

Abstract
The WNT-signaling pathway plays a major role during mammalian embryogenesis. We report a novel autosomal-recessive syndrome that consists of female to male sex reversal and renal, adrenal, and lung dysgenesis and is associated with additional developmental defects. Using a candidate-gene approach, we identified a disease-causing homozygous missense mutation in the human WNT4 gene. The mutation was found to result in markedly reduced WNT4 mRNA levels in vivo and in vitro and to downregulate WNT4-dependant inhibition of β-catenin degradation. Taken together with previous observations in animal models, the present data attribute a pivotal role to WNT4 signaling during organogenesis in humans.
Introduction
The deciphering of the molecular machinery underlying a developmental process can have far-reaching consequences, given that many physiological (e.g., wound repair) and pathological (e.g., cancer) events in the adult organism often grossly recapitulate major phases of embryonic development.1,2 Gonadal differentiation is considered as one of the most complex and puzzling developmental processes in humans. The presence or absence of a Y chromosome is known to determine the subsequent formation of the primordial gonad into a testis or an ovary, respectively.3 Genetically engineered mice have revealed the role of a large number of molecules involved in the formation of the reproductive system, among which WNT4 is considered as most important for the proper differentiation of female gonadal tissues.4 WNT4 has been shown to play a critical role not only in the development of the reproductive system but also in the formation of the kidneys, adrenals, pituitary gland, and mammary tissues.5 WNT4 belongs to the WNT family, a large group of secreted glycoproteins encoded by 19 distinct genes in the vertebrate genome, which are expressed and function in a tissue-specific fashion and have been shown to play key roles in the development of multicellular animals.6,7
In the present report, we delineate for the first time (to our knowledge) the clinical features and molecular abnormalities associated with a homozygous null mutation in WNT4 in humans. Our data demonstrate that the protein encoded by this gene plays an essential role in human sex-determination and organogenesis.
Material and Methods
Patients and Biological Materials
Blood samples were obtained from each living participant after informed and written consent (according to a protocol reviewed and approved by the local Helsinki Committee and by the National Committee for Genetic Studies of the Israeli Ministry of Health) was received. Fifteen milliliters of blood were drawn from each individual, and genomic DNA was isolated from blood samples via the salt-chloroform extraction method. Autopsies were performed and tissues samples were obtained for histology or DNA and/or RNA extraction after informed and written consent from both parents of each aborted fetus was received. DNA was extracted from paraffin-embedded specimens with QiAmp DNA kit (QIAGEN, Valencia, CA) according to the manufacturer's instructions.
Microsatellite Analysis
Polymorphic microsatellite markers spanning the WNT4 locus were selected from the GDB database, Genotypes were established by PCR amplification of genomic DNA with Supertherm Taq polymerase (Eisenberg Brothers, Givat Schmuel, Israel) and fluorescently labeled primer pairs (Research Genetics, InVitrogen, Carlsbad, CA) according to the manufacturer's recommendations. PCR conditions were 5 min at 95 C° followed by 35 cycles for 30 s at 95 C°, 30 s at 56 C°, 30 s at 72 C°, and a final extension step at 72 C° for 5 min. PCR products were separated by PAGE on an ABI 310 sequencer system, and allele sizes were determined with Genescan 3.1 and Genotyper 2.0 software. Parsimonious haplotypes were subsequently established for each individual.
Mutation Analysis
Genomic DNA was PCR-amplified with primer pairs encompassing all exons and exon-intron boundaries of the WNT4 gene (accession number NC000001) (Table 1). Gel-purified (QIAquick gel extraction kit) amplicons were subjected to bidirectional DNA sequencing with the BigDye terminator system on an ABI Prism 3100 sequencer (PE Applied Biosystems).
Table 1.
Oligonucleotide Sequences for WNT4 Sequencing
| Exon | Primer | Sequence | Expected Size |
|---|---|---|---|
| 1 | Forward | GCGGCGCTGACAGCTGGTCCG | 413 bp |
| 1 | Reverse | GGTGTGCAGAGGGACGTTCG | 413 bp |
| 2 | Forward | CAGTCACTGGGACGAGTCAG | 444 bp |
| 2 | Reverse | GTTGCTCACGAGCGTCTCATTTC | 444 bp |
| 3–4 | Forward | GTGCCTAGCACATGATTTCTGC | 699 bp |
| 3–4 | Reverse | GTAACGCCTGCCTGCCTGTCTG | 699 bp |
| 5 | Forward | GCAAATCTGACTGCAGCGTCAG | 678 bp |
| 5 | Reverse | GGGTAGGTGGTGGGAGACTG | 678 bp |
Mutation c.C341T creates a recognition site for endonuclease HpyCH4IV. Thus, to confirm and screen for mutation c.C341T, we PCR-amplified a 699 bp fragment with forward primer 5′-GTGCCTAGCACATGATTTCTGC-3′ and reverse primer 5′-GTAACGCCTGCCTGCCTGTCTG-3′; the resulting amplicon was digested with endonuclease HpyCH4IV. To identify the mutation in paraffin-embedded derived DNA samples, we PCR-amplified a 231 bp fragment with forward primer 5′-GCAGGAGGCTCCATATGC-3′ and reverse primer 5′-CACCTACCCTGTGGGCTG-3′; the resulting amplicon was digested with the same endonuclease.
Plasmids
The full-length WNT4 cDNA was cloned into pT-Rex-DEST30 (Invitrogen, CA). c.C314T was introduced into pT-Rex-DEST30-WNT4 by site-directed mutagenesis with the QuickChange II XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) and with primers mutwnt4F 5′-GCCTTCGTGTACGTCATCTCTTCGGCAG-3′ and mutwnt4R 5′-CTGCCGAAGAGATGACGTACACGAAGGC-3′. Plasmid sequence was verified by direct sequencing as described above.
Cell Cultures
OVCAR3 cells were plated at a density of 4 × 106 in 35 mm 6-well dishes and cultured in RPMI 1640 supplemented with 20% FCS, 1% L-glutamine, 10 mM HEPES, 1 mM sodium pyruvate (Biological Industries, Bet Haemek, Israel). Cells were transfected with 4 μg plasmid by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions and collected for further analysis 24 hr later.
Amniocytes were cultured for 48 hr in DMEM supplemented with 15% FCS (Biological Industries).
Reverse-Transcription Polymerase Chain Reaction
Total RNA was extracted from cell cultures with the High Pure RNA Isolation Kit (Roche Diagnostics, Mannheim, Germany). Total cDNA was synthesized with the Reverse-iT 1st Strand Synthesis Kit (ABgene, Surrey, UK) and amplified with Taq polymerase, Q solution (QIAGEN, Valencia, CA), and intron-crossing WNT4-specific primers (forward 5′-CTTCGCCGTCTTCTCAG-3′ and reverse 5′-GTTCCGCTTGCACATCT-3′; expected size 138 bp). Cycling conditions were 95°C 5 min followed by 35 cycles at 95°C 30 s, 57°C 45 s, and 72°C 90 s and a final extension step at 72°C for 7 min. As a control, we amplified total RNA as described above with ACTB-specific primers (forward 5′-CCAAGGCCAACCGCGAGAAGATGAC-3′ and reverse 5′-AGGGTACATGGTGGTGCCGCCAGAC-3′; expected size 587 bp).
For quantitative real-time PCR, cDNA PCR amplification was carried out with the SYBR Green Jump Start Taq ReadyMix for quantitative PCR (Sigma, Rehovot, Israel) on a real-time quantitative MX3000P PCR system (Strategene, La Jolla, CA) with primer pairs specific for WNT4 (forward 5′-GCCTCGTCCAGCAGAGC-3′ and reverse 5′-GAACTGTGCGTTGCGTG-3′) or ACTB (forward 5′-TTGCCGACAGGATGCAGAAGGA-3′ and reverse 5′-AGGTGGACAGCGAGGCCAGGAT-3′). To ensure the specificity of the reaction conditions, at the end of the individual runs, we measured the melting temperature (Tm) of the amplified products to confirm its homogeneity. Cycling conditions were as follows: 95°C for 10 min; 95°C for 15 s, 60°C for 15 s, and 72°C for 15 s for a total of 40 cycles. Each sample was analyzed in triplicate. For quantification, standard curves were obtained with serially diluted cDNA amplified in the same real-time PCR run. Results were normalized to ACTB mRNA levels. After the quantification procedure, the products were resolved by 2.5% agarose gel electrophoresis to confirm that the reaction had amplified the correct DNA fragments of known size.
Western Blotting
Cells were homogenized in lysis buffer (25 mM HEPES, 300 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.1% Triton X-100, and protease inhibitor mix including 1 mM PMSF, 1 mg/ml aprotinin and leupeptin; Sigma). Following centrifugation at 10,000 × g for 10 min at 4°C, proteins were electrophoresed through a 10% SDS-PAGE and transferred onto a nitrocellulose membrane (Trans-Blot, Bio-Rad, Hercules, CA). After 1 hr blocking with 1× TBS (20 mM Tris, 150 mM NaCl) with 5% skim milk and 0.01% Tween 20, blots were incubated with anti-β-catenin polyclonal antibody (Cell Signaling Technology, Beverly, MA). The blots were washed three times with 1× TBS, 0.05% Tween-20. After incubation with secondary HRP-conjugated antibodies (Sigma) and subsequent washings, proteins were detected with the EZ-ECL chemiluminescence detection kit (Biological Industries). In order to compare the amount of protein in the different samples, we reprobed the blots with an anti-β-actin monoclonal antibody (Abcam, Cambridge, MA).
Gas-Chromatography Mass-Spectrometry (GC-MS) Profiling of Urinary Steroids
Urinary-steroid profiles were determined from spot urine specimens as previously reported with few changes.8 An aliquot of urine was processed by solid-phase extraction, hydrolysis, re-extraction, and derivatization and purified by gel chromatography. Gas chromatography was performed with an Optima-1 fused column, and helium was used as carrier gas at a flow rate of 1 ml/min. The gas chromatograph (Agilent 6890) was directly interfaced to a mass selective detector (Agilent 5972A) operated in selected ion monitoring (SIM) mode.
Bioinformatics
Sequence alignment was performed with MAFFT, a multiple-sequence alignment program. mRNA secondary structure was predicted with the GeneBee RNA secondary-structure prediction software.
Results
Clinical and Pathological Features
We studied a consanguineous kindred of Arab Muslim origin (Figure 1A). This family has been followed since the birth of a full-term male (II-10) who died of hyperammonemia due to citrullinemia type I (MIM 215700) at age 4 days. The parents of this patient (I-3 and I-4) are 2nd-degree cousins. The family chose to interrupt a first pregnancy at gestation age 24 weeks (II-6) and a second pregnancy at 19 weeks of gestation (II-12) after ultrasonic finding of renal agenesis in both cases. This couple reported three other first-trimester spontaneous miscarriages (II-7, II-8, II-11). Reportedly, another member of the family (I-2) underwent termination of pregnancy in the 23rd week of gestation (II-2) because of similar ultrasonic findings of renal agenesis. In all three cases (II-2, II-6, II-12), a presumptive diagnosis of Potter syndrome was entertained.
Figure 1.
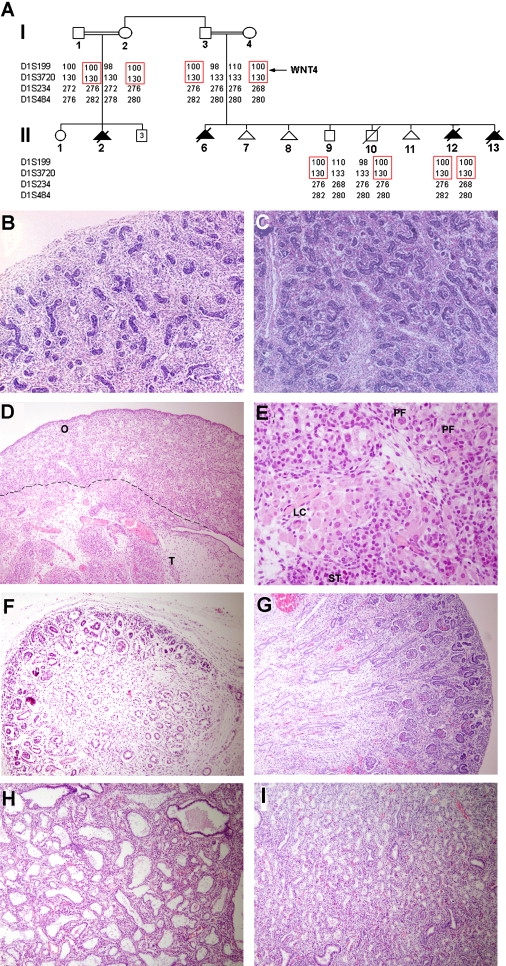
Pedigree Structure and Pathological Features
(A) Family pedigree and haplotype analysis using polymorphic microsatellite markers on 1p36.23-p35.1. The disease-associated haplotype is boxed in red. The position of the WNT4 gene is indicated.
(B–I) Histopathogical examination of tissues obtained from affected fetuses (B, D, F, and H) and from a normal 19-week-old fetus (C, E, G, and I). Affected 46,XX fetus II-12 displays (B) a normal-looking testis (sex reversal) with coiled seminiferous tubules and numerous Leydig cells throughout the interstitium as compared with the testis of an 46,XY healthy fetus (C); affected fetus II-6 demonstrates both testicular (“T”) and ovarian (“O”) tissues (D) including seminiferous tubules (“ST”), Leydig cells (“LC”), and primordial follicles (“PF”) (E); the kidney of affected fetus II-12 (F) showed a very thin rim of nephrogenic zone, with decreased numbers of mature glomeruli, and very few proximal tubules with immature interstitial tissue resembling undifferentiated mesenchyme of dysplastic kidney as compared with a normal fetal kidney at 19 weeks of gestation (G), where the numerous glomeruli identify the renal cortex; examination of lung tissues of affected fetus II-2 (H) revealed focal marked dilatation of distal airways compared with control fetal lung tissue (I) (magnification, 50× [B and C], 100× [D, F, G, H, and I], 400× [E]).
The macroscopic pathological findings of autopsies of three fetuses (II-2, II-6, II-12) are summarized in Table 2. Examination of fetus II-12 revealed a small-for-gestational-age male fetus, with cleft lip and palate, cardiac ventricular septal defect, pulmonary artery stenosis, and small lungs. Neither kidneys nor adrenal glands were identified on gross pathologic examination. Histopathological studies of this fetus revealed apparently normal-looking testes (Figure 1B), minute kidneys with markedly decreased numbers of normal mature glomeruli and paucity of proximal tubules (Figure 1F), a hypoplastic urinary bladder with thin ureters, and exceedingly small adrenals with seemingly normal morphology (not shown). Despite the presence of male gonads, cytogenetic analysis revealed a normal 46,XX female karyotype, suggestive of female to male sex reversal. PCR amplification of genomic DNA derived from this fetus revealed absence of SRY (not shown), a Y chromosome-encoded gene; this result supports the cytogenetic diagnosis.
Table 2.
SERKAL Syndrome: Phenotypic Features
| Affected Fetuses (year of birth) |
II-2 (1990) |
II-6 (2000) |
II-12 (2006) |
|---|---|---|---|
| Termination of pregnancy (age weeks) | 23 | 24 | 19 |
| Ultrasound | |||
| Growth retardation | + | + | + |
| Renal agenesis | + | + | + |
| Oligohydramnion | + | + | + |
| Presumptive diagnosis: Potter syndrome | + | + | + |
| Maternal serum estriol | ND∗ | low | low |
| Macroscopic Pathological Findings | |||
| Renal agenesis/dysgenesis | + | + | + |
| Hypoplastic lungs | + CCAM∗ | + | + |
| Adrenal | Small | NR | not found |
| Cleft lip/palate | + | − | + |
| Congenital heart disease | − | − | VSD∗ and pulmonary stenosis |
| Diaphragmatic hernia | − | only remnant of diaphragma; liver surrounding the heart | − |
| Intestine malrotation | + | − | − |
| Urinary bladder | NR | hypoplastic | hypoplastic |
| External genitalia (testis/ovum) | Female NR | Male: distorted penis and hypospadias (ovotestis) | Male genitalia (testis) |
The following abbreviations (∗) are used: ND, not done; NR, not reported; CCAM, congenital cystic adenomatoid malformation; and VSD, ventricular septal defect.
The two other fetuses assessed displayed similar features. Pathological examination of fetus II-6 (Table 2) disclosed a 24-week-old fetus, with a curved penis and hypospadias, bilateral diaphragmatic hernia with the liver surrounding the heart, small lungs and a single kidney remnant with disorganized glomeruli and tubuli, and rudimentary urinary bladder and ovotestes (Figure 1D). Fetus II-2 was a 23-week-old fetus with low-set ears, facial features resembling Potter syndrome, bilateral cleft lip and palate, hypoplastic lungs, renal agenesis, and undersized adrenal glands with normal microscopic morphology. This fetus also displayed severe focal dilation of distal airways in one lung, reminiscent of congenital cystic adenomatoid malformation (Figure 1H).
Given that affected fetuses featured largely overlapping features including (1) female sex reversal and dysgenesis of (2) kidneys, (3) adrenals, and (4) lungs, we named this novel autosomal-recessive phenotype SERKAL syndrome.
Identification of a Causative Mutation in WNT4
The high degree of inbreeding characterizing the family under study and the clinical and pathological findings reviewed above were suggestive of a lethal recessive disorder caused by a mutation in a gene coding for a protein involved in both sex determination and organogenesis. Given the similarities between the present cases (II-2,II-6, II-12) and a WNT4 knocked-out mouse model previously described,5,9–11 we genotyped all available family members for microsatellite markers spanning the WNT4 locus. The affected fetus from which DNA was available (II-12) displayed a homozygous haplotype, which was found to be carried in a heterozygous state by all parents and siblings (Figure 1A), suggesting the existence of a homozygous mutation in WNT4 in the affected fetus.
Accordingly, direct sequencing of the entire coding sequence of the WNT4 gene in this fetus revealed the presence of a homozygous C → T transition at cDNA position 341 (c.C341T) (Figure 2A). The mutation was found to be carried in a heterozygous state by all parents and siblings. The mutation is predicted to result in the substitution of a valine residue for an alanine residue at amino acid position 114 (p.A114V). Using a PCR-RFLP assay, we confirmed segregation of the mutation in the family (Figure 2B) and excluded it from a panel of 300 population-matched healthy unrelated individuals (600 chromosomes), suggesting that this sequence variant does not represent a common nonconsequential polymorphism. We confirmed the presence of homozygous c.C341T mutation in the two affected fetuses from whom cells or blood samples were not available by using DNA extracted from paraffin-embedded blocks and a PCR-RFLP assay (not shown).
Figure 2.

Mutation Analysis
(A) Sequence analysis revealed in an affected fetus (II-12) a homozygous transition (c.C341T) (blackened letter, upper panel), resulting in amino acid substitution A114V. The wild-type sequence is given for comparison (lower panel).
(B) c.C314T creates a novel recognition site for HpyCH4IV endonuclease. A 699 bp fragment was amplified as described in Material and Methods and digested with HpyCH4IV. The affected fetus displays two fragments, of 172 bp and 527 bp, that are carried in a heterozygous state by all other family members.
(C) The amino acid (upper panel) and cDNA (lower panel) sequences of human WNT4 were compared to those of other human WNT proteins and WNT4 orthologs in other species. Note the high degree of conservation of both amino acid A114 (upper panel) and nucleotide C341 (lower panel).
Delineation of the Consequences of c.C341T Mutation
The c.C341T mutation affects a highly conserved alanine residue found in all human WNT proteins as well as in WNT4 orthologs in all living organisms for which WNT4 sequence is known, including zebrafish and Drosophila (Figure 2C), suggesting that it might be of functional importance. Upon secretion, WNT proteins bind to the frizzled family of receptors and activate a number of molecular pathways—an activation that might result in β-catenin stabilization, and, as a consequence, increased transcriptional activity.6 WNT4 has been shown to be able to activate this canonical pathway.12 We therefore transfected OVCAR3 cells with wild-type and p.A114V-carrying full-length WNT4 cDNA-expression vectors. Whereas the wild-type vector led to a significant increase in β-catenin protein levels, as expected and as previously reported,12 no change, compared to baseline, was observed in β-catenin levels in cells transfected with the mutant vector, suggesting that p.A114V mutation indeed abolishes WNT4 activity (Figure 3A).
Figure 3.
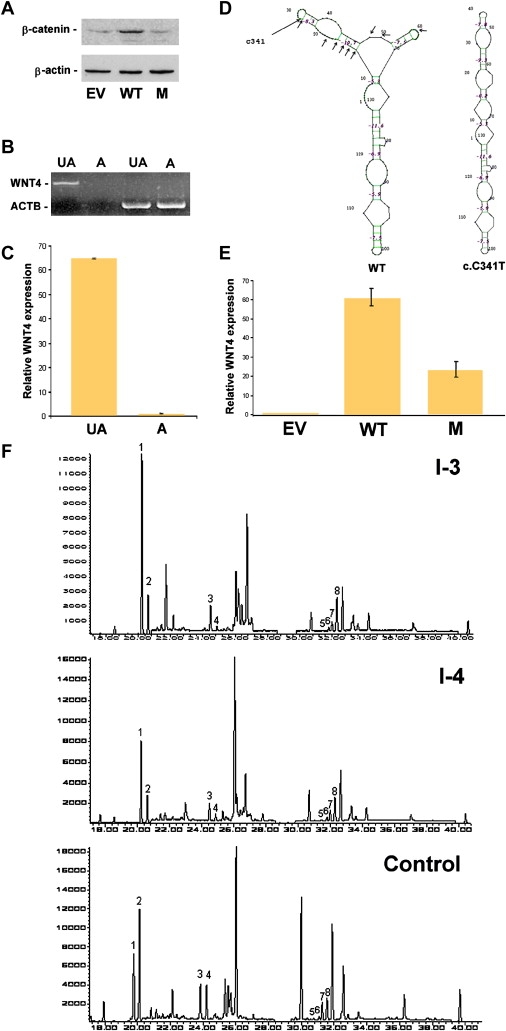
Consequences of Mutation c.C341T
(A) Western-blot analysis of β-catenin and β-actin expression in OVCAR3 cells transfected with empty vector (EV), wild-type WNT4 cDNA (WT), and c.C341T mutant cDNA (M).
(B) Expression of WNT4 and ACTB, coding for β-actin, assessed with RT-PCR in amniocytes obtained from an affected embryo (“A”) as compared with control amniocytes (“UA”).
(C) Quantitative real-time PCR analysis for gene expression of WNT4 in amniocytes obtained from an affected embryo (“A”) as compared with control amniocytes (“UA”) (bars represent standard errors). Expression levels are expressed as absolute values normalized relative to ACTB RNA levels.
(D) Predicted secondary structures of wild-type (“WT”) and c.C314T mutant WNT4 RNA spanning exon 3. The position of c.C341T and mutations resulting in a similar change in mRNA secondary structure are marked with arrows.
(E) Quantitative real-time PCR analysis for gene expression of WNT4 in OVCAR3 cells transfected with empty vector (“EV”), wild-type WNT4 cDNA (“WT”), and c.C341T mutant cDNA (“M”). Expression levels are expressed as absolute values normalized relative to ACTB RNA levels (bars represent standard errors).
(F) Chromatograms (GC-MS, SIM mode) of urinary-steroid hormones. In both parents, 5-α/5-β isomer ratios [Androsterone/Etiocholanolone(1,2);11-OH-Androsterone/11-OH-etiocholanolone (3,4); α-tetrahydrocorticosterone (THB)/β-THB (5,6); α-tetrahydrocortisol (THF)/β-THF (7,8)], reveal an excess of activity of the enzyme 5-alpha-reductase. The 5α-isomers are 1, 3, 6, and 8; the 5β-isomers are 2, 4, 5, and 7.
To determine the mechanism underlying decreased WNT4 activity in this cell-based assay, we assessed WNT4 gene expression. In the course of this study, individual I-4 became pregnant and underwent chorionic villus sampling (CVS) in the 11th week of gestation. Direct sequencing of genomic DNA extracted from CVS revealed that the fetus (II-13) carried a homozygous mutation in ASS encoding argininosuccinate synthetase (p.E191K), known to be associated with citrullinemia in this family, as well as homozygous c.C341T mutation in WNT4. After parental informed consent was obtained in writing, total RNA was extracted from fetal amniocytes obtained during termination of pregnancy at week 13. Surprisingly, using either standard RT-PCR (Figure 3B) or quantitative RT-PCR (Figure 3C), we were unable to detect WNT4 RNA in fetal amniocytes. This could not have been due to nonspecific RNA degradation because ACTB RNA levels were comparable in wild-type and mutant amniocytes.
These results suggested that c.C341T might affect mRNA stability. A number of mRNA-surveillance mechanisms have been reported, including nonsense-mediated mRNA decay, leading to degradation of mRNA species carrying nonsense mutations; non-stop decay, associated with absence of native stop codons; and a recently described form of mRNA decay called no-go decay, triggered by ribosome stalling, which probably results from its inability to disrupt conserved mRNA secondary structures.13 As shown in Figure 2C, C341 and neighboring nucleotides are extremely well-preserved throughout evolution to an extent that highly suggests conservation at the RNA level. This prompted us to examine the effect of c.C341T on the putative secondary structure of WNT4 mRNA. Computational analysis indicates that c.C341T significantly affects WNT4 mRNA predicted structure (Figure 3D). Other mutations in the region surrounding C341 have a similarly deleterious effect on the predicted structure of WNT4 mRNA, whereas mutations located distally to this region have only a marginal influence on the mRNA structure (data not shown). These results are consistent with the possibility that the c.C341T mutation destabilizes WNT4 mRNA.
To further assess this hypothesis, we measured RNA level in OVCAR3 cells transfected with wild-type and c.C341T-mutation-carrying vectors. Although both genes were transcribed by the same CMV promoter, supposedly driving the same degree of transcriptional activity, the level of the mutant transcript was 2.8- to 3-fold lower than that of the wild-type transcript (Figure 3E).
Steroid Profiling of Healthy Carriers of a Deleterious WNT4 Mutation
Despite being carriers of a deleterious mutation in WNT4, all four parents displayed a normal phenotype and a normal serum-steroid profile (not shown). To obtain in vivo evidence relating the c.C341T mutation to the XX-male phenotype displayed by the affected fetuses, we studied the profile of 39 urinary-steroid metabolites in individual I-3 and I-4. In both asymptomatic heterozygous parents, we found increased ratios of four 5α/5β isomers (Figure 3F), indicating elevated activity of 5α-reductase, responsible for conversion of testosterone to 5α-dihydrotestosterone (5α-DHT).8
Discussion
Over the past decade, rare congenital disorders have emerged as a unique source of information on the biological pathways determining organogenesis in humans.14–16 In the present study, we delineate the clinical manifestations and the molecular pathology associated with a WNT4 null phenotype in humans. Through ascertainment of gene expression in human embryonic cells in vivo and using an in vitro functional assay, we show that WNT4 deficiency results in severe multisystem developmental anomalies, demonstrating the crucial role played by this molecule in orchestrating important aspects of mammalian organogenesis.
Despite its intraexonic position and the fact it affects a preserved amino acid residue, the WNT4 missense mutation associated with this novel syndrome was shown to adversely affect mRNA stability, most probably as a result of disruption of a highly conserved mRNA secondary structure. This phenomenon has been previously reported in a small number of cases,17,18 including a recent report demonstrating that a prevalent polymorphism in the catechol-O-methyltransferase gene, associated with pain perception, alters this gene mRNA secondary structure, thereby regulating enzyme activity and levels by inducing mRNA degradation.19
Although it is possible that the new phenotype reported here is rare, it might have been overlooked because of the probably lethal nature of biallelic recessive mutations in WNT4 as suggested by the present study. Careful pathological assessment of the three affected fetuses demonstrated for the first time (to our knowledge) the wide range of developmental anomalies associated with WNT4 deficiency (Table 2). Defective WNT4 activity was found to affect the development of three major organs (gonads, kidneys, and adrenal glands), all of which originate from the primordial urogenital ridge,20 suggesting a pivotal role for WNT4 at an early embryological stage of development.
Similar to previous observations in WNT4-knockout mice,9,10 we found out that WNT4 deficiency resulted in severe dysgenesis of the kidneys and female to male sex reversal; in contrast to the murine model, adrenal development and external genitalia were also abnormal in human fetuses. Dominant heterozygous deleterious mutations in WNT4 have recently been reported in two female individuals with androgen excess, Mullerian duct abnormal development but normal female genitalia, and in one case, unilateral renal agenesis;12,21 these two previously reported missense mutations in WNT4 (R83C and E226G) were shown to exert a dominant-negative effect on the expression of two steroidogenic genes, HSD3B2 and CYP17A1,12,21 which we did not observe when assessing the effect of c.C341T in the same in vitro system (not shown). In the present study, we detected elevated ratios of four urinary 5α/5β isomers in heterozygous parents indicating increased activity of the 5α-reductase (Figure 3F). These data therefore suggest that WNT4 suppresses 5α-reductase activity in humans. The most important role of 5α-reductase in humans is the conversion of testosterone to 5α-DHT, which is much more potent than testosterone and is responsible for the morphogenesis of the human male external genitalia.21 We hypothesize that the increased activity of 5α-reductase in heterozygous individual I-4 was too low to result in the development of male external genitalia.
In mammals, WNT4 function was initially studied during nephrogenesis because mice lacking WNT4 die shortly after birth as a result of kidney failure.22 Renal agenesis was common to all studied fetuses (Table 2). These findings are in line with recent studies suggesting in a mouse model that suboptimal WNT signaling results in renal hypoplasia.23 The present observations might indicate that WNT4-mutation analysis should be considered as part of the evaluation of bilateral renal dysgenesis, oligohydramnion, and pulmonary hypoplasia, known as Potter syndrome.24 Abnormal lung development in Potter syndrome is considered to be a direct consequence of oligohydramnion. However, it has recently been shown that WNT/β-catenin signaling is a critical upstream regulator of proximal-distal patterning of the lungs and of respiratory epithelial cell differentiation,25,26 which might underlie the marked abnormalities identified in lung tissues in affected fetus II-2. An important clinical clue that might help in differentiating the present syndrome from other causes of Potter syndrome is very low levels of unconjugated estriol (E3) in maternal serum, a reliable marker for primary or secondary adrenal hypoplasia,27 which was observed in two cases (Table 2). The incidence of developmental disorders of the adrenal gland is probably underestimated because autopsies do not routinely include thorough investigation for adrenal pathologies. Whereas no distinct deviation in size or shape of adrenal glands was demonstrated in knockout mice,28 c.C341T resulted in marked reduction in human adrenal size, although histology was preserved.
In summary, through the delineation of a novel autosomal-recessive phenotype, the present observations demonstrate the pleiotropic pathological consequences of WNT4 deficiency. Given the steadily growing spectrum of physiological pathways regulated through WNT-mediated signaling,6,7 careful clinical assessment and molecular analysis of additional cases in the future are likely to further expand the spectrum of developmental defects associated with WNT4 mutations.
Web Resources
The URLs for data presented herein are as follows:
GDB database, http://www.gdb.org
MAFFT software, http://align.bmr.kyushu-u.ac.jp/mafft/software/
GeneBee software, http://www.genebee.msu.su/services/rna2_reduced.html
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for citrullinemia type I)
Acknowledgments
We are grateful to the family members for their generous participation in our study. We wish to thank Karl Skorecki and Maty Tzukerman for the gift of the OVCAR3 cell line, Yaacov Bechar for providing the pathological slides of case II-6, Hanna Bar-El for assistance in amniotic cell culture, Raya Shalginov for help with DNA extraction from paraffin-embedded tissues, and Vered Friedman and Rita Fuhrer-Mor for assistance in nucleic acid analysis.
References
- 1.Page-McCaw A., Ewald A.J., Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007;8:221–233. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blanpain C., Horsley V., Fuchs E. Epithelial stem cells: Turning over new leaves. Cell. 2007;128:445–458. doi: 10.1016/j.cell.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yao H.H. The pathway to femaleness: current knowledge on embryonic development of the ovary. Mol. Cell. Endocrinol. 2005;230:87–93. doi: 10.1016/j.mce.2004.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim Y., Capel B. Balancing the bipotential gonad between alternative organ fates: A new perspective on an old problem. Dev. Dyn. 2006;235:2292–2300. doi: 10.1002/dvdy.20894. [DOI] [PubMed] [Google Scholar]
- 5.Bernard P., Harley V.R. Wnt4 action in gonadal development and sex determination. Int. J. Biochem. Cell Biol. 2007;39:31–43. doi: 10.1016/j.biocel.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 6.Logan C.Y., Nusse R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 7.Hoppler S., Kavanagh C.L. Wnt signalling: Variety at the core. J. Cell Sci. 2007;120:385–393. doi: 10.1242/jcs.03363. [DOI] [PubMed] [Google Scholar]
- 8.Wudy S.A., Hartmann M.F. Gas chromatography-Mass spectrometry profiling of steroids in times of molecular biology. Horm. Metab. Res. 2004;36:415–422. doi: 10.1055/s-2004-814565. [DOI] [PubMed] [Google Scholar]
- 9.Vainio S., Heikkila M., Kispert A., Chin N., McMahon A.P. Female development in mammals is regulated by Wnt-4 signalling. Nature. 1999;397:405–409. doi: 10.1038/17068. [DOI] [PubMed] [Google Scholar]
- 10.Heikkila M., Prunskaite R., Naillat F., Itaranta P., Vuoristo J., Leppaluoto J., Peltodeto H., Vainio S. The partial female to male sex reversal in Wnt-4-deficient females involves induced expression of testosterone biosynthetic genes and testosterone production, and depends on androgen action. Endocrinology. 2005;146:4016–4023. doi: 10.1210/en.2005-0463. [DOI] [PubMed] [Google Scholar]
- 11.Jeays-Ward K., Hoyle C., Brennan J., Dandonneau M., Alldus G., Capel B., Swain A. Endothelial and steroidogenic cell migration are regulated by WNT4 in the developing mammalian gonad. Development. 2003;130:3663–3670. doi: 10.1242/dev.00591. [DOI] [PubMed] [Google Scholar]
- 12.Biason-Lauber A., Konrad D., Navratil F., Schoenle E.J. A WNT4 mutation associated with Mullerian-duct regression and virilization in a 46,XX woman. N. Engl. J. Med. 2004;351:792–798. doi: 10.1056/NEJMoa040533. [DOI] [PubMed] [Google Scholar]
- 13.Garneau N.L., Wilusz J., Wilusz C.J. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 2007;8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- 14.Sudbeck P., Schmitz M.L., Baeuerle P.A., Scherer G. Sex reversal by loss of the C-terminal transactivation domain of human SOX9. Nat. Genet. 1996;13:230–232. doi: 10.1038/ng0696-230. [DOI] [PubMed] [Google Scholar]
- 15.Hawkins J.R. Mutational analysis of SRY in XY females. Hum. Mutat. 1993;2:347–350. doi: 10.1002/humu.1380020504. [DOI] [PubMed] [Google Scholar]
- 16.Parma P., Radi O., Vidal V., Chaboissier M.C., Dellambra E., Valentini S., Guerra L., Schedl A., Camerino G. R-spondin1 is essential in sex determination, skin differentiation and malignancy. Nat. Genet. 2006;38:1304–1309. doi: 10.1038/ng1907. [DOI] [PubMed] [Google Scholar]
- 17.Shen L.X., Basilion J.P., Stanton V.P. Single-nucleotide polymorphisms can cause different structural folds of mRNA. Proc. Natl. Acad. Sci. USA. 1999;96:7871–7876. doi: 10.1073/pnas.96.14.7871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duan J., Wainwright M.S., Comeron J.M., Saitou N., Sanders A.R., Gelernter J., Gejman P.V. Synonymous mutations in the human dopamine receptor D2 (DRD2) affect mRNA stability and synthesis of the receptor. Hum. Mol. Genet. 2003;12:205–216. doi: 10.1093/hmg/ddg055. [DOI] [PubMed] [Google Scholar]
- 19.Nackley A.G., Shabalina S.A., Tchivileva I.E., Satterfield K., Korchynskyi O., Makarov S.S., Maixner W., Diatchenko L. Human catechol-O-methyltransferase haplotypes modulate protein expression by altering mRNA secondary structure. Science. 2006;314:1930–1933. doi: 10.1126/science.1131262. [DOI] [PubMed] [Google Scholar]
- 20.Else T., Hammer G.D. Genetic analysis of adrenal absence: Agenesis and aplasia. Trends Endocrinol. Metab. 2005;16:458–468. doi: 10.1016/j.tem.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 21.Biason-Lauber A., De Filippo G., Konrad D., Scarano G., Nazzaro A., Schoenle E.J. WNT4 deficiency–a clinical phenotype distinct from the classic Mayer-Rokitansky-Kuster-Hauser syndrome: A case report. Hum. Reprod. 2007;22:224–229. doi: 10.1093/humrep/del360. [DOI] [PubMed] [Google Scholar]
- 22.Stark K., Vainio S., Vassileva G., McMahon A.P. Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by WNT4. Nature. 1994;372:679–683. doi: 10.1038/372679a0. [DOI] [PubMed] [Google Scholar]
- 23.Iglesias D.M., Hueber P.A., Chu L., Campbell R., Patenaude A.M., Dziarmaga A.J., Quinlan J., Mohamed O., Dufort D., Goodyer P.R. Canonical WNt signaling during kidney developmet. Am. J. Physiol. Renal Physiol. 2007;293:F494–F500. doi: 10.1152/ajprenal.00416.2006. [DOI] [PubMed] [Google Scholar]
- 24.Sanna-Cherchi S., Caridi G., Weng P.L., Scolari F., Perfumo F., Gharavi A.G., Ghiggeri G.M. Genetic approaches to human renal agenesis/hypoplasia and dysplasia. Pediatr. Nephrol. 2007 doi: 10.1007/s00467-007-0479-1. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mucenski M.L., Nation J.M., Thitoff A.R., Besnard V., Xu Y., Wert S.E., Harada N., Taketo M.M., Stahlman M.T., Whitsett J.A. Beta-catenin regulates differentiation of respiratory epithelial cells in vivo. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005;289:L971–L979. doi: 10.1152/ajplung.00172.2005. [DOI] [PubMed] [Google Scholar]
- 26.Shu W., Guttentag S., Wang Z., Andl T., Ballard P., Lu M.M., Piccolo S., Birchmeier W., Whitsett J.A., Millar S.E., Morrisey E.E. Wnt/beta-catenin signaling acts upstream of N-mcy, BMP4, and EGF signaling to regulate proximal-distal patterning in the lung. Dev. Biol. 2005;283:226–239. doi: 10.1016/j.ydbio.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 27.Weintrob N., Drouin J., Vallette-kasic S., Taub E., Marom D., Lebenthal Y., Klinger G., Bron-Harlev E., Shohat M. Low estriol levels in the maternal triple-marker screen as a predictor of isolated adrenocorticotropic hormone deficiency caused by new mutation in the TPIT gene. Pediatrics. 2006;117:e322–e327. doi: 10.1542/peds.2005-1973. [DOI] [PubMed] [Google Scholar]
- 28.Heikkila M., Peltoketo H., Leppäluoto J., Ilves M., Vuolteenaho O., Vainio S. Wnt-4 deficiency alters mouse adrenal cortex function, reducing aldosterone production. Endocrinology. 2002;143:4358–4365. doi: 10.1210/en.2002-220275. [DOI] [PubMed] [Google Scholar]