

Abstract
We describe a distinct retinal disorder, autosomal-recessive bestrophinopathy (ARB), that is consequent upon biallelic mutation in BEST1 and is associated with central visual loss, a characteristic retinopathy, an absent electro-oculogram light rise, and a reduced electroretinogram. Heterozygous mutations in BEST1 have previously been found to cause the two dominantly inherited disorders, Best macular dystrophy and autosomal-dominant vitreoretinochoroidopathy. The transmembrane protein bestrophin-1, encoded by BEST1, is located at the basolateral membrane of the retinal pigment epithelium in which it probably functions as a Cl− channel. We sequenced BEST1 in five families, identifying DNA variants in each of ten alleles. These encoded six different missense variants and one nonsense variant. The alleles segregated appropriately for a recessive disorder in each family. No clinical or electrophysiological abnormalities were identified in any heterozygotes. We conducted whole-cell patch-clamping of HEK293 cells transfected with bestrophin-1 to measure the Cl− current. Two ARB missense isoforms severely reduced channel activity. However, unlike two other alleles previously associated with Best disease, cotransfection with wild-type bestrophin-1 did not impair the formation of active wild-type bestrophin-1 channels, consistent with the recessive nature of the condition. We propose that ARB is the null phenotype of bestrophin-1 in humans.
Introduction
The retinal pigment epithelium (RPE) is the cellular monolayer that interdigitates with the outer segments of photoreceptors and is critical in homeostatic maintenance of the outer retina.1 Furthermore, it is critical for the phagocytosis of shed outer segments and is the site for the regeneration of the chromophore 11-cis-retinal. Several inherited retinal disorders are caused by mutations in RPE-expressed genes including both macular and generalized photoreceptor dystrophies (RetNet). A classic example is Best disease or vitelliform macular dystrophy (VMD, MIM 153700), an autosomal-dominant disorder associated with a reduced or absent electro-oculogram (EOG) and an accumulation of yellowish material between the RPE and photoreceptors in the macula. The condition leads to visual loss in late adolescence or adulthood and is caused by mutations in the BEST1 gene.2
The protein product of BEST1, bestrophin-1, is a 585 amino acid transmembrane protein located at or close to the basolateral membrane of the RPE.3 Bestrophin-1 functions as a Cl− channel when expressed in HEK293 cells,4–6 and dominant mutations in putative transmembrane domains of the murine ortholog alter anion permeability.6 Bestrophin-1 also alters the activity of L type Ca2+ channels when expressed in RPE-derived cells, suggesting that it might also act to regulate entry of Ca2+ into RPE cells.7 Bestrophin-1 is thought to exist as an oligomer, and when purified from porcine RPE, exists as a dimer.8
Over 100 different BEST1 mutations have been described in families affected by Best disease (University of Regensberg VMD2 database; e.g.,9–12). Almost all are missense mutations located within the first 310 residues, often within or close to transmembrane domains. Some mutations induce smaller whole-cell currents than wild-type isoforms when expressed in HEK293 cells and act in a dominant-negative manner because their coexpression with wild-type bestrophin-1 severely reduces or abolishes wild-type currents.4,13,14 Phenotypic variability among patients carrying these mutations includes those with adult-onset vitelliform dystrophy as well as atypical forms of Best disease, including a single report by Schatz et al. (2006) of a case associated with compound heterozygous mutations.15 A second distinct dominant disorder, autosomal-dominant vitreoretinochoroidopathy (ADVIRC, MIM 193220), has also been reported to be caused by mutation of BEST1.16 These mutations disrupt BEST1 pre-mRNA splicing and are predicted to cause in-frame deletions.
Here we report a third distinct disorder, autosomal-recessive bestrophinopathy (ARB), due to BEST1 mutation. We characterize the detailed clinical features of 5 families, determine mutations in each of the alleles and show segregation as a recessive disorder. We conduct whole-cell patch-clamping of bestrophin-1 transfected HEK293 cells to measure the Cl− current and show a physiological difference between ARB alleles and two others associated previously with Best disease. We subsequently propose that ARB represents the human null phenotype of bestrophin-1.
Material and Methods
Patient Details
The study was approved by Research Ethics Committees at Moorfields, Manchester Eye, and Ghent University Hospitals. Informed consent was obtained from all participants prior to conducting investigations. Clinical examination of members of the affected families included Snellen visual acuity testing, slit lamp biomicroscopy, fundoscopy, ISCEV-standard full-field electroretinogram (ERG), pattern ERG (PERG) and EOG,17–19 and infrared, autofluorescence, and red free imaging. Blood was taken from all family members by venesection, and genomic DNA was extracted according to standard procedures.
DNA Sequencing
The exonic regions of BEST1 were analyzed by sequencing in families 1–5. The absence of any disease causing mutations for the three novel mutations p.P152A, p.R200X, and p.V317M in 210 control chromosomes was confirmed by SSCP analysis or restriction digest. For SSCP/heteroduplex analysis (p.V317M), 5 μl PCR product was mixed with 5 μl of formamide loading dye, denatured, and separated on an 8% acrylamide/bis-acrylamide gel (350 V for 16 hr at 4°C) and silver stained in accordance with standard methods. Restriction digests for the presence of an additional Cac8I site in a 156 bp amplicon (p.P152A) or absence of a TaqI site in a 250 bp amplicon (p.R200X) were carried out according to the manufacturer's instructions.
Primers were designed to amplify all BEST1 coding exons including 50–100 bp of flanking intron sequence (obtained from the UCSC Genome Browser; primers available on request). PCR reactions with Abgene Reddymix Taq (Abgene, Epsom, UK) contained 50 ng genomic DNA in a volume of 30 μl and were cycled as follows: 95°C for 5 min and then 30 cycles of 94°C for 30 s; “x” °C for 30 s; 72°C for 1 min (for which x °C is primer annealing temperature) with a final extension of 72°C for 10 min. PCR products were purified with Microcon columns (Millipore, Watford, UK) according to the manufacturer's instructions. Standard cycle-sequencing reactions with BigDye terminator mix v1.1 (Applied Biosystems, Warrington, UK) contained 3–10 ng purified PCR product in 10 μl and were performed with forward and reverse primers used for initial amplification. The sequencing reactions were precipitated, dried, and analyzed on an ABI 3700 capillary sequencer.
Cloning
The wild-type BEST1, p.W93C, and p.R218C cDNA clones in pADlox3 were a kind gift from Prof A Marmorstein (University of Arizona). The wild-type clone was used for generating the p.R141H and p.P152A mutants with the QuickChange II site-directed mutagenesis kit (Stratagene, Amsterdam, Netherlands) per the manufacturer's protocol.
In Vitro Electrophysiology
HEK293 Cell Culture
HEK293 cells were grown in minimum essential medium with Earle's salts and L-glutamine (Invitrogen, Paisley, UK), containing 9% (v/v) fetal-calf serum (Globepharm, Guildford, UK), 1% (v/v) nonessential amino acids, gentamicin (0.25 mg/ml), penicillin G (100 U/ml), streptomycin (100 μg/ml), and amphotericin (0.25 μg/ml (all Invitrogen) at 37°C in a humidified atmosphere of air:CO2 (19:1).
Transient-Transfection Studies
In studies examining bestrophin-1 Cl−-channel function, HEK293 cells were transiently transfected with pADlox containing p.W93C, p.R218C (both Best disease mutations), p.R141H, p.P152A (both ARB mutations), or wild-type BEST1. Cells were cotransfected at a 5:1 ratio with the empty vector, pEGFP-C1, as previously described.20 Positively transfected cells were visualized by GFP fluorescence, and recordings made only from these cells. We performed in vitro assay for each vector to show that each was transcribed to the same level. Recordings of voltage-gated calcium currents (CaV) were made in HEK293 cells stably expressing L type α1C subunits.21 Cells were transiently cotransfected with an auxiliary β2a subunit linked to the red fluorescent protein (RFP) and either wild-type BEST1 or the p.R141H mutant, as previously described.20 Cells were transfected in a 5:1 ratio (BEST1:RFP-β2a), and determination of successful cotransfection was made by visualization of RFP. The β2a subunit was a kind gift from Dr. E. Perez-Reyes (University of Virginia). The RFPβ2a construct and the α1C cell line were created by Mr. S. Brown (McMaster University).
Whole-Cell Patch Clamp
Patch pipettes were borosilicate capillary tubes (WPI) and were pulled on a two-stage vertical puller (PC10, Narishige) and fire polished with tip resistances between 2–4 MΩ. Conventional whole-cell recordings were made after establishment of gigaohm seals and measured with a Multiclamp 700B amplifier (Axon) in the voltage clamp mode. Voltage protocols were driven from an IBM-compatible computer equipped with a Digidata interface (Axon) with pClamp 9 software (Axon).
In experiments that would determine Cl−-selective currents, the extracellular bath solution contained 140 mM NaCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, 30 mM mannitol, and 10 mM HEPES, and pH was 7.4 with NaOH. The pipette solution was a low Cl−, high Ca2+ solution containing 20 mM CsCl, 110 mM Cs aspartate, 2 mM MgCl2, 10 mM glucose, 10 mM HEPES, 10 mM EGTA, and 7.2 mM CaCl2 and pH was adjusted to 7.2 with CsOH. Free Ca2+ concentration was calculated to be approximately 0.5 μM with the WEBMAXC program. Cs+-containing/K+-free solutions eliminated any potential contamination of the whole-cell currents by endogenous K+ channels. Asymmetrical Cl− concentrations were used (bath = 146 mM; pipette = 38.4 mM) to help distinguish anion currents (which should reverse toward ECl = −34 mV) from leak and nonspecific cation currents that should reverse at 0 mV. Voltage was stepped from a holding potential of −50 mV to between −120 to +80 mV in +20 mV steps, and each step duration was 450 ms. Whole-cell capacitance was compensated with the analog circuitry of the Multiclamp amplifier.
In experiments that would determine the effects of bestrophin-1 on CaV function, 20 mM Ba2+ was used as charge carrier (for solution composition, see Hudasek et al.21). Cells were voltage clamped at −80 mV, and currents were evoked by step-depolarizing cells to +10 mV for 100 ms. Currents were filtered at 5 kHz and sampled at 10 kHz. Activating sections of current records were fit with a double exponential function of the form:
in which A1 and A2 are the amplitudes of the fast and slow components of channel activation, respectively, τ2 and τ1 are the time constants of these components, K is the time at the start of the fit, and C is the offset.
Results are presented as mean ± SEM. Data were analyzed by ANOVA and Student's t test where appropriate.
Results
Clinical Findings and Molecular Data
Seven affected individuals from five unrelated families of European ethnicity were examined. The individual clinical details are summarized in Table 1, and the pedigrees are shown in Figure 1. Representative images from color photography, fluorescein angiography, autofluorescence imaging, and OCT imaging are shown in Figure 2. In all affected individuals, clinical electrophysiology demonstrated abnormal full-field ERGs in addition to a severe reduction in the EOG light rise analogous to that seen with dominant BEST1 mutations that cause both Best disease and ADVIRC (see Figures 3 and 4). Sequencing of the coding fragments of BEST1 identified pathogenic sequence variants in all cases (Figure 1).
Table 1.
Clinical Details of Affected Individuals from Five Families with ARB
| Family Individual | Age of Visual Deterioration | VA (Snellen) / Refraction | Angle Closure Glaucoma | Retina | Fluorescein Angiography | ERG | EOG | Mutation cDNA (protein) |
|---|---|---|---|---|---|---|---|---|
| 1.1 (male) | 30 years | OD 20/60 +4/+1.75 @ 95°OS 20/200 +4/+1.5 @ 90° | no | irregular pale reflex from RPE, peripheral sparing; small pale subretinal deposits | early patchy hyperfluorescence | undetectable pattern ERG; reduced rod and cone full-field ERGs | absent EOG light rise | c.598C > T (p.R200X)c.598C > T (p.R200X) |
| 1.2 (female) | 18 years | OD hand movement +1.75/+1.0 @ 110° OS 20/120 +2.25/+0.75 @ 110° | yes | bilateral widespread RPE irregularity with small subretinal deposits at the posterior poles | widespread early patchy hyperfluorescence | reduced rod and cone full-field ERGs | absent EOG light rise | c.598C > T (p.R200X)c.598C > T (p.R200X) |
| 2.1 (female) | 40 years | OD 20/80 +0.5DSOS 20/120 +1.0/+1.75 @ 30° | no | widespread irregularity of the RPE pigmentation, subretinal deposits at the maculae with presumed bilateral macular edema | patchy choroidal fluorescence | pattern ERG amplitudes mildly reduced; rod and cone full-field ERGs delayed and reduced | absent EOG light rise | c.442G > A (p.R141H)c.949G > A (p.V317M) |
| 3.1 (female) | 7 years | OD 20/80 +1.25DS/−1 @ 40° OS 20/120 +1.25DS/0.75@ 130° | no | widespread RPE irregularities and subretinal deposits | not done | pattern ERG amplitudes mildly reduced; rod and cone full-field ERGs delayed and reduced | EOG light rise profoundly reduced | c.122T > C (p.L41P)c.454C > G (p.P152A) |
| 4.1 (female) | 4 years | OD 20/120 +4/+0.5 @ 80°OS 20/200 +3/+0.5 @ 120° | no | scars at each macula and widespread RPE irregularity | patchy hyperfluorescence | pattern ERGs undetectable; reduced amplitude rod ERGs; cone ERGs amplitude reduced and delayed | absent EOG light rise | c.442G > A (p.R141H)c.122T > C (p.L41P) |
| 5.1 (male) | 31 years | OS 20/60 +0.5/−0.5 @ 90°OS 20/60 +0.5/−0.5 @ 90° | yes | macular edema, perifoveal yellow-white deposits; multiple small white dots in periphery | early patchy hyperfluorescence | rod and cone full-field ERGs reduced | EOG light rise profoundly reduced | c.934G > A (p.D312N)c.974T > C (p.M325T) |
| 5.2 (female) | 28 years | OD/OS 20/20 at presentation | yes | bilateral macular edema with white subretinal deposits in the midperiphery | early patchy hyperfluorescence | severely reduced pattern ERG; rod and cone full-field ERGs reduced | EOG light rise profoundly reduced | c.934G > A (p.D312N)c.974T > C (p.M325T) |
OD, right eye; OS, left eye.
Figure 1.
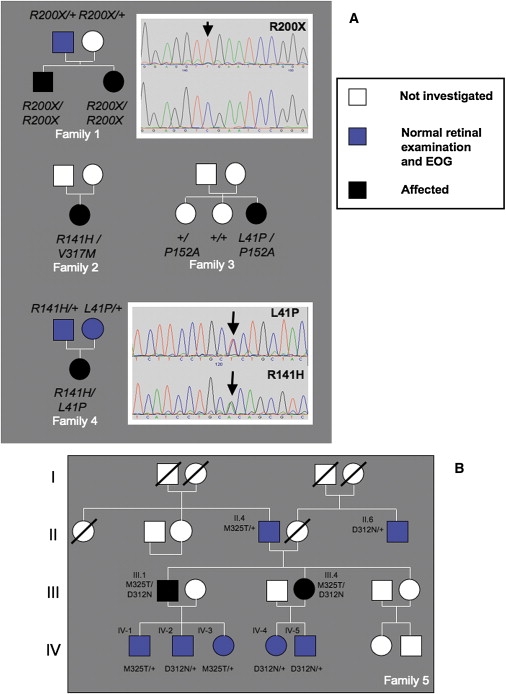
Pedigrees and Mutation Analysis
(A) Pedigrees from families 1–4. Sequencing electropherograms showing homozygous c.598c > t p.R200X variant (top) and normal sequence (underneath) are shown to the right of family 1. Electropherograms from the family 4 patient showing the c.122t > c p.L41P and c.442 g > a p.R141H variants are shown to the right of the pedigree for family 4.
(B) Family 5 pedigree. Pathogenic variants are inherited separately and have no apparent deleterious phenotypic consequences in the heterozygous state.
Figure 2.
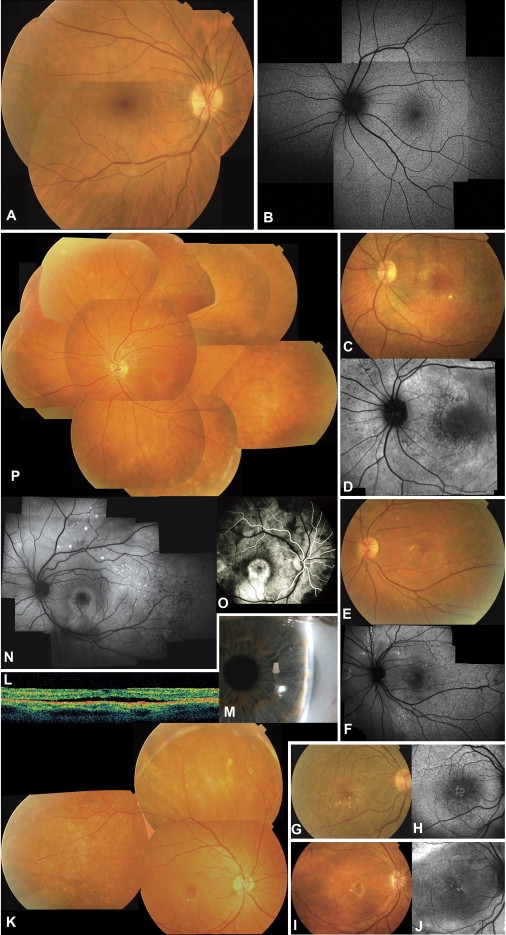
Clinical Features of Autosomal-Recessive Bestrophinopathy
(A–P) Clinical features of normal controls and the five ARB families.
(A) Normal fundus of right eye (RE) of 26-year old woman.
(B) Normal autofluorescence of left eye (LE) of 43-year old man.
(C and D) Fundus picture (C) and autofluorescence (AF) image (D) of LE of proband of family 1. Note presence of diffuse alterations of RPE and white subretinal deposits in macular area and midperiphery; changes are better visible on AF imaging as areas of hypoautofluorescence and hyperautofluorescence.
(E and F) Fundus picture (E) and AF (F) of LE of proband of family 2. Retinal phenotype is similar to that in (C) with white subretinal deposits alternating with areas of RPE atrophy (dark areas on AF).
(G and H) Fundus picture (G) and AF (H) of RE of proband of family 3, showing widespread RPE irregularities with white subretinal deposits, some of which are better seen on AF.
(I and J) Fundus picture (I) and AF (J) of RE of proband of family 4, with central macular scar visible on fundus picture and white deposits better seen on AF.
(K–M) Proband of family 5. (K) shows the composite of fundus of RE with fine white subretinal deposits surrounding area of neurosensory detachment in the macula; additional deposits are visible in retinal midperiphery and far periphery; (L) shows optical coherence tomography (horizontal line scan OCT) of RE showing subretinal fluid between RPE and neurosensory retina; and (M) shows slit-lamp image of extremely shallow anterior chamber of temporal part of LE.
(N–P) Affected sister of proband of family 5. (N) shows the composite AF of LE showing similar, albeit milder, phenotype than that of her brother; in (O), early-stage fluorescein angiography of RE shows immediate hyperfluorescence in central macula probably due to RPE atrophy and retinal edema; and (P) shows the composite of fundus of LE.
Figure 3.
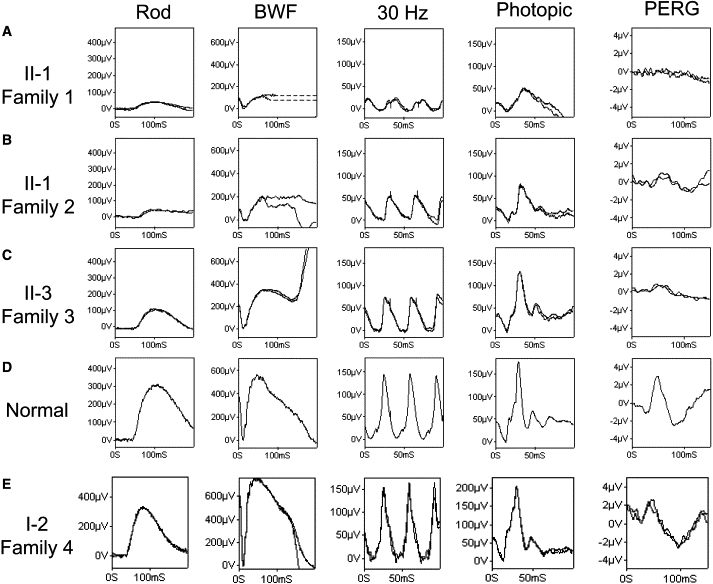
ERG Findings of Autosomal-Recessive Bestrophinopathy
Findings are based on ISCEV (the International Society for Clinical Electrophysiology of Vision) standard. Data from the right eye of four affected patients (A, B, C, and E) and a representative normal control (D) are shown. BWF represents ISCEV standard flash +0.6 log units (better to reveal rod-photoreceptor function, ∼11.5 cd.s/m2); in (A), patient II-1, family 1, shows a similar degree of rod-system dysfunction to patient II-1, family 2, but a greater cone-system dysfunction with lower amplitude and increased implicit time. The almost undetectable PERG is in keeping with severe macular dysfunction. In (B), patient II-1, family 2, shows generalized rod- and cone-system dysfunction with all ERGs being subnormal and delayed. In (C), patient II-3, family 3, shows a subnormal rod-specific ERG, a mildly subnormal BWF ERG a-wave, a borderline subnormal 30 Hz flicker ERG, a photopic ERG within the normal range, and a markedly subnormal PERG. EOGs in all patients (data not shown) contained either a severely reduced or an undetectable light rise. (E) shows ERG (RE) on an unaffected 50-year-old female (I-2, family 4) heterozygous for the p.L41P mutation. Note the normal ERG amplitude and 30 Hz flicker response delay. EOG light rise was 250%.
Figure 4.
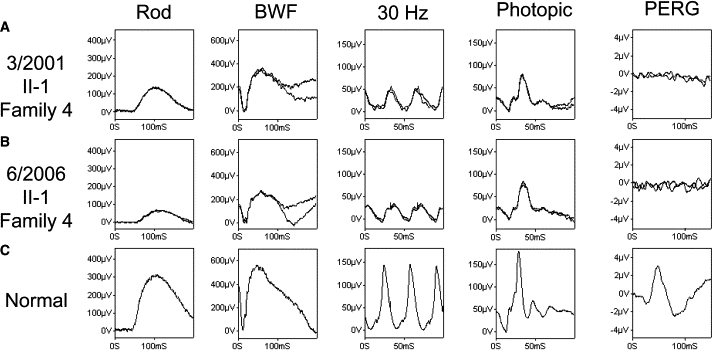
Longitudinal ERG Findings of Autosomal-Recessive Bestrophinopathy
(A and B) Data from one patient (II-1, family 4), showing deterioration over a 5 year period. Initial findings show mildly subnormal rod-system ERGs with more marked cone-system abnormalities (reduced and delayed). All full-field ERGs show amplitude reduction and implicit time increase over a 5 year period, except the single-flash photopic ERG that only shows implicit time increase. The undetectable PERG is in keeping with severe macular dysfunction.
(C) Representative normal control. ERGs were recorded in accordance with the recommendations of ISCEV. The rod-specific ERG arises in the rod ON bipolar cells and acts as a measure of rod-system sensitivity. The BWF ERG has an initial a-wave predominantly arising in relation to photoreceptor function. This response is dominated by the rod system, but there is a small cone contribution. The 30 Hz flicker ERG (recorded with a photopic rod-suppressing background) is a cone-system response arising in inner retina and is the most sensitive measure of generalized cone dysfunction. The single-flash cone has a photopic a-wave to which there is some contribution from the cone photoreceptors (also the OFF bipolar cells). The PERG P50 component acts as a measure of macular function.
From the five families, 5/7 affected individuals were female and the age range at examination was from 28 to 51 years. Six of the individuals presented with symptomatic central visual loss (range 4–40 years), whereas a younger sibling of an affected brother presented before the onset of symptoms (patient 5.2, Table 1). Some older patients (2.1, 3.1) suggested that their central vision had remained stable in recent years. All, on whom refraction was available, were hyperopic, and three individuals from two families required surgery for acute angle-closure glaucoma.
The retinopathy was present in all seven cases and included an irregularity of the RPE throughout the posterior fundus, often with scattered punctate flecks. This was most easily seen during autofluorescence imaging, which showed a widespread variability of autofluorescence. Retinal edema and subretinal fluid was common and was confirmed on OCT imaging. No patients were ever noted to have vitelliform lesions during the course of their follow-up. Fluorescein angiography showed widespread patchy hyperfluorescence, and EOG consistently showed an absent or severely reduced light rise. All seven affected individuals showed significant reduction in the PERG (subserved by macular function) and reduction and delay of the rod and cone ERGs (Figure 3).
None of the families had other affected family members. Ten heterozygotes were examined in detail, and all were asymptomatic, with a normal retinal examination, ERG responses, and EOG light rise. These individuals were heterozygous for the following alleles: p.R200X (aged 68 years), p.R141H (aged 55 years), p.L41P (aged 55 years), p.M325T (three individuals, aged 16–78 years), and p.D312N (four individuals, aged 16–73 years).
Goldmann-Favre syndrome had previously been diagnosed in families 1 and 5, and fundus flavimaculatus had initially been diagnosed in family 3. As part of their clinical workup, families 3 and 5 had molecular analysis of the ABCA4 gene performed. No disease-associated variants were detected in either family.
Molecular analysis revealed that all ten BEST1 alleles were variant (Table 1) encoding six missense (c.122t>c p.L41P, c.442g>a p.R141H, c.454c>g p.P152A, c.934g>a p.D312N, c.949g>a p.V317M, and c.974t>c p.M325T) and one nonsense (c.598c > t p.R200X) mutation. These were in trans in each affected individual (unable to test in family 2) and segregated with disease in the two sets of affected siblings (families 1 and 5).
All novel BEST1 variants were absent from 210 control chromosomes derived from subjects of European descent. A ClustalW analysis of the bestrophin-1 protein shows complete conservation of two regions that harbor 5/6 of the missense variants (p.R141H, p.P152A, p.D312N, p.V317M, and p.M325T), suggesting they have a critical functional role within these regions. The sixth sequence variant (p.L41P) lies outside these regions in an area of decreased conservation (Figure 5). It is of note that 3/6 missense alterations described in these five families are present between residues 312 and 325. These are more C-terminal than the majority of known mutations in bestrophin-1, and this finding suggests that they might lie in a domain with a distinct function. The residues in this region are also highly conserved through evolution and between different members of the bestrophin-1 family of proteins, further suggesting a critical functional role.
Figure 5.

Multiple Alignment of Bestrophin-1 around the Mutated Bestrophin-1 Residues Associated with ARB
Multiple alignment (ClustalW) of bestrophin-1 around the mutated residues (p.L41P, p.R141H, p.P152A, p.D312N, p.V317M, and p.M325T) associated with ARB for nine species (Xenopus tropicalis, Gallus gallus, Tetraodon nigroviridis, Takifugu rubripes, Danio rerio, Pan troglodytes, Homo sapiens, Canis familiarise, and Rattus norvegicus). The boxes represent transmembrane (TM) domains in the model proposed by Milenkovic et al.14 Solid lines represent membrane-embedded TM domains, and dashed lines represent “TM” domains outside the membrane.
Whole-Cell Patch-Clamp Analysis of ARB Mutant Isoforms
All Best-disease-causing mutations reported to date have been autosomal dominant, although with varying penetrance.22,23 However, the mutations reported here were found to be inherited in a nondominant manner, and we investigated the characteristics of two by using whole-cell patch-clamp methods including the p.R141H mutation that had been found to be associated with ARB on two separate occasions. Currents were recorded from HEK293 cells transfected with wild-type bestrophin-1 (Figure 6B) or with the p.R141H mutant (Figure 6C). Large currents were observed with the wild-type channel, particularly at positive (depolarising) potentials, compared to the currents observed in the untransfected HEK293 cells (Figure 6A). The plot of current as function of voltage for the wild-type channel (Figure 6D) is similar to that previously reported for bestrophin-14 and gives a reversal potential of −17.7 ± 1.7 mV (n = 16). This is toward the expected value for an anion-selective current (ECl = −34 mV with 38.4 mM and 146 mM Cl− in the pipette and bath solutions, respectively). This reversal potential indicates that the currents carried by bestrophin-1 are predominantly anion selective and are not due to the activation of nonselective cation channels or an increase in leak current in the transfected cells. Assuming that the channels are impermeant to cations, a value for the relative permeability to the two anions present (Cl− and aspartate) can be calculated, Paspartate: PCl = 0.31. A similar value has been quoted for many other anion-selective channels including other bestrophin channels.13
Figure 6.
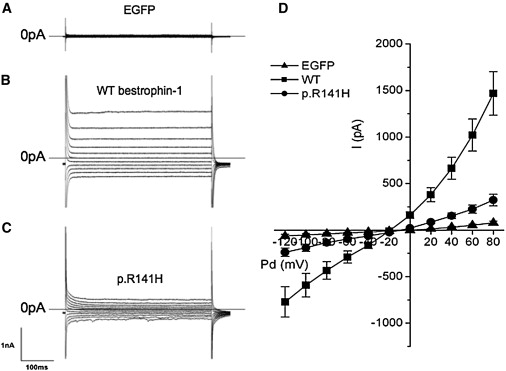
Whole-Cell Currents from Normal HEK293 Cells Transiently Transfected with Wild-Type Bestrophin-1 and p.R141H Bestrophin-1 cDNA
Representative traces of HEK293 cells transiently transfected with the following: (A) EGFP cDNA alone or plus (B) wild-type bestrophin-1 or (C) p.R141H bestrophin-1 cDNA. Voltage was stepped from a holding potential of −50 mV to between −120 and +80 mV in +20 mV steps. Step duration was 450 ms. (D) shows the mean steady-state current-voltage relationship for mock transfected HEK293 cells (n = 9) and cells transfected with wild-type bestrophin-1 (n = 16) or p.R141H bestrophin-1 (n = 15). Results are presented as mean ± SEM.
The currents observed in the cells transfected with the p.R141H mutant were much smaller than the wild-type currents (Figures 6C and 6D). The reversal potential (−17.8 ± 3.3 mV, n = 15), however, was not significantly different from that of the wild-type channel (p > 0.1 by unpaired t test). These data indicate that both wild-type bestrophin-1 and the p.R141H mutant function as anion-selective channels in HEK293 cells but that the size of the whole-cell conductance is significantly reduced with the p.R141H compared to the wild-type channel (p < 0.01; Figure 7C).
Figure 7.
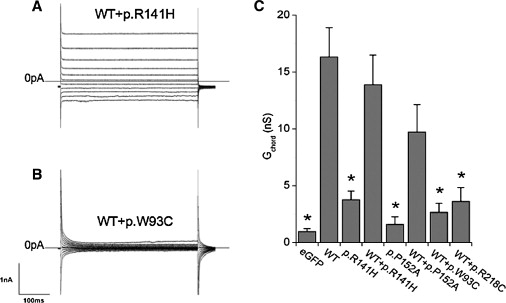
Cotransfection of Normal HEK293 Cells with Wild-Type Bestrophin-1 plus Either ARB or Best Disease Mutations Produces Markedly Different Current Phenotypes
Representative traces of HEK293 cells transiently transfected with wild-type bestrophin-1 plus either (A) p.R141H or (B) p.W93C bestrophin-1. The voltage protocol was as in Figure 6A. (C) shows the mean outward chord conductance (Gchord) in transfected HEK293 cells, calculated over 0 mV to +80 mV. ∗p < 0.05 compared to wild-type. Results are presented as mean ± SEM.
To investigate the apparent autosomal-recessive nature of the p.R141H mutant, we cotransfected it with wild-type bestrophin-1 into HEK293 cells and examined the effect on Cl−-channel activity. Figure 7A shows typical currents measured in these experiments. The currents were similar to those observed with the wild-type channel alone. The reversal potential also indicated that cotransfection generated a predominantly Cl−-selective current (−16.3 ± 2.6 mV). The magnitude of the mean whole-cell conductance from 20 recordings was not significantly different from that observed with the wild-type channel alone (p > 0.1 by ANOVA, Figure 7C). Similar results were obtained with another ARB mutation p.P152A. Much reduced currents were observed in cells transfected only with this mutant (n = 10; p < 0.05 compared to wild-type; Figure 7C). In cells cotransfected with wild-type and p.P152A, however, the conductances of the currents observed were not significantly different from that of the wild-type currents (n = 14; p > 0.1; Figure 7C).
In order to establish whether the effects of cotransfection of mutant and wild-type bestrophin-1 were restricted to ARB mutations, we also performed cotransfection experiments with wild-type bestrophin-1 and two Best-disease-causing mutants, p.W93C and p.R218C. Currents in cells cotransfected with the wild-type and pW93C mutant were similar to those observed in untransfected cells (Figure 7B). Figure 7C shows that the mean conductance measured (n = 16) was not significantly different from that in untransfected cells (p > 0.1) and was significantly less than that observed in cells transfected with wild-type bestrophin-1 alone. Similar results were obtained with p.R218C (n = 14, Figure 7C).
Bestrophin-1 has previously been reported to modulate the activation kinetics of α1D subunits of the L type, voltage-gated Ca2+ channel (CaV) expressed in RPE cells.7,24 To investigate whether these data represent a general effect of bestrophin-1 on CaV channels or is specific to the CaV α1D, we performed experiments to investigate the actions of bestrophin-1 in HEK293 cells stably expressing L type CaV α1C channel subunits. Figure 8 shows data from these cells when they are transiently transfected with the CaV auxiliary β2A subunit (required for normal CaV activity) and either wild-type bestrophin-1 or p.R141H. Figure 8A shows CaV activation when the membrane potential was depolarized from −80 to +10 mV, in cells transfected with CaV in the absence (solid line) and presence (dotted line) of wild-type bestrophin-1. The rate of CaV activation was accelerated in the presence of bestrophin-1. To quantify this effect, we fitted channel activation with a double exponential function. Figure 8C shows that wild-type bestrophin-1 significantly reduced the slow rate constant for activation (p < 0.05), and although the fast rate constant was also reduced, this effect was not statistically significant (p > 0.05). By contrast, coexpression of the p.R141H mutant had no detectable effect on the rate of CaV activation (Figures 8B and 8C; p > 0.05 compared to currents recorded in the absence of bestrophin-1).
Figure 8.
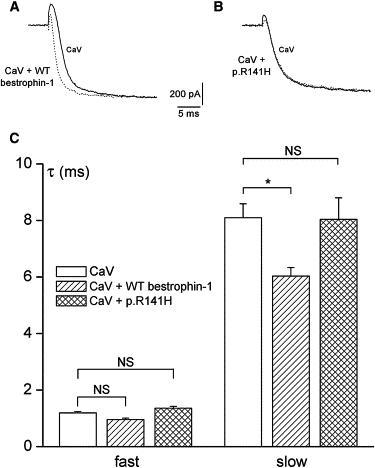
Ca2+-Channel Activation Is Accelerated by Wild-Type Bestrophin-1, but Not the p.R141H Mutant
Recordings were made in HEK293 cells stably expressing L type calcium channel α1C subunits.21 Cells were also transiently transfected with an RFP-β2a construct, which also acted as an expression marker. (A) shows representative traces taken from cells expressing CaV channels alone (unbroken line) and coexpressing wild-type bestrophin-1 (dashed line). Currents were evoked by step-depolarizing cells +10 mV from a holding potential of −80 mV. Currents have been scaled such that peak current amplitudes are the same in each case. (B) is as shown in (A), except cells coexpressed the p.R141H bestrophin-1 mutant. (C) shows the mean time constants for channel activation, derived from recordings such as those in (A) and (B). Activating sections of current traces were fit with a double exponential function, yielding time constants (τ) for the fast and slow components of channel activation. Data are means (±SEM) taken from eight or nine cells in each case. NS, not significant. ∗p < 0.05.
Discussion
Until now sequence alterations in BEST1 have been associated with two distinct autosomal-dominant ocular phenotypes, Best macular dystrophy and ADVIRC. Here, we demonstrate that homozygous or compound heterozygous sequence variants in BEST1 also cause a third retinal phenotype that we have termed autosomal-recessive bestrophinopathy (ARB).
ARB, a Novel Retinal Phenotype Caused by Compound Heterozygous or Homozygous BEST1 Mutation
The ARB phenotype is distinct from that seen in Best disease. None of the patients showed the vitelliform lesions characteristic of Best disease25 at any time during their follow-up, and none of the five families had a family history suggestive of dominant disease. Instead, patients showed a diffuse irregularity of the reflex from the RPE, including dispersed punctate flecks, which are distinct from the extramacular vitelliform lesions that have been described in some cases of Best disease. This was most clearly demonstrated by autofluorescent imaging (Figure 1). All patients showed an accumulation of fluid within and/or beneath the neurosensory retina in the macular region, and such an accumulation was most likely to be caused the reduction of visual acuity. All patients were hyperopic, and three (from two families) also had angle-closure glaucoma. In all patients there was a severe reduction in the EOG light rise similar to that seen in both Best disease and ADVIRC.
Uniquely, full-field ERGs were reduced and delayed for both cone and rod responses (Figure 3) relatively early in the evolution of the disease phenotype and, at least in one case, showed some reduction with time (Figure 4). The absent or severely reduced light rise on the EOG is not explained by the magnitude of the ERG abnormalities and thus confirms a generalized RPE dysfunction. In most photoreceptor dystrophies, such as the various forms of retinitis pigmentosa and related disorders, the degree of EOG light-rise reduction approximates to the degree of rod-photoreceptor dysfunction demonstrated by ERG. Hence, a disproportionate EOG reduction might be used to direct mutational screening of BEST1 in Best disease, ADVIRC, and ARB.
Electrophysiology is crucial to diagnosis. Ten proven heterozygous carriers from four families including parents (n = 4), an uncle, an unaffected sibling, and children (n = 4) were examined clinically and tested with ERG and EOG. None of these individuals had symptoms of retinal disease, and all had normal cone and rod ERG responses and a normal EOG light rise. This strongly suggests that heterozygosity for alleles causative of ARB does not cause clinical or subclinical human disease in the majority of cases. This differs from the report of Shatz et al.15 who have described an “atypical” form of Best disease associated with compound heterozygous mutations (p.R141H and p.Y29X), in which one 59-year-old heterozygote had a reduction of ERG amplitudes, but a normal EOG light rise. Although the ERGs are difficult to compare with our own (because of the different standards used), it might be significant that this individual had a normal EOG light rise, a response that is the most sensitive test for bestrophinopathies. Our data suggest that this “atypical” form of Best disease is likely to be ARB.15
We sequenced BEST1 in five ARB families, identifying seven homozygous or compound heterozygous variants. Six were missense variants, and there was a one nonsense alteration. All novel variants were absent from 210 control chromosomes and were in conserved residues. Therefore, the large majority of BEST1 mutations that cause Best disease, ADVIRC, and ARB are missense (Figure 9). However, the identification of the p.R141H and p.L41P variants in two families suggests a correlation between genotype and phenotype. It might also be functionally significant that 3/6 missense alterations (p.D312N, p.V317M, and p.M325T) are located closer to the C terminus than previously described missense mutations. The region is conserved within the family of bestrophins and suggests it might have a critical functional role. The C-terminal region of bestrophin-1 interacts with protein phophatase2A,26 and it might be that these residues are important in this regulatory interaction.
Figure 9.
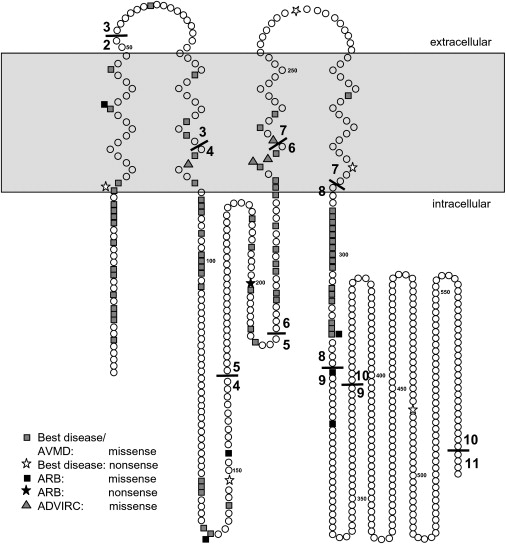
Diagram of Bestrophin-1 Summarizing Known Mutations Associated with Best Disease, Adult Vitelliform Macular Degeneration, ADVIRC, and ARB
Topology is based on that determined by Milenkovic et al.14
Cellular Electrophysiological Properties of Mutant Bestrophin-1 Variants that Cause ARB
Previous studies have investigated the cellular electrophysiological properties of dominant mutant bestrophin-1 isoforms. In one, transient transfection of missense isoforms resulted in normal targeting to the cell membrane but an abolition of Cl− current. Additionally, the mutant isoforms associated normally with wild-type protein and inhibited its chloride-channel activity.4,22 We confirmed this for two Best-disease-associated mutations, p.W93C and p.R218C (Figure 8). In contrast, we have shown that the ARB variants p.R141H and p.P152A severely reduce Cl−-channel activity in HEK293 cells (Figure 6) when expressed alone but do not significantly alter the conductance when coexpressed with wild-type bestrophin-1 (Figure 8). Therefore, we have demonstrated that channels containing only mutant isoforms of bestrophin-1 are likely to produce little or no activity when present in the homozygous, or presumably compound heterozygous, states (Figure 8). Because heteromeric mutant wild-type channels are much less reduced in function, we suggest that carriers with one wild-type and one mutant allele retain sufficient channel activity to be phenotypically normal, unlike Best disease patients who only require the expression of one mutant allele. In all of our in vitro experiments, we demonstrated comparable transfection efficiencies and demonstrated that transcription of the constructs was equivalent. It will be important to determine therefore whether the reduced activity associated with ARB mutations is due to a decrease in the number of membrane channels (for example as a result of reduced protein stability or altered membrane targeting) or to a change in single-channel properties.
In a separate series of experiments, the expression of wild-type bestrophin-1 in HEK293 cells expressing the α1C subunit of CaV was found to modulate the activation kinetics of the CaV channel (Figure 7). Similar effects have previously been observed with the α1D subunit in RPE cells.7 These data suggest that bestrophin-1 might modulate the activity of all L-type CaV channels and that the phenomenon is not confined to RPE cells. Coexpression studies with the p.R141H channel found no effect on CaV activity (Figure 7). Similar observations have previously been made with the p.W93C and p.R218C mutant isoforms.7 Whether this effect is due to a decrease in the mutant channel activity, or because of reduced protein-protein interaction between the mutant bestrophin-1 and CaV subunits, remains to be defined.
Phenotypic heterogeneity associated with recessive or dominant mutations within the same gene is well recognized.27 Three of the variants we describe have previously been shown to cause dominantly inherited phenotypes. Two, p.L41P and p.R141H, have been reported in Best disease,9,10,15,28 whereas the p.D312N variant has been described in a family with adult vitelliform macular dystrophy.10 Because full clinical details were not given, it is difficult to be certain of the significance of these observations, although it is interesting that all three variants demonstrated reduced penetrance. Although incomplete penetrance (even for electrophysiological abnormalities) is well recognized in Best disease,2,22,23 the clinical picture, lack of a dominant family history, and absence of electrophysiological abnormalities in heterozygous carriers lead us to conclude that it is not incomplete penetrance that we are observing here. It remains possible that certain variants that have pathogenic manifestations in homotzygous or compound heterozygous states might also occasionally be disease causing in the heterozygous state.
ARB Is the Null Human Phenotype of Bestrophin-1
In family 1, we describe a homozygous nonsense alteration, p.R200X. It is likely that this variant (in exon 5 of an 11 exon gene) undergoes nonsense-mediated decay (NMD), although it was not possible to prove this in vivo because of BEST1 expression being restricted to the RPE. The cellular electrophysiological data and our observation of a similar phenotype in all five ARB families suggest that either homozygosity or compound heterozygosity for the variants we describe severely reduce bestophin-1 function. This leads us to conclude that the ARB phenotype represents the true bestrophin-1 null phenotype in humans. A recessive Best1 retinopathy in the dog, termed canine multifocal retinopathy, was recently described. The two reported homozygous alterations (p.R25X and p.G161D) did not cause a fundus phenotype in obligate heterozygotes, and we think it is likely that these models will be useful in modeling ARB rather than Best disease, as the authors suggest.29
This report adds to the spectrum of phenotypes associated with BEST1 mutations, in which bestrophin-1 channel activity is a major determinant of phenotype. Our recognition of the human null phenotype helps in the understanding of the molecular mechanisms underlying the pathogenesis of the expanding range of disorders or “bestrophinopathies” that are caused by BEST1 mutation. Alongside the identification of large-animal models, this might lead to the development of novel treatment modalities for this important group of disorders.
Web Resources
The URLs for data presented herein are as follows:
ClustalW, http://www.ebi.ac.uk/ClustalW/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for VMD and VRCP)
UCSC Genome Browser, http://genome.ucsc.edu/
University of Regensberg VMD2 database, http://www-huge.uni-regensburg.de/VMD2_database/index.php?select_db=VMD2
WEBMAXC program, http://www.stanford.edu/∼cpatton/webmaxc/webmaxcS.htm
Acknowledgments
G.C.M.B. is a Wellcome Senior Research Fellow in Clinical Science. I.M.F. is supported by the British Heart Foundation (PG/05/042). I.D.M. is supported by Wellcome grant 070139/Z/02. This study is further supported by FWO Flanders grants 1.2.843.07.N.01, 1.5.244.05 (EDB), OZP 3G004306 (E.D.B., B.P.L.). The authors would like to thank the European Vision Institute Genoret (European Union), Foundation Fighting Blindness (USA), and The Special Trustees of Moorfields Eye Hospital for financial support. The authors also gratefully acknowledge the patients participating in this study.
References
- 1.Boulton M., Dayhaw-Barker P. The role of the retinal pigment epithelium: Topographical variation and ageing changes. Eye. 2001;15:384–389. doi: 10.1038/eye.2001.141. [DOI] [PubMed] [Google Scholar]
- 2.Petrukhin K., Koisti M.J., Bakall B., Li W., Xie G.C., Marknell T., Sandgren O., Forsman K., Holmgren G., Andreasson S. Identification of the gene responsible for Best macular dystrophy. Nat. Genet. 1998;19:241–247. doi: 10.1038/915. [DOI] [PubMed] [Google Scholar]
- 3.Marmorstein A.D., Marmorstein L.Y., Rayborn M., Wang X.X., Hollyfield J.G., Petrukhin K. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA. 2000;97:12758–12763. doi: 10.1073/pnas.220402097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun H., Tsunenari T., Yau K.W., Nathans J. The vitelliform macular dystrophy protein defines a new family of chloride channels. Proc. Natl. Acad. Sci. USA. 2002;99:4008–4013. doi: 10.1073/pnas.052692999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsunenari T., Sun H., Williams J., Cahill H., Smallwood P., Yau K.W., Nathans J. Structure-function analysis of the bestrophin family of anion channels. J. Biol. Chem. 2003;278:41114–41125. doi: 10.1074/jbc.M306150200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qu Z.Q., Fischmeister R., Hartzell C. Mouse bestrophin-2 is a bona fide Cl- channel: Identification of a residue important in anion binding and conduction. J. Gen. Physiol. 2004;123:327–340. doi: 10.1085/jgp.200409031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenthal R., Bakall B., Kinnick T., Peachey N., Wimmers S., Wadelius C., Marmorstein A., Strauss O. Expression of bestrophin-1, the product of the VMD2 gene, modulates voltage-dependent Ca2+ channels in retinal pigment epithelial cells. FASEB J. 2005;19:178–180. doi: 10.1096/fj.05-4495fje. [DOI] [PubMed] [Google Scholar]
- 8.Stanton J.B., Goldberg A.F.X., Hoppe G., Marmorstein L.Y., Marmorstein A.D. Hydrodynamic properties of porcine bestrophin-1 in triton X-100. Biochim. Biophys. Acta. 2006;1758:241–247. doi: 10.1016/j.bbamem.2006.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lotery A.J., Munier F.L., Fishman G.A., Weleber R.G., Jacobson S.G., Affatigato L.M., Nichols B.E., Schorderet D.F., Sheffield V.C., Stone E.M. Allelic variation in the VMD2 gene in best disease and age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2000;41:1291–1296. [PubMed] [Google Scholar]
- 10.Kramer F., Mohr N., Kellner U., Rudolph G., Weber B.H. Ten novel mutations in VMD2 associated with Best macular dystrophy (BMD) Hum. Mutat. 2003;22:418. doi: 10.1002/humu.9189. [DOI] [PubMed] [Google Scholar]
- 11.Caldwell G.M., Kakuk L.E., Griesinger I.B., Simpson S.A., Nowak N.J., Small K.W., Maumenee I.H., Rosenfeld P.J., Sieving P.A., Shows T.B. Bestrophin gene mutations in patients with best vitelliform macular dystrophy. Genomics. 1999;58:98–101. doi: 10.1006/geno.1999.5808. [DOI] [PubMed] [Google Scholar]
- 12.Downs K., Zacks D.N., Caruso R., Karoukis A.J., Branham K., Yashar B.M., Haimann M.H., Trzupek K., Meltzer M., Blain D. Molecular testing for hereditary retinal disease as part of clinical care. Arch. Ophthalmol. 2007;125:252–258. doi: 10.1001/archopht.125.2.252. [DOI] [PubMed] [Google Scholar]
- 13.Qu Z., Wei R.W., Mann W., Hartzell H.C. Two bestrophins cloned from Xenopus laevis oocytes express Ca(2+)-activated Cl(−) currents. J. Biol. Chem. 2003;278:49563–49572. doi: 10.1074/jbc.M308414200. [DOI] [PubMed] [Google Scholar]
- 14.Milenkovic V.M., Rivera A., Horling F., Weber B.H.F. Insertion and topology of normal and mutant bestrophin-1 in the endoplasmic reticulum membrane. J. Biol. Chem. 2007;282:1313–1321. doi: 10.1074/jbc.M607383200. [DOI] [PubMed] [Google Scholar]
- 15.Schatz P., Klar J., Andreasson S., Ponjavic V., Dahl N. Variant phenotype of best vitelliform macular dystrophy associated with compound heterozygous mutations in VMD2. Ophthalmic Genet. 2006;27:51–56. doi: 10.1080/13816810600677990. [DOI] [PubMed] [Google Scholar]
- 16.Yardley J., Leroy B.P., Hart-Holden N., Lafaut B.A., Loeys B., Messiaen L.M., Perveen R., Reddy M.A., Bhattacharya S.S., Traboulsi E. Mutations of VMD2 splicing regulators cause nanophthalmos and autosomal dominant vitreoretinochoroidopathy (ADVIRC) Invest. Ophthalmol. Vis. Sci. 2004;45:3683–3689. doi: 10.1167/iovs.04-0550. [DOI] [PubMed] [Google Scholar]
- 17.Bach M., Hawlina M., Holder G.E., Marmor M.F., Meigen T., Vaegan, Miyake Y. Standard for pattern electroretinography. International society for clinical electrophysiology of vision. Doc. Ophthalmol. 2000;101:11–18. doi: 10.1023/a:1002732114721. [DOI] [PubMed] [Google Scholar]
- 18.Holder G.E., Brigell M.G., Hawlina M., Meigen T., Vaegan, Bach M. ISCEV standard for clinical pattern electroretinography-2007 update. Doc. Ophthalmol. 2007;114:111–116. doi: 10.1007/s10633-007-9053-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marmor M.F., Zrenner E. Standard for clinical electrooculography. Doc. Ophthalmol. 1993;85:115–124. doi: 10.1007/BF01371127. [DOI] [PubMed] [Google Scholar]
- 20.Campanucci V.A., Brown S.T., Hudasek K., O'Kelly I.M., Nurse C.A., Fearon I.M. O-2 sensing by recombinant twik-related halothane-inhibitable K+ channel-1 background K+ channels heterologously expressed in human embryonic kidney cells. Neuroscience. 2005;135:1087–1094. doi: 10.1016/j.neuroscience.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 21.Hudasek K., Brown S.T., Fearon I.M. H2O2 regulates recombinant Ca2+ channel alpha(1c) subunits but does not mediate their sensitivity to acute hypoxia. Biochem. Biophys. Res. Commun. 2004;318:135–141. doi: 10.1016/j.bbrc.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 22.Marchant D., Yu K., Bigot K., Roche O., Germain A., Bonneau D., Drouin-Garraud V., Schorderet D., Munier F., Schmidt D. New VMD2 gene mutations identified in patients affected by Best Vitelliform Macular Dystrophy. J. Med. Genet. 2007;44:e70. doi: 10.1136/jmg.2006.044511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wabbels B., Preising M.N., Kretschmann U., Demmler A., Lorenz B. Genotype-phenotype correlation and longitudinal course in ten families with Best vitelliform macular dystrophy. Graefes Arch. Clin. Exp. Ophthalmol. 2006;244:1453–1466. doi: 10.1007/s00417-006-0286-6. [DOI] [PubMed] [Google Scholar]
- 24.Marmorstein L.Y., Wu J., McLaughlin P., Yocom J., Karl M.O., Neussert R., Wimmers S., Stanton J.B., Gregg R.G., Strauss O. The light peak of the electroretinogram is dependent on voltage-gated calcium channels and antagonized by bestrophin (Best-1) J. Gen. Physiol. 2006;127:577–589. doi: 10.1085/jgp.200509473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mullins R.F., Oh K.T., Heffron E., Hageman G.S., Stone E.M. Late development of vitelliform lesions and flecks in a patient with Best disease - Clinicopathologic correlation. Arch. Ophthalmol. 2005;123:1588–1594. doi: 10.1001/archopht.123.11.1588. [DOI] [PubMed] [Google Scholar]
- 26.Marmorstein L.Y., McLaughlin P.J., Stanton J.B., Yan L., Crabb J.W., Marmorstein A.D. Bestrophin interacts physically and functionally with protein phosphatase 2A. J. Biol. Chem. 2002;277:30591–30597. doi: 10.1074/jbc.M204269200. [DOI] [PubMed] [Google Scholar]
- 27.Afzal A.R., Jeffery S. One gene, two phenotypes: ROR2 mutations in autosomal recessive Robinow syndrome and autosomal dominant brachydactyly type B. Hum. Mutat. 2003;22:1–11. doi: 10.1002/humu.10233. [DOI] [PubMed] [Google Scholar]
- 28.Kramer F., White K., Pauleikhoff D., Gehrig A., Passmore L., Rivera A., Rudolph G., Kellner U., Andrassi M., Lorenz B. Mutations in the VMD2 gene are associated with juvenile-onset vitelliform macular dystrophy (Best disease) and adult vitelliform macular dystrophy but not age-related macular degeneration. Eur. J. Hum. Genet. 2000;8:286–292. doi: 10.1038/sj.ejhg.5200447. [DOI] [PubMed] [Google Scholar]
- 29.Guziewicz K.E., Zangerl B., Lindauer S.J., Mullins R.F., Sandmeyer L.S., Grahn B.H., Stone E.M., Acland G.M., Aguirre G.D. Bestrophin gene mutations cause canine multifocal retinopathy: A novel animal model for best disease. Invest. Ophthalmol. Vis. Sci. 2007;48:1959–1967. doi: 10.1167/iovs.06-1374. [DOI] [PMC free article] [PubMed] [Google Scholar]