsynonymous variant17 and one demonstrating an association with common variants and the language component of autism,18 also point to CNTNAP2 as an autism-susceptibility gene.
Given that Alarcón and colleagues also identified a common noncoding variant in CNTNAP2,18 and there is some overlap in samples, it is important to highlight the independent yet complementary nature of our two findings. First, different phenotypes were studied, with Alarcón and colleagues focusing on a quantitative trait, “age at first word,” whereas we used a qualitative strict autism diagnosis. Second, although 70/72 families from our stage I sample were included in the Alarcón study, 2/3 of our stage II samples were nonoverlapping. Even after exclusion of all overlapping samples from our stage II data, rs779475 was still significantly associated with autism (p < 0.05), demonstrating the independence of this association. Third, and most importantly, the variants identified in both studies are more than 1 Mb apart and show no evidence of linkage disequilibrium, indicating that despite being in the same gene and exhibiting a male-specific bias, these are truly independent loci providing independent evidence for association of common noncoding variants in CNTNAP2 with autism susceptibility. Indeed, the combined evidence from all three studies strongly suggests the existence of alleleic heterogeneity that needs to be addressed fully by genotyping all samples with the same sets of markers and sequencing all coding exons.
In conclusion, we identified a common variant in CNTNAP2 that is associated with increased risk for autism in two independent family-based samples and exhibits a parent-of-origin bias. Furthermore, given the strength of our initial linkage signal, it is likely that additional genetic variants in this gene that contribute to autism susceptibility remain to be discovered.
Acknowledgments
We thank all of the families who have participated in and contributed to the public resource that we have used in these studies. We thank Drs. Andrew West and Dan Geschwind for discussions of autism genetics and the role of CNTNAP2. This research was funded by grants from the National Institute of Mental Health (MH60007).
The collection of data and biomaterials in one project that participated in the National Institute of Mental Health (NIMH) Autism Genetics Initiative has been supported by National Institutes of Health grants MH52708, MH39437, MH00219, and MH00980; National Health Medical Research Council grant 0034328; and by grants from the Scottish Rite, the Spunk Fund, Inc., the Rebecca and Solomon Baker Fund, the APEX Foundation, the National Alliance for Research in Schizophrenia and Affective Disorders (NARSAD), the endowment fund of the Nancy Pritzker Laboratory (Stanford); and by gifts from the Autism Society of America, the Janet M. Grace Pervasive Developmental Disorders Fund, and families and friends of individuals with autism. The principal investigators and coinvestigators were: Neil Risch, Ph.D., Richard M. Myers, Ph.D., Donna Spiker, Ph.D., Linda J. Lotspeich, M.D., Joachim Hallmayer, M.D., Helena C. Kraemer, Ph.D., Roland D. Ciaranello, M.D., and Luca L. Cavalli-Sforza, M.D. (Stanford University, Stanford, CA); and William M. McMahon, M.D., and P. Brent Petersen (University of Utah, Salt Lake City, UT). The Stanford team is indebted to the parent groups and clinician colleagues who referred families. The Stanford team extends our gratitude to the families with individuals with autism who were our partners in this research.
The collection data and biomaterials come from the Autism Genetic Resource Exchange (AGRE) collection. This program has been supported by a National Institutes of Health grant MH64547 and the Cure Autism Now Foundation. The principal investigator is Daniel H. Geschwind, M.D., Ph.D. (UCLA). The coprincipal investigators include Stanley F. Nelson, M.D., and Rita Cantor, Ph.D. (UCLA), Christa Lese Martin, Ph.D. (U. Chicago), and T. Conrad Gilliam, Ph.D. (Columbia). Coinvestigators include Maricela Alarcon, Ph.D., Kenneth Lange, Ph.D., and Sarah J. Spence, M.D., Ph.D. (UCLA); David H. Ledbetter, Ph.D. (Emory); and Hank Juo, M.D., Ph.D. (Columbia). Scientific oversight of the AGRE program is provided by the AGRE steering committee (chair, Daniel H. Geschwind, M.D., Ph.D; members: W. Ted Brown, M.D., Ph.D., Maja Bucan, Ph.D., Joseph Buxbaum, Ph.D., T. Conrad Gilliam, Ph.D., David Greenberg, Ph.D., David Ledbetter, Ph.D., Bruce Miller, M.D., Stanley F. Nelson, M.D., Jonathan Pevsner, Ph.D., Carol Sprouse, Ed.D., Gerard Schellenberg, Ph.D., and Rudolph Tanzi, Ph.D.).
The collection of data and biomaterials in another project has been supported by a supplement to National Institutes of Health grant MH61009 (“Molecular Genetics of 15q11–q13 Defects in Autism”) and by Development Funds from the Vanderbilt Centers for Human Genetics Research and Kennedy Center for Research on Human Development. The principal investigator was James S. Sutcliffe, Ph.D. (Vanderbilt University, Nashville, TN). The coinvestigator was Jonathan L. Haines, Ph.D., and the Clinical and Phenotypic Coordinator for this project was Genea Crocket, M.S.
The collection of data and biomaterials in another project has been supported by National Institutes of Health grant MH55135 (“Collaborative Linkage Study of Autism”). The principal investigator was Susan E. Folstein, M.D. (Tufts University/New England Medical Center, Boston, MA), and her key Clinical and Phenotypic Coordinators were Brian Winklosky and Beth Rosen-Sheidley, M.S., C.G.C. Coinvestigators included James S. Sutcliffe, Ph.D. and Jonathan L. Haines, Ph.D. (Vanderbilt University, Nashville, TN).
The collection of data and biomaterials in another project has been supported by National Institutes of Health grant MH55284. The principal investigator and coinvestigators were Joseph Piven, M.D. (University of North Carolina, Chapel Hill); Val Sheffield, M.D., Ph.D., Veronica Vieland, Ph.D., and Thomas Wassink, M.D. (University of Iowa, Iowa City).
Web Resources
The URLs for data presented herein are as follows:
Haploview, http://www.broad.mit.edu/mpg/haploview
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
References
- 1.Kanner L. Autistic disturbances of affective contact. Nervous Child. 1943;2:217–250. [PubMed] [Google Scholar]
- 2.Chakrabarti S., Fombonne E. Pervasive developmental disorders in preschool children: confirmation of high prevalence. Am. J. Psychiatry. 2005;162:1133–1141. doi: 10.1176/appi.ajp.162.6.1133. [DOI] [PubMed] [Google Scholar]
- 3.Folstein S.E., Rosen-Sheidley B. Genetics of autism: complex aetiology for a heterogeneous disorder. Nat. Rev. Genet. 2001;2:943–955. doi: 10.1038/35103559. [DOI] [PubMed] [Google Scholar]
- 4.Risi S., Lord C., Gotham K., Corsello C., Chrysler C., Szatmari P., Cook E.H., Jr., Leventhal B.L., Pickles A. Combining information from multiple sources in the diagnosis of autism spectrum disorders. J. Am. Acad. Child Adolesc. Psychiatry. 2006;45:1094–1103. doi: 10.1097/01.chi.0000227880.42780.0e. [DOI] [PubMed] [Google Scholar]
- 5.Mitchell A.A., Cutler D.J., Chakravarti A. Undetected genotyping errors cause apparent overtransmission of common alleles in the transmission/disequilibrium test. Am. J. Hum. Genet. 2003;72:598–610. doi: 10.1086/368203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin S., Chakravarti A., Cutler D.J. Exhaustive allelic transmission disequilibrium tests as a new approach to genome-wide association studies. Nat. Genet. 2004;36:1181–1188. doi: 10.1038/ng1457. [DOI] [PubMed] [Google Scholar]
- 7.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 8.Alarcon M., Yonan A.L., Gilliam T.C., Cantor R.M., Geschwind D.H. Quantitative genome scan and ordered-subsets analysis of autism endophenotypes support language QTLs. Mol. Psychiatry. 2005;10:747–757. doi: 10.1038/sj.mp.4001666. [DOI] [PubMed] [Google Scholar]
- 9.Poliak S., Gollan L., Martinez R., Custer A., Einheber S., Salzer J.L., Trimmer J.S., Shrager P., Peles E. Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron. 1999;24:1037–1047. doi: 10.1016/s0896-6273(00)81049-1. [DOI] [PubMed] [Google Scholar]
- 10.Poliak S., Salomon D., Elhanany H., Sabanay H., Kiernan B., Pevny L., Stewart C.L., Xu X., Chiu S.Y., Shrager P. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J. Cell Biol. 2003;162:1149–1160. doi: 10.1083/jcb.200305018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Falconer D.S. The inheritance of liability to certain diseases, estimated from the incidence among relatives. Ann. Hum. Genet. 1965;29:51–76. [Google Scholar]
- 12.Morton N.E., McLean C.J. Analysis of family resemblance. III. Complex segregation of quantitative traits. Am. J. Hum. Genet. 1974;26:318–330. [PMC free article] [PubMed] [Google Scholar]
- 13.Verkerk A.J., Mathews C.A., Joosse M., Eussen B.H., Heutink P., Oostra B.A. CNTNAP2 is disrupted in a family with Gilles de la Tourette syndrome and obsessive compulsive disorder. Genomics. 2003;82:1–9. doi: 10.1016/s0888-7543(03)00097-1. [DOI] [PubMed] [Google Scholar]
- 14.Emison E.S., McCallion A.S., Kashuk C.S., Bush R.T., Grice E., Lin S., Portnoy M.E., Cutler D.J., Green E.D., Chakravarti A. A common sex-dependent mutation in a RET enhancer underlies Hirschsprung disease risk. Nature. 2005;434:857–863. doi: 10.1038/nature03467. [DOI] [PubMed] [Google Scholar]
- 15.Strauss K.A., Puffenberger E.G., Huentelman M.J., Gottlieb S., Dobrin S.E., Parod J.M., Stephan D.A., Morton D.H. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N. Engl. J. Med. 2006;354:1370–1377. doi: 10.1056/NEJMoa052773. [DOI] [PubMed] [Google Scholar]
- 16.Smith D.I., Zhu Y., McAvoy S., Kuhn R. Common fragile sites, extremely large genes, neural development and cancer. Cancer Lett. 2006;232:48–57. doi: 10.1016/j.canlet.2005.06.049. [DOI] [PubMed] [Google Scholar]
- 17.Bakkaloglu B., O'Roak B.J., Louvi A., Gupta A.R., Abelson J.F., Morgan T.M., Chawarska K., Klin A., Ercan-Sencicek A.G., Stillman A.A. Molecular cytogenetic analysis and resequencing of Contactin Associated Protein-Like 2 in autism spectrum disorders. Am. J. Hum. Genet. 2007;82:165–173. doi: 10.1016/j.ajhg.2007.09.017. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alarcón M., Abrahams B.S., Stone J.L., Duvall J.A., Perederiy J.V., Bomar J.M., Sebat J., Wigler M., Martin C.L., Ledbetter D.H. Linkage, association, and gene expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Hum. Genet. 2007;82:150–159. doi: 10.1016/j.ajhg.2007.09.005. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]