

Abstract
Multiple pterygium syndromes (MPS) comprise a group of multiple congenital anomaly disorders characterized by webbing (pterygia) of the neck, elbows, and/or knees and joint contractures (arthrogryposis). MPS are phenotypically and genetically heterogeneous but are traditionally divided into prenatally lethal and nonlethal (Escobar) types. Previously, we and others reported that recessive mutations in the embryonal acetylcholine receptor g subunit (CHRNG) can cause both lethal and nonlethal MPS, thus demonstrating that pterygia resulted from fetal akinesia. We hypothesized that mutations in acetylcholine receptor-related genes might also result in a MPS/fetal akinesia phenotype and so we analyzed 15 cases of lethal MPS/fetal akinesia without CHRNG mutations for mutations in the CHRNA1, CHRNB1, CHRND, and rapsyn (RAPSN) genes. No CHRNA1, CHRNB1, or CHRND mutations were detected, but a homozygous RAPSN frameshift mutation, c.1177-1178delAA, was identified in a family with three children affected with lethal fetal akinesia sequence. Previously, RAPSN mutations have been reported in congenital myasthenia. Functional studies were consistent with the hypothesis that whereas incomplete loss of rapsyn function may cause congenital myasthenia, more severe loss of function can result in a lethal fetal akinesia phenotype.
Main Text
Multiple pterygia are found infrequently in children with arthrogryposis and in fetuses with fetal akinesia syndrome.1 Autosomal-recessive inheritance is most common, but autosomal-dominant and X-linked inheritance has been described. Lethal multiple pterygium syndrome (LMPS [MIM 253290]) is an autosomal-recessively inherited disorder characterized by intrauterine growth retardation, multiple pterygia and flexion contractures causing severe arthrogryposis, and fetal akinesia. In severe cases, subcutaneous edema may be severe, causing fetal hydrops with cystic hygroma and lung hypoplasia. Developmental defects including cleft palate, cryptorchidism, intestinal malrotation, cardiac hypoplasia, diaphragmatic hernia, obstructive uropathy, microcephaly, or cerebellar and pontine hypoplasia can also be present.2,3 The etiology of MPS is heterogeneous. In a few cases, a diagnosis of a specific primary myopathy, metabolic, or neurodevelopmental disorder is made, but until recently, in many cases the underlying etiology was unknown.4 Recently, defects in the embryonal acetylcholine receptor (AChR) were discovered to account for a significant proportion of patients with lethal MPS and fetal akinesia. Thus, mutations in the acetylcholine receptor γ subunit gene (CHRNG) were detected in approximately 30% of lethal MPS cases, and also in nonlethal Escobar variant MPS.5,6 The nicotinic AChR of skeletal muscle is a pentameric transmembrane protein composed of four different subunits and exists in two forms. The adult form is predominant in innervated adult muscle and the embryonic form predominates in fetal and denervated muscle. The embryonic AChR consists of two alpha and one each beta, gamma, and delta subunit (α2βδγ), whereas in the adult AChR, the gamma subunit is replaced by an epsilon subunit (α2βδɛ).7,8 In humans, the switch from γ to ɛ occurs during fetal life and is apparently complete by 31 weeks gestation (in rodents the switch occurs postnatally).9–11 The identification of CHRNG mutations in MPS patients suggested that pterygia resulted from early-onset fetal akinesia. Although the embryonal AChR has a key role in normal prenatal muscle development, the γ subunit is not required postnatally and so Escobar variant MPS patients did not demonstrate marked muscle weakness.6 Mutations in other AChR subunits (except CHRNE) would be predicted to produce both pre- and postnatal neuromuscular transmission deficits, and we speculated that mutations in genes implicated in embryonal AChR function might cause fetal akinesia and pterygia in patients without CHRNG mutations.6 In addition, because we have found that intrafamilial phenotypic variation in kindreds with CHRNG mutations such that some affected children had fetal akinesia without detectable pterygia, we also included patients referred for CHRNG mutation analysis without pterygia.
Mutation analysis of CHRNG was undertaken in patients with MPS and/or fetal akinesia as described previously (unpublished data).6 Fifteen probands with no evidence of a germline CHRNG mutation were then selected for mutation analysis of the coding sequence and flanking intronic sequence of CHRNA1, CHRNB1, CHRND, and RAPSN. The clinical features of the 15 families are summarized in Table 1.
Table 1.
Clinical Features of 15 Fetuses Analyzed
| Family ID | Ethnic Origin | Parental Consanguinity | Phenotype |
|---|---|---|---|
| MPS013 | Pakistani | yes | LMPS |
| MPS012 | Bengali | yes | AMC, no pterygia |
| MPS014 | white | no | LMPS |
| MPS016 | white | no | LMPS |
| MPS017 | white | no | LMPS |
| MPS018 | white | no | LMPS |
| MPS019 | Indian | yes | LMPS |
| MPS020 | white | no | LMPS |
| MPS021 | white | no | LMPS |
| MPS022 | Afrikaner | no | LMPS |
| MPS023 | white | no | LMPS |
| MPS010 | Pakistani | yes | AMC, no pterygia |
| MPS009 | Pakistani | yes | LMPS |
| MPS024 | Palestinian | yes | LMPS |
| MPS025 | Ethiopian | no | LMPS |
LMPS, lethal multiple pterygium syndrome.
DNA was extracted and mutation analysis was performed by standard methods and conditions (details of primer sequences available on request). No pathogenic mutations were detected in CHRNA1, CHRNB1, or CHRND. However, a RAPSN frameshift mutation was detected in a consanguineous family with three affected children (see Figure 1). Both parents were heterozygous, and all three affected children were homozygous, for a c.1177-1178delAA mutation (see Figure 1). The family presented when fetal akinesia sequence was detected at 19 weeks gestation in twin male fetuses. On ultrasound examination, both fetuses had micrognathia and fixed position of the hands, elbows, and feet. There were no respiratory movements, the thorax appeared small, and there was mild hydrops. The pregnancy was terminated at 23 weeks of gestation. Postmortem examination revealed monochorionic, monoamniotic twins with no evidence of growth retardation (twin 1, 0.587 kg [∼50 percentile]; twin 2, 0.665 kg [>50 percentile]). Both twins had a flat facial profile, hypertelorism, moderate micrognathia, and a short broad neck. They also had mild hydrops with hydrothoraces and subcutaneous edema of the head, neck, back of the shoulders, and proximal extremities (Figures 2A and 2B). The lungs were hypoplastic and the diaphragm was thin and membranous. Both twins displayed an abnormal posture, with hyperextension at the wrists and flexion of the fingers. Ankles were fixed in a varus position and the toes were hyperextended. No pterygia were detected. Macroscopic and microscopic examination of the brain showed no abnormality. Muscle histopathology revealed muscle fibers of varying diameters (considered to be normal for the gestational age) with a normal striated pattern and no fibrosis. Chromosome analysis was normal and molecular testing for congenital myotonic dystrophy was negative. The couple went on to have another similarly affected female singleton pregnancy. Fetal akinesia sequence was detected on ultrasound at 19 weeks of gestation and the pregnancy was terminated at 23 weeks gestation. Postmortem examination showed fetal dysmorphology (micrognathia, low-set ears, a short nose, a short philtrum, thin lips, and a short broad neck) and subcutaneous edema of the head, neck, and shoulders (Figure 2C). The elbows were abnormally flexed, the wrists were hyperextended, and the second and third digits overlapped on both hands. No pterygia were identified. The lungs were hypoplastic. Neuromuscular histopathological examination failed to identify a specific underlying cause for the fetal akinesia. The brain and spinal cord were normal on macroscopic and microscopic examination. The muscle fibers were of normal diameter for gestational age. No abnormalities were detected on enzyme analysis. On electron microscopy, the striated muscle appeared well developed. All three cases showed occasional muscle fibers with centrally located nuclei, but this appearance was observed in <5% of the muscle fibers examined and was not diagnostic of a myotubular dystrophy. The couple also have two healthy children.
Figure 1.
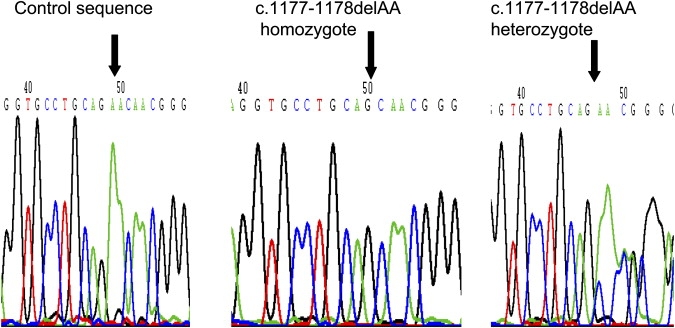
Sequence Traces for Normal Control, Homozygote, and Heterozygote Carrier of RAPSN Frameshift Mutation
Figure 2.
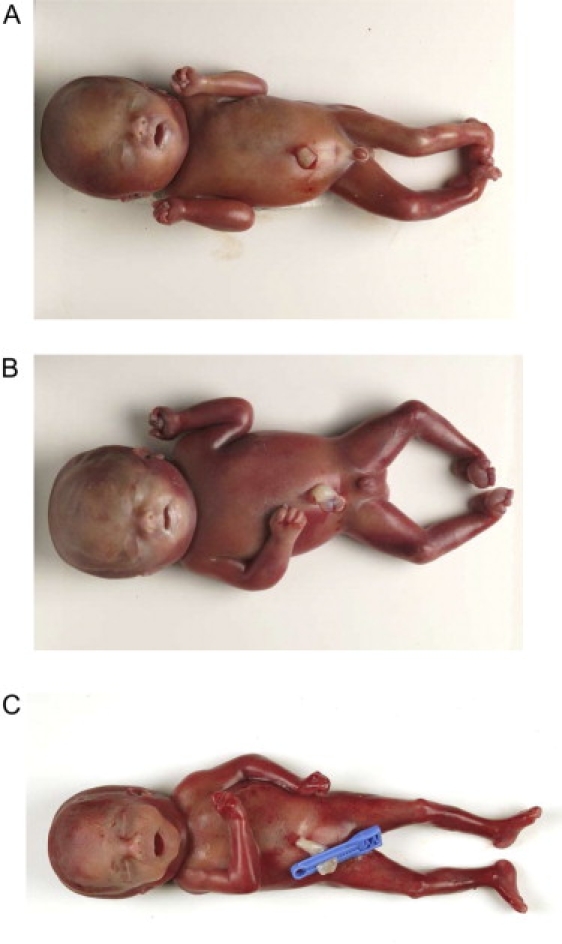
Clinical Features of Fetal Akinesia Associated with Homozygous Rapsyn Mutation
(A and B) Affected twin fetuses.
(C) Affected singleton fetus with clinical features of the fetal akinesia sequence.
Previously, mutations in AChR subunits have been associated with congenital myasthenic syndromes (CMS). These are a heterogeneous group of inherited disorders of neuromuscular transmission characterized by fatiguable muscle weakness. CMS affecting the AChR may alter AChR channel kinetics or severely reduce the number of AChRs in the postsynaptic membrane and can result in AChR deficiency from mutations in CHRNA1, CHRNB1, CHRND, or CHRNE.12,13 However, mutations in CHRNE generally result in a mild phenotype, whereas AChR deficiency mutations in CHRNA1, CHRNB1, and CHRND are rare and more severe. Although the CMS phenotypes are distinct from that of MPS, one CMS patient who was a compound heterozygote for a mutation that gives rise to fast-channel AChR kinetics and a null mutation in CHRND had arthrogryposis.14
CMS may also result from mutations in other postsynaptic proteins involved in neuromuscular transmission or in the formation and maintenance of the neuromuscular junction. Thus, recessive germline mutations in RAPSN have been reported in patients with CMS and AChR deficiency.15–18 The RAPSN gene product (rapsyn, receptor-associated protein of the synapse) has a key role in the clustering of AChRs at the neuromuscular synapse.19 The most frequent RAPSN mutation associated with CMS is the founder p.N88K missense mutation that is thought to have arisen in an ancient Indo-European population.20 Genotype-phenotype correlations have been described for CMS-associated RAPSN mutations, such that individuals who are homozygous for a p.N88K disease have less severe disease than compound heterozygotes with a single p.N88K allele.21 These observations suggested that p.N88K may cause a less severe defect than mutations that produce a truncated protein or altered membrane attachment. To date, homozygosity (or compound heterozygosity) for mutations in RAPSN predicted to cause a truncated protein have not been described in CMS. The c.1177-1178delAA mutation we identified was not detected in 340 control chromosomes (140 in the current study and 200 reported previously16) and has previously been reported (in combination with the p.N88K allele) in patients with CMS. Affected patients had decreased fetal movements, flexion contractures, craniofacial anomalies, hypotonia, and intermittent respiratory failure.16,22 We postulated that the c.1177-1178delAA mutation caused a more severe deficit in rapsyn function than p.N88K and so homozygosity for c.1177-1178delAA caused a fetal akinesia, rather than a CMS phenotype. To test this hypothesis, the effect of the c.1177-1178delAA mutation on rapsyn function was investigated.
The c.1177-1178delAA mutation predicts a frameshift after residue 392 of rapsyn resulting in 82 C-terminal missense amino acids (Figure 3A). Wild-type and mutant rapsyn were tagged with EGFP in-frame at their C termini and cotransfected with cDNAs encoding the human AChR into the muscle cell line TE671. TE671 cells were maintained at 37°C in DMEM (Sigma-Aldrich) supplemented with 10% FCS (TCS Cellworks) and 100 units/ml each of penicillin G and streptomycin (PS) purchased from Invitrogen. TE671 cells do not express detectable levels of endogenous rapsyn polypeptide. After transfection, effects on AChR clustering were studied. Thus, TE671 cells were seeded at 2 × 105 cells per well on coverslips in 6-well plates and the following day were transfected with a total of 3 μg DNA per well via calcium phosphate precipitation. Amounts of DNA used per well was 1 μg AChR α-subunit DNA, 0.5 μg each of AChR β-, δ-, and ɛ-subunit cDNA, and 0.5 μg of rapsyn cDNA or pcDNA3.1-hygro for “no rapsyn” control transfections. Transfections with only rapsyn and no AChR were carried out with 3 μg/well of rapsyn expression plasmids. The next day, the media was replaced with fresh medium. Whereas transfection with wild-type rapsyn resulted in the colocalization of rapsyn and AChR on the cell surface in small dense clusters, transfection with rapsyn-392+82-EGFP resulted in low levels of EGFP fluorescence within the cells and unclustered AChR expressed uniformly along the cell surface. To examine this observation further, we performed a time course of rapsyn expression after transfection by western blots on cell extracts. Confirming the observation from fluorescence microscopy, the mutant rapsyn was present at much lower levels and by 3 days after transfection could not be detected. The results suggest that the mutant rapsyn is rapidly degraded. It is likely that in the patient fetal AChR are not clustered because of the loss of rapsyn, which results in the disruption of the neuromuscular synapse formation and neuromuscular transmission, which in turn results in severe fetal akinesia.
Figure 3.
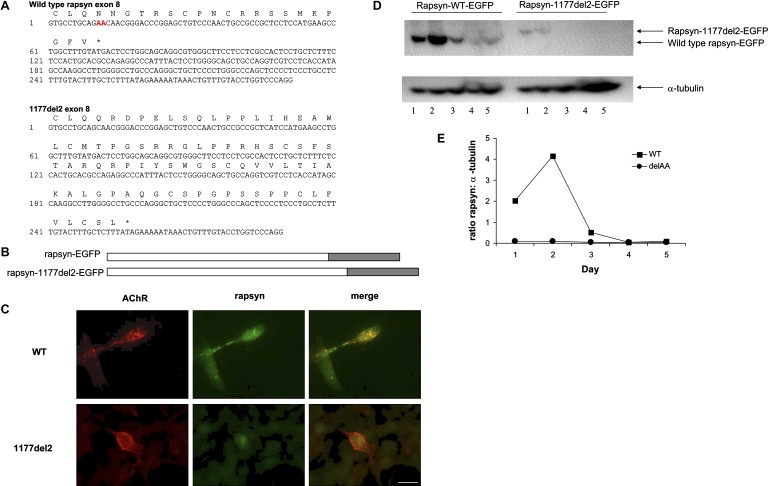
Functional Analysis of RAPSN Mutation Associated with Fetal Akinesia Sequence
(A) DNA sequences and translations of exon 8 for wild-type and mutant human rapsyn. In the wild-type sequence, the two nucleotides that are deleted in the mutant are shown in red. As a consequence of the frameshift, mutant rapsyn is 62 amino acid residues longer than wild-type (asterisk indicates termination codon).
(B) Diagram of wild-type and mutant rapsyn tagged with EGFP at the carboxyl terminus. Wild-type or mutant rapsyn cDNA without a stop codon were cloned into pEGFP-N1 (Clontech) so that EGFP was in-frame with rapsyn. Mutagenesis to delete the two adenine residues was carried out with the QuikChange mutagenesis kit purchased from Stratagene and was confirmed by DNA sequencing. The frameshift in the c.1177-1178delAA mutant extends the length of the protein by 62 amino acid residues, and so the corresponding 3′UTR was included in this construct.
(C) Rapsyn mutation c.1177-1178delAA (also referred to as 1177delAA) does not cluster the AChR. TE671 cells were cotransfected with cDNAs encoding rapsyn-EGFP or rapsyn-1177delAA-EGFP and the human AChR subunits. AChR was detected with mAb B3 directed against the AChR β subunit.28 Cells were fixed with 3% paraformaldeyde at room temperature for 20 min, washed three times with PBS, and incubated with secondary antibody Alexa Fluor 594 goat anti-mouse IgG (H+L) diluted 1:1000 in PBS containing 1% BSA (Molecular Probes). Cells were washed 3× in PBS and mounted in fluorescent mounting media (Dako Cytomation). Microscopy was performed on an Olympus BX60 wide-field fluorescence microscope, and images were captured with Openlab software (Improvision).
(D) Western blot of rapsyn-EGFP and rapsyn-1177delAA-EGFP expressed in TE671 muscle cells. TE671 cells were transfected with wild-type or mutant rapsyn-EGFP, and 48 hr later, total cell lysate was analyzed by Western blotting. Rapsyn was detected by mAb clone 1234 (Abcam) followed by anti-mouse-HRP and ECL (Amersham). As a control, α-tubulin was detected on the same western blots with a mAb (Sigma-Aldrich) followed by anti-mouse-HRP and ECL. The experiment was performed twice; one example is shown.
(E) The ratio of rapsyn:α-tubulin was obtained by densitometric scanning of western blots. Whereas rapsyn-EGFP gave robust expression on days 1 and 2, rapsyn-1177delAA-EGFP was barely detectable throughout the time course.
We found that homozygosity for a frameshift RAPSN mutation that severely impaired protein stability was associated with a fetal akinesia phenotype. Previously it was reported that rapsyn missense mutations p.L361R and p.N88K showed a partial loss of function with reduced colocalization. Both mutations could effectively mediate agrin-induced AChR clusters in myotubes but they were less stable than clusters generated with wild-type rapsyn.23 This observation is consistent with the hypothesis that RAPSN mutations may cause a spectrum of phenotypes ranging from later onset (third decade) CMS with “mild mutations” through neonatal CMS with arthrogryposis, to fetal akinesia with lethality (homozygosity for severe truncating mutations).24 The c.1177-1178delAA mutation identified in our family had previously been reported in CMS, although it is not known whether this represents a recurrent or founder mutation. Although the development of neuromuscular junction differs in mice and men, the phenotype of Chrng and Rapsn gene inactivation in mice are similar. Thus, homozygous mutant transgenic mice with targeted disruption of the Rapsn gene died within hours of birth with respiratory insufficiency and profound muscle weakness.25 The mutant mice neuromuscular junction showed no detectable AChR clusters along the length of muscle fibers. Together with the previous reports of CHRNG in MPS, it suggests that functional AChR deficiency will be a significant cause of MPS/fetal akinesia.5,6 Although we did not identify germline mutations in CHRNA1, CHRNB1, and CHRND in our cohort of patients, further analysis of these genes and of other genes implicated in CMS, such as MUSK26 or DOK7,27 may further expand the role of defective signal transmission at the neuromuscular junction in the MPS/fetal akinesia sequence.
Acknowledgments
We thank WellChild and the Wellcome Trust, MRC and Myasthenia Gravis Association/Muscular Dystrophy Campaign for financial support, and we thank the families and referring clinicians for their help with this research.
Web Resources
The URLs for data presented herein are as follows:
Ensembl Genome Browser, http://www.ensebl.org/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for LMPS and EVMPS)
Accession Numbers
Data have been deposited in the Ensemble Genome Browser with the following accession numbers: for human CHRNA1, ENSG00000138435; for CHRNB1, ENSG00000170175; for CHRND, ENSG00000135902; for CHRNG, ENSG00000196811; and for RAPSN, ENSG00000165917.
References
- 1.Hall J.G., Reed S.D., Rosenbaum K.N., Gershanik J., Chen H., Wilson K.M. Limb pterygium syndromes: a review and report of eleven patients. Am. J. Med. Genet. 1982;12:377–379. doi: 10.1002/ajmg.1320120404. [DOI] [PubMed] [Google Scholar]
- 2.Hall J.G. The lethal multiple pterygium syndromes. Am. J. Med. Genet. 1984;4:803–807. doi: 10.1002/ajmg.1320170410. [DOI] [PubMed] [Google Scholar]
- 3.Froster U.G., Stallmach T., Wisser J., Hebisch G., Robbiani M.B., Huch R., Huch A. Lethal multiple pterygium syndrome: suggestion for a consistent pathological workup and review of reported cases. Am. J. Med. Genet. 1997;68:82–85. doi: 10.1002/(sici)1096-8628(19970110)68:1<82::aid-ajmg16>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 4.Cox P.M., Brueton L.A., Bjelogrlic P., Pomroy P., Sewry C.A. Diversity of neuromuscular pathology in lethal multiple pterygium syndrome. Pediatr. Dev. Pathol. 2003;6:59–68. doi: 10.1007/s10024-002-0042-9. [DOI] [PubMed] [Google Scholar]
- 5.Morgan N.V., Brueton L.A., Cox P., Greally M.T., Tolmie J., Pasha S., Aligianis I.A., van Bokhoven H., Marton T., Al-Gazali L. Mutations in the embryonal subunit of the acetylcholine receptor (CHRNG) cause lethal and Escobar variants of multiple pterygium syndrome. Am. J. Hum. Genet. 2006;79:390–395. doi: 10.1086/506256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoffmann K., Muller J.S., Stricker S., Megarbane A., Rajab A., Lindner T.H., Cohen M., Chouery E., Adaimy L., Ghanem I. Escobar syndrome is a prenatal myasthenia caused by disruption of the acetylcholine receptor fetal gamma subunit. Am. J. Hum. Genet. 2006;79:303–312. doi: 10.1086/506257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mishina M., Takai T., Imoto K., Noda M., Takahashi T., Numa S. Molecular distinction between fetal and adult forms of muscle acetylcholine receptor. Nature. 1986;321:406–411. doi: 10.1038/321406a0. [DOI] [PubMed] [Google Scholar]
- 8.Bouzat C., Bren N., Sine S.M. Structural basis of the different gating kinetics of fetal and adult acetylcholine receptors. Neuron. 1994;6:1395–1402. doi: 10.1016/0896-6273(94)90424-3. [DOI] [PubMed] [Google Scholar]
- 9.Hesselmans L.F., Jennekens F.G., Van den Oord C.J., Veldman H., Vincent A. Development of innervation of skeletal muscle fibers in man: relation to acetylcholine receptors. Anat. Rec. 1993;236:553–562. doi: 10.1002/ar.1092360315. [DOI] [PubMed] [Google Scholar]
- 10.Kues W.A., Sakmann B., Witzemann V. Differential expression patterns of five acetylcholine receptor subunit genes in rat muscle during development. Eur. J. Neurosci. 1995;7:1376–1385. doi: 10.1111/j.1460-9568.1995.tb01129.x. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi M., Kubo T., Mizoguchi A., Carlson C.G., Endo K., Ohnishi K. Spontaneous muscle action potentials fail to develop without fetal-type acetylcholine receptors. EMBO Rep. 2002;3:674–681. doi: 10.1093/embo-reports/kvf128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Engel A.G., Ohno K., Shen X.M., Sine S.M. Congenital myasthenic syndromes: multiple molecular targets at the neuromuscular junction. Ann. N Y Acad. Sci. 2003;998:138–160. doi: 10.1196/annals.1254.016. [DOI] [PubMed] [Google Scholar]
- 13.Jurkat-Rott K., Lehmann-Horn F. Muscle channelopathies and critical points in functional and genetic studies. J. Clin. Invest. 2005;115:2000–2009. doi: 10.1172/JCI25525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brownlow S., Webster R., Croxen R., Brydson M., Neville B., Lin J.P., Vincent A., Newsom-Davis J., Beeson D. Acetylcholine receptor delta subunit mutations underlie a fast-channel myasthenic syndrome and arthrogryposis multiplex congenita. J. Clin. Invest. 2001;108:125–130. doi: 10.1172/JCI12935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohno K., Engel A.G., Shen X.M., Selcen D., Brengman J., Harper C.M., Tsujino A., Milone M. Rapsyn mutations in humans cause endplate acetylcholine-receptor deficiency and myasthenic syndrome. Am. J. Hum. Genet. 2002;70:875–885. doi: 10.1086/339465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burke G., Cossins J., Maxwell S., Owens G., Vincent A., Robb S., Nicolle M., Hilton-Jones D., Newsom-Davis J., Palace J., Beeson D. Rapsyn mutations in hereditary myasthenia: distinct early- and late-onset phenotypes. Neurology. 2003;61:826–828. doi: 10.1212/01.wnl.0000085865.55513.ae. [DOI] [PubMed] [Google Scholar]
- 17.Maselli R.A., Dunne V., Pascual-Pascual S.I., Bowe C., Agius M., Frank R., Wollmann R.L. Rapsyn mutations in myasthenic syndrome due to impaired receptor clustering. Muscle Nerve. 2003;28:293–301. doi: 10.1002/mus.10433. [DOI] [PubMed] [Google Scholar]
- 18.Muller J.S., Mildner G., Muller-Felber W., Schara U., Krampfl K., Petersen B., Petrova S., Stucka R., Mortier W., Bufler J. Rapsyn N88K is a frequent cause of congenital myasthenic syndromes in European patients. Neurology. 2003;60:1805–1810. doi: 10.1212/01.wnl.0000072262.14931.80. [DOI] [PubMed] [Google Scholar]
- 19.Apel E.D., Roberds S.L., Campbell K.P., Merlie J.P. Rapsyn may function as a link between the acetylcholine receptor and the agrin-binding dystrophin-associated glycoprotein complex. Neuron. 1995;15:115–126. doi: 10.1016/0896-6273(95)90069-1. [DOI] [PubMed] [Google Scholar]
- 20.Muller J.S., Abicht A., Burke G., Cossins J., Richard P., Baumeister S.K., Stucka R., Eymard B., Hantai D., Beeson D., Lochmuller H. The congenital myasthenic syndrome mutation RAPSN N88K derives from an ancient Indo-European founder. J. Med. Genet. 2004;41:e104. doi: 10.1136/jmg.2004.021139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dunne V., Maselli R.A. Identification of pathogenic mutations in the human rapsyn gene. J. Hum. Genet. 2003;48:204–207. doi: 10.1007/s10038-003-0005-7. [DOI] [PubMed] [Google Scholar]
- 22.Banwell B.L., Ohno K., Sieb J.P., Engel A.G. Novel truncating RAPSN mutations causing congenital myasthenic syndrome responsive to 3,4-diaminopyridine. Neuromuscul. Disord. 2004;14:202–207. doi: 10.1016/j.nmd.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 23.Cossins J., Burke G., Maxwell S., Spearman H., Man S., Kuks J., Vincent A., Palace J., Fuhrer C., Beeson D. Diverse molecular mechanisms involved in AChR deficiency due to rapsyn mutations. Brain. 2006;129:2773–2783. doi: 10.1093/brain/awl219. [DOI] [PubMed] [Google Scholar]
- 24.Burke G., Cossins J., Maxwell S., Robb S., Nicolle M., Vincent A., Newsom-Davis J., Palace J., Beeson D. Distinct phenotypes of congenital acetylcholine receptor deficiency. Neuromuscul. Disord. 2004;14:356–364. doi: 10.1016/j.nmd.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 25.Gautam M., Noakes P.G., Mudd J., Nichol M., Chu G.C., Sanes J.R., Merlie J.P. Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice. Nature. 1995;377:232–236. doi: 10.1038/377232a0. [DOI] [PubMed] [Google Scholar]
- 26.Chevessier F., Faraut B., Ravel-Chapuis A., Richard P., Gaudon K., Bauché S., Prioleau C., Herbst R., Goillot E., Ioos C. MuSK, a new target for mutations causing congenital myasthenic syndrome. Hum. Mol. Genet. 2004;13:3229–3240. doi: 10.1093/hmg/ddh333. [DOI] [PubMed] [Google Scholar]
- 27.Beeson D., Higuchi O., Palace J., Cossins J., Spearman H., Maxwell S., Newsom-Davis J., Burke G., Fawcett P., Motomura M. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science. 2006;313:1975–1978. doi: 10.1126/science.1130837. [DOI] [PubMed] [Google Scholar]
- 28.Jacobson L., Beeson D., Tzartos S., Vincent A. Monoclonal antibodies raised against human acetylcholine receptor bind to all five subunits of the fetal isoform. J. Neuroimmunol. 1999;98:112–120. doi: 10.1016/s0165-5728(99)00086-7. [DOI] [PubMed] [Google Scholar]