

Abstract
Mitochondrial DNA (mtDNA) deletions have been investigated in a number of neurodegenerative diseases. This study aimed to investigate the characteristics of mtDNA deletions found in single substantia nigra neurons from three patient groups: controls, Parkinson disease patients, and a patient with Parkinsonism due to multiple mtDNA deletions. We have identified 89 deletions from these neurons and examined the breakpoint characteristics of them. There was no difference in the types of mtDNA deletions detected in these neurons. These results suggest that the mechanism leading to the formation of these deletions in these three distinct groups could be the same.
Main Text
Recently, very high levels of somatic mitochondrial DNA (mtDNA) deletions (≈50%) have been described in substantia nigra neurons from both elderly control subjects and patients with Parkinson disease (PD [MIM 168600]).1,2 Although this is one of the most striking examples of high levels of somatic mtDNA mutations in human aging, somatic mtDNA mutations have been identified in several different tissues from elderly humans. In the substantia nigra and other tissues, these mtDNA mutations clonally expand within individual cells, and if high levels of an individual mutation are reached, then this results in a biochemical defect. This defect is detected by the histochemical presence of low activity of cytochrome c oxidase (COX) in single cells, an enzyme complex with three subunits encoded by the mitochondrial genome.3,4
An important feature of the analysis of aging postmitotic tissues is that in the COX-deficient cells the clonally expanded mtDNA mutations are usually (if not exclusively) deletions of the mitochondrial genome. Important questions are raised about the mechanism both of mtDNA deletion formation in these postmitotic cells and, once formed, how they clonally expand to high levels in individual cells (often over 60% to cause a biochemical defect).4 Also, because these somatic mutations and COX-deficient cells are observed in certain neurodegenerative diseases [e.g., Alzheimer's disease (AD [MIM 104300]) and PD],1,5 is there a similar mechanism involved? To get a greater understanding of these mechanisms, we have looked at the types of mtDNA deletions detected in substantia nigra neurons from both elderly control subjects and patients with PD. In addition, we have also studied substantia nigra neurons from a patient with multiple deletions in muscle and a mutation in the mitochondrial polymerase (POLG1) who had Parkinsonism and extensive COX-deficient neurons and cell loss in the substantia nigra.
The use of all human tissue has been consented to by the appropriate Local Research Ethics Committee and conforms to the United Kingdom Medical Research Council (UK MRC) guidelines on the use of tissue in medical research. Frozen midbrain tissue samples were collected from five PD cases (mean age 77.4 years, mean postmortem [PM] delay 27.3 hr), five age-matched controls (mean age 78.4 years, mean PM delay 24.6 hr), and from a single patient (age 59) with a multiple-mtDNA-deletion disorder. This patient presented with progressive external ophthalmoplegia at age 22 but developed Parkinsonism at age 51. Muscle biopsy from this patient contained 14.5% COX-deficient muscle fibers and multiple mtDNA deletions. The substantia nigra of this patient contained 21% COX-deficient neurons and multiple mtDNA deletions. Serial transverse frozen midbrain sections were cut from rostral to caudal at 20 μm thickness.
Mitochondrial dysfunction was detected by sequential histochemical staining for the activities of COX and succinate dehydrogenase (SDH). Single neurons were captured with a Leica laser microdissection microscope (Leica LMD) and stored at −20°C for molecular genetic analysis.
Total DNA was isolated from single neurons with a QIAamp DNA Micro Kit (QIAGEN) and eluted in 20 μl of water. To detect possible mtDNA deletions in individual cells, we used a two-step long-range polymerase chain reaction (PCR) strategy (Expand Long Template PCR System, Roche) to amplify a 10 kb fragment spanning the major arc of the genome. First-round PCR reactions used 1 μl of DNA as template and comprised 1× reaction buffer (Roche), 10 mM each deoxynucleotide triphosphate (dNTP), 10 μg bovine serum albumin (BSA) (10×), 0.75 μl enzyme mix, and 30 pmol of primer (nt 5855–5875 [according to Cambridge reference sequence] and nt 15896–15877) in a total volume of 50 μl. Amplifications were carried out with a GeneAmp PCR System 9600 (Applied Biosystems) with the following cycling conditions: 3 min at 93°C; 10 cycles of 93°C for 30 s, 58°C for 30 s, and 68°C for 12 min; 20 cycles of 93°C for 30 s, 58°C for 30 s, and 68°C for 12 min plus 5 s per additional cycle; and a final extension of 11 min at 68°C. This reaction product was then diluted to 1:50, and 1 μl was used as template in a second round of PCR amplification, with the same reagents as above except the primers used were nt 6358–6377 and nt 15368–15349 for the generation of a 9 kb fragment and nt 7272–7293 and nt 13859–13839 for the generation of a 6.5 kb fragment. In view of the previous observations for a hotspot for deletions at 16070 bp,6,7 we also performed long-range PCR to encompass this region. The PCR conditions were identical to those described above except the primers used were nt 5855–5875 and nt 129–110 for the first round and nt 6358–6377 and nt 019–001 for the second round. Amplified products were separated through 0.7% agarose gels containing ethidium bromide and visualized over ultraviolet (UV) light. These amplified products were then gel extracted with the QIAGEN Gel Extraction Kit per the manufacturer's instructions. The concentration of the extracted DNA was then measured with a NanoDrop spectrophotometer. Amplified samples were purified (ExoSapIT, Amersham Pharmacia), sequenced with BigDye (v3.1) terminator cycle sequencing chemistries on an ABI 3100 Genetic Analyzer (Applied Biosystems), and directly compared to the revised Cambridge reference sequence (rCRS) with SeqScape software (Applied Biosystems).
We detected a total of 89 mtDNA deletions by using nested, long-range PCR assays that amplified between the regions nt 7293 and nt 13859 (n = 59) and between nt 6377 and nt 019 (n = 30) in individual neurons from the substantia nigra of three different groups: patients with PD, age-matched controls, and a patient with multiple-deletion disorder. In the majority of neurons in which we were able to identify a long-range PCR product, we never detected a full-length product (Figure 1). Although long-range PCR is not quantitative, this result highlights how frequent mtDNA deletions are within individual neurons. We detected deletions of various sizes ranging from 1763 bp to 9445 bp within the major arc of the mitochondrial genome (Tables 1 and 2). All detected deletions resulted in the loss of both transfer RNA (tRNA) and protein-encoding genes. For the purposes of this report, the breakpoint closest to the origin of light-strand replication (nt 5721–5798) was denoted 5′ and the breakpoint closest to the origin of heavy-strand replication was denoted 3′. Each neuron harboured a different mtDNA deletion. Twelve mtDNA deletions were detected in more than one neuron, and these deletions were detected in neurons from the same and different individuals. The 4977 bp “common” deletion was detected a total of seven times in the neurons studied. We also detected a 7920 bp (nt 6423–14344) and a 8558 bp (nt 6585–15144) deletion in four neurons and a 6877 (nt 7114–13992), a 6278 bp (nt 7409–13688), and a 9128 bp (nt 6790–15918) deletion three times each. Six other deletions were detected in two neurons each. By using the primers that spanned the 16070 region, we detected three deletions with a breakpoint within this region; two were detected in aged neurons with 3′ breakpoints at nt 16074 and nt 16035, and one was detected in a neuron from the patient with multiple-deletion disorder 3′ breakpoint at nt 16071.
Figure 1.
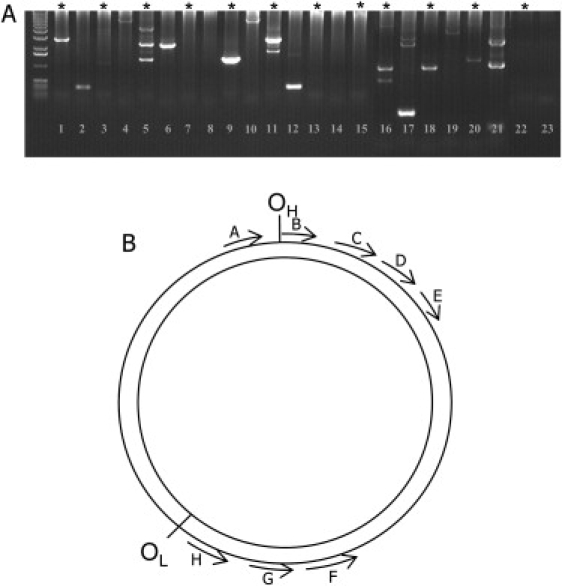
An Agarose Gel Displaying a Typical Long-Range PCR of Substantia Nigra Neurons
(A) A typical agarose gel displaying the different sized mtDNA deletions that have been amplified with long-range PCR on DNA from single neurons. This gel shows the products from a second-round PCR, and each lane represents a PCR from a single cell. In some cells, it is possible to detect multiple deletions. Lanes 1, 2, 9, and 10 show deletions detected in a cell from an aged individual, lanes 5 and 6 show deletions detected in a cell from a PD patient, lanes 11,12, and 16–21 show deletion detected within cells from the patient with multiple mtDNA deletions, and lanes 22 and 23 show negative controls. Those cells marked with an ∗ indicate products from a 9 kb PCR, whereas the remaining lanes are the product of a 6 kb PCR.
(B) A representation of the positions of the primer pairs used in this study. Primer pairs C (15896–15877) and H (5855–5875) were used for the generation of the 10 kb first round PCR, primer pairs D (15368–15348) and G (6358–6377) the 9 kb second round, and primer pairs E (13859–13839) and F (7272–7293) the 6 kb second round. Primer pairs A (129–110) and H (5855–5875) were used for the generation of the first-round PCR products for the 16070 bp assay and primer pairs B (019–001) and G (6358–6377) the second round products of this assay.
Table 1.
MtDNA Deletion Breakpoints from Substantia Nigra Neurons with Primers within the Major Arc
| Deletion | Break Points | Repeat | 5′ Sequence | 3′ Sequence | Class of Deletion | Size (bp) |
|---|---|---|---|---|---|---|
| PD | ||||||
| 1 | 9729–15338 | no repeat | 5′-TCTCTATTTTACCC-3′ | 5′-CTATTCTTGCACG-3′ | Class III | 5608 |
| 2 | 7805–13844 | 5′-CCTAG-3′ | 5′-CCGCCATCAT(CCTAG)-3′ | 5′-ACCTCAACTACCTAA-3′ | Class I | 6038 |
| 3 | 8280–13782 | 5′-CCCCCTCTAC-3′ | 5′-CACCCCCTCTA(CCCCCTCTAG)-3′ | 5′-(CCCCCTCTAC)CTAAAACTC-3′ | Class II | 5501 |
| 4 | 6423–14344 | 5′-ACCCC-3′ | 5′-TCAATATAAA(ACCCC)-3′ | 5′-ATCATACTCTTTC-3′ | I | 7920 |
| 5 | 8482–13460 | 5′-ACCTCCCTCACCA-3′ | 5′-AAACTACCACCT(ACCTCCCTCACCA)-3′ | 5′-TTGGCAGCCTAGCATT-3′ | I | 4977 |
| 6 | 8566–13991 | 5′-TCCTAGACCT-3′ | 5′-TGCCCCCACAA(TCCTAGGCCT)-3′ | 5′-(TCCTAGACCT)AACCTGACTA-3′ | II | 5424 |
| 7 | 7114–13992 | 5′-CCTAGACCTAACCT-3′ | 5′-TCAGGCTACAC(CCTAGACCAAACCT)-3′ | 5′-(CCTAGACCTAACCT)GACTAGAAAA-3′ | II | 6877 |
| 8 | 7114–13992 | 5′-CCTAGACCTAACCT-3′ | 5′-TCAGGCTACAC(CCTAGACCAAACCT)-3′ | 5′-(CCTAGACCTAACCT)GACTAGAAAA-3′ | II | 6877 |
| 9 | 6423–14344 | 5′-ACCCC-3′ | 5′-TCAATATAAA(ACCCC)-3′ | 5′-ATCATACTCTTTC-3′ | I | 7920 |
| 10 | 9659–11536 | 5′-CATAGTCTA-3′ | 5′-AATCACCTGAGCTCAC(CATAGTCTA)-3′ | 5′-(CATAGCCTA)CCCCTTCC-3′ | II | 1876 |
| 11 | 8034–11434 | 5′-GAAGCCCCCAT-3′ | 5′-TACTCCCGATT(GAAGCCCCCAT)-3′ | 5′-CGCTGGGTCAATAGTAC-3′ | I | 3399 |
| 12 | 6423–14344 | 5′-ACCCC-3′ | 5′-TCAATATAAA(ACCCC)-3′ | 5′-ATCATACTCTTTC-3′ | I | 7920 |
| 13 | 10104–11868 | 5′-GCCTTAC-3′ | 5′-AACACCCTCCTA(GCCTTAC)-3′ | 5′-CCCCCACTATTAACCT-3′ | I | 1763 |
| 14 | 8482–13460 | 5′-ACCTCCCTCACCA-3′ | 5′-AAACTACCACCT(ACCTCC>CTCACCA)-3′ | 5′-TTGGCAGCCTAGCATT-3′ | I | 4977 |
| 15 | 8049–14115 | no repeat | 5′-TTCGTATAATAATTAC-3′ | 5′-CCCACTCATCCTAACCC-3′ | III | 6065 |
| 16 | 7677–15013 | no repeat | 5′-CCCTCATAATCAT-3′ | 5′-ATTCTTTATCTGCCTCTT-3′ | III | 7335 |
| 17 | 8482–13460 | 5′-ACCTCCCTCACCA-3′ | 5′-AAACTACCACCT(ACCTCCCTCACCA)-3′ | 5′-TTGGCAGCCTAGCATT-3′ | I | 4977 |
| 18 | 8428–15482 | 5′-ACACTATTC-3′ | 5′-CCCCCATACTCCTT(ACACTATTC)-3′ | 5′-TCACCAGACCTCCTAGG-3′ | I | 7054 |
| 19 | 7409–13688 | 5′-CCCCACCCTAC-3′ | 5′-TATATGGATGCC(CCCCACCCTAC)-3′ | 5′-TAAACCCCATTAACG-3′ | I | 6278 |
| 20 | 7409–13688 | 5′-CCCCACCCTAC-3′ | 5′-TATATGGATGCC(CCCCACCCTAC)-3′ | 5′-TAAACCCCATTAACG-3′ | I | 6278 |
| 21 | 7635–15310 | 5′-TCAT-3′ | 5′-GACGCTACTTCCCCTA(TCAT)-3′ | 5′-TATTGCAGCCCTAGCAAC-3′ | I | 7674 |
| 22 | 8482–13460 | 5′-ACCTCCCTCACCA-3′ | 5′-AAACTACCACCT(ACCTCCCTCACCA)-3′ | 5′-TTGGCAGCCTAGCATT-3′ | I | 4977 |
| Aged | ||||||
| 1 | 7805–13844 | 5′-CCTAG-3′ | 5′-CCGCCATCAT(CCTAG)-3′ | 5′-ACCTCAACTACCTAA-3′ | I | 6038 |
| 2 | 6555–14345 | 5′-CCA-3′ | 5′-GCAACCTCAACA(CCA)-3′ | 5′-TCATACTCTTTCACCC-3′ | I | 7789 |
| 3 | 6437–14077 | 5′-CCATAACCCAA-3′ | 5′-CCCCCTG(CCATAACCCAA)-3′ | 5′-(CCTCAACCCAA)AAAGGCATA-3′ | II | 7639 |
| 4 | 6427–15269 | no repeat | 5′-AAACCCCCTGC-3′ | 5′-CTCACACGATTCTTT-3′ | III | 8841 |
| 5 | 8418–14127 | 5′-TCCT-3′ | 5′-TTACCCCCATAC(TCCT)-3′ | 5′-AACCCTACTCCTAAT-3′ | I | 5708 |
| 6 | 9924–15194 | no repeat | 5′-GAAGCCGCCGCCT-3′ | 5′-CTATCCGCCATCCC-3′ | III | 5269 |
| 7 | 6453–14288 | 5′-CCCCTCT-3′ | 5′-TACCAAACG(CCCCTCT)-3′ | 5′-CCTTCATAAATTATTC-3′ | I | 7834 |
| 8 | 8408–14118 | 5′-CCC-3′ | 5′-ACCATAATTA(CCC)-3′ | 5′-ACTCATCCTAACCC-3′ | I | 5709 |
| 9 | 8482–13460 | 5′-ACCTCCCTCACCA-3′ | 5′-AAACTACCACCT(ACCTCCCTCACCA)-3′ | 5′-TTGGCAGCCTAGCATT-3′ | I | 4977 |
| 10 | 6423–14344 | 5′-ACCCC-3′ | 5′-TCAATATAAA(ACCCC)-3′ | 5′-ATCATACTCTTTC-3′ | I | 7920 |
| 11 | 6416–14182 | 5′-CAATTATCAATATA-3′ | 5′-CACAA(CAATTATCAATATA)-3′ | 5′-(CAATTA_CAATATA)TACACCAACAAA-3′ | II | 7765 |
| 12 | 8049–14115 | no repeat | 5′-TTCGTATAATAATTAC-3′ | 5′-CCCACTCATCCTAACCC-3′ | III | 6065 |
| 13 | 6476–14146 | 5′-TCCTAATCACA-3′ | 5′-CTGATCCG(TCCTAATCACA)-3′ | 5′-TAACCTATTCCCCCG-3′ | I | 7669 |
| 14 | 7815–13581 | 5′-CTCATCGC-3′ | 5′-CATCCTAGTC(CTCATCGC)-3′ | 5′-TACCTCCCTGACAA-3′ | I | 5765 |
| 15 | 7658–15548 | 5′-ATCAAGCCC-3′ | 5′-ATCACCTTTCATG(ATCACGCCC)-3′ | 5′-(ATCAAGCCC)GAATGATATTT-3′ | II | 7889 |
| 16 | 6585–15144 | 5′-GACC-3′ | 5′-CCGGAGGAGGA(GACC)-3′ | 5′-(GTCC)TCCCGTGAGGCCAAA-3′ | II | 8558 |
| 17 | 6585–15144 | 5′-GACC-3′ | 5′-CCGGAGGAGGA(GACC)-3′ | 5′-(GTCC)TCCCGTGAGGCCAAA-3′ | II | 8558 |
| 18 | 6585–15144 | 5′-GACC-3′ | 5′-CCGGAGGAGGA(GACC)-3′ | 5′-(GTCC)TCCCGTGAGGCCAAA-3′ | II | 8558 |
| Multiple-Deletion Patient | ||||||
| 1 | 6577–15154 | 5′-GAGG-3′ | 5′-TTCTTCGACCCCGCCG(GAGG)-3′ | 5′-CCAAATATCATTCTGAG-3′ | I | 8576 |
| 2 | 6587–14285 | 5′-GACCCC-3′ | 5′-CGCCGGAGGAGGA(GACCCC)-3′ | 5′-TCTCCTTCATAAA-3′ | I | 7697 |
| 3 | 9417–13684 | 5′-CCACC-3′ | 5′-CATACCAAGG(CCACC)-3′ | 5′-CTACTAAACCCC-3′ | I | 4266 |
| 4 | 7406–12395 | 5′-CCCCCCACCCTTACCAC-3′ | 5′-TATGGATG(CCCCCCACCCT_ACCAC)-3′ | 5′-(CCCCCCATCCTTACCAC)CCTCGT-3′ | II | 4988 |
| 5 | 6457–14867 | 5′-CTGATCCTCC-3′ | 5′-CCCCTCTTCGT(CTGATCCGTCC)-3′ | 5′-(CTGATCCTCC)AAATCACCACAGG-3′ | II | 8409 |
| 6 | 6587–14285 | 5′-GACCCC-3′ | 5′-CGCCGGAGGAGGA(GACCCC)-3′ | 5′-TCTCCTTCATAAA-3′ | I | 7697 |
| 7 | 6572–12304 | 5′-GCC-3′ | 5′-TTCTTCGACCCC(GCC)-3′ | 5′-CCAAAAATTTTGGT-3′ | I | 5731 |
| 8 | 6585–15144 | 5′-GACC-3′ | 5′-CCGGAGGAGGA(GACC)-3′ | 5′-(GTCC)TCCCGTGAGGCCAAA-3′ | II | 8558 |
| 9 | 8482–13460 | 5′-ACCTCCCTCACCA-3′ | 5′-AAACTACCACCT(ACCTCCCTCACCA)-3′ | 5′-TTGGCAGCCTAGCATT-3′ | I | 4977 |
| 10 | 7367–12063 | no repeat | 5′-TCCTAATAGTAGAAGAACCC-3′ | 5′-CCCTCATGTTCATACACCT-3′ | III | 4694 |
| 11 | 6422–15208 | 5′-AACCC-3′ | 5′-TTATCAATATAA(AACCC)-3′ | 5′-(ATCCC)ATACATTGGG-3′ | II | 8785 |
| 12 | 6551–13852 | 5′-ACCTCAAC-3′ | 5′-CTAACAGACCGCA(ACCTCAAC)-3′ | 5′-TACCTAACCAACAAA-3′ | I | 7300 |
| 13 | 7114–13992 | 5′-CCTAGACCTAACCT-3′ | 5′-TCAGGCTACAC(CCTAGACCAAACCT)-3′ | 5′-(CCTAGACCTAACCT)GACTAGAAAA-3′ | II | 6877 |
| 14 | 8482–13460 | 5′-ACCTCCCTCACCA-3′ | 5′-AAACTACCACCT(ACCTCCCTCACCA)-3′ | 5′-TTGGCAGCCTAGCATT-3′ | I | 4977 |
| 15 | 7409–3688 | 5′-CCCCACCCTAC-3′ | 5′-TATATGGATGCC(CCCCACCCTAC)-3′ | 5′-TAAACCCCATTAACG-3′ | I | 6278 |
| 16 | 7204–13903 | 5′-TATC-3′ | 5′-TTTCTCGGCC(TATC)-3′ | 5′-(TTTC)TCCAACATAC-3′ | II | 6698 |
| 17 | 10071–14622 | no repeat | 5′-AAACTTCGCCT-3′ | 5′-CCACAAACCCCATT-3′ | III | 4550 |
| 18 | 7998–12300 | no repeat | 5′-GACTCCTTGACGT-3′ | 5′-GGCCCCAAAAATTTT-3′ | III | 4301 |
| 19 | 7201–14809 | 5′-TCGGCCTCCCC-3′ | 5′-CACTTTC(TCGGCCTATCC) -3′ | 5′-(TCGACCTCCCC)ACCCCAT-3′ | II | 7607 |
This table displays all 59 mtDNA deletions detected in single substantia nigra neurons between primers at nt 15896–15877 and nt 5855–5875. Repeat sequences are shown in brackets and, where known, the deleted repeat shown in italics, imperfect bases within repeats are underlined, and missing bases are indicated by an underscore.
Table 2.
MtDNA Deletion Breakpoints from Substantia Nigra Neurons with Primers within the Major Arc that Include the D Loop
| Deletion | Breakpoints | Repeat | 5′ Sequence | 3′ Sequence | Class of Deletion | Size (bp) |
|---|---|---|---|---|---|---|
| PD | ||||||
| 1 | 6790–15918 | no repeat | 5′-TTACAGTAGG-3′ | 5′-TTGTAAAGCCG-3′ | III | 9128 |
| 2 | 7667–14359 | no repeat | 5′-GATCACGCCC-3′ | 5′-CCACAGCACCA-3′ | III | 6692 |
| 3 | 6537–13838 | 5′-ACTACTAACAG-3′ | 5′-CACTAT(ACTACTAACAG)-3′ | 5′-(ACTTCTAACAG)CCCTAGACC-3′ | II | 7301 |
| 4 | 7108–14688 | 5′-CTACAACC-3′ | 5′-TTCTCAGG(CTACACCC) -3′ | 5′-(CTACAACC)ACGACCAAT-3′ | II | 7580 |
| 5 | 6790–15918 | no repeat | 5′-TTACAGTAGG-3′ | 5′-TTGTAAGCCG-3′ | III | 9128 |
| 6 | 7128–14006 | no repeat | 5′-GACCAAACCT-3′ | 5′-GACTAGAAAA-3′ | III | 6889 |
| 7 | 6476–13777 | 5′-ACA-3′ | 5′-GTCCTAATC(ACA)-3′ | 5′-ACAATCCCCC-3′ | I | 7301 |
| 8 | 7188–14335 | 5′-TTCCCACAAC-3′ | 5′-CTTTC(TTCCCACAAC)-3′ | 5′-(TTACCACAAC)CACCA-3′ | II | 7147 |
| Aged | ||||||
| 1 | 6625–16074 | 5′-TCACCC-3′ | 5′-ATTTTTCGG(TCACCC)-3′ | 5′-ATCAACAACA-3′ | I | 9445 |
| 2 | 6759–15865 | no repeat | 5′-CTAGGGTTTA-3′ | 5′-AAACAAAATA-3′ | III | 9106 |
| 3 | 8232–15542 | 5′-CCCCT-3′ | 5′-AGAATTAATT(CCCCT)-3′ | 5′-CCCCACATCAAG-3′ | I | 7312 |
| 4 | 7821–13760 | 5′-CCTCCC-3′ | 5′-TCATCGC(CCTCCC)-3′ | 5′-(CCCCCG)CATCCCCCTTC-3′ | II | 5939 |
| 5 | 7265–16035 | no repeat | 5′-CATCCTATCA-3′ | 5′-GGAAGCAGATT-3′ | III | 8770 |
| 6 | 6790–15918 | no repeat | 5′-TTTACAGTAGG-3′ | 5′-TTGTAAGCC-3′ | III | 9128 |
| 7 | 6835–14380 | 5′-CATCGCTAACCCCAC-3′ | 5′-CCATAAT(CATCGCTATCCCCAC)-3′ | 5′-(CATCGCTAACCCCAC)TAAAA-3′ | II | 7545 |
| 8 | 7629–14813 | 5′-ACTTCCCC-3′ | 5′-AAGACGCT(ACTTCCCC)-3′ | 5′-(ACCTCCCC)ACCCCATCC-3′ | II | 7183 |
| 9 | 7808–14799 | 5′-TCATCGACCTCCC-3′ | 5′-CTAGTCC(TCATCGCCCTCCC)-3′ | 5′-(TCATCGACCTCCC)CACCCC-3′ | II | 6991 |
| 10 | 8904–14903 | 5′-CCTAGCC-3′ | 5′-TTAAAAATGC(CCTAGCC)-3′ | 5′-ATGCACTACTCA-3′ | I | 5999 |
| Multiple-Deletion Patient | ||||||
| 1 | 6501–13802 | 5′-CAG_CCCT-3′ | 5′-CTATCTCTCC(CAGTCC_T)-3′ | 5′-(CAG_CCCT)CGCTGTCACT-3′ | II | 7300 |
| 2 | 7125–14391 | no repeat | 5′-CTAGACCAAA-3′ | 5′-CCACTAAAAC-3′ | III | 7264 |
| 3 | 8232–15542 | 5′-CCCCT-3′ | 5′-AATTAATT(CCCCT)-3′ | 5′-CCCCACATCA-3′ | I | 7310 |
| 4 | 7414–15609 | 5′-CCCTACCACAC-3′ | 5′-GCCCCCCA(CCCTACCACAC)-3′ | 5′-(CCCTAA_CAAAC)TAGGAGGCG-3′ | II | 8195 |
| 5 | 6501–13802 | 5′-CAG_CCCT-3′ | 5′-CTATCTCTCC(CAGTCC_T)-3′ | 5′-(CAG_CCCT)CGCTGTCACT-3′ | II | 7300 |
| 6 | 6851–14816 | 5′- TCATCGCTATCCCCACC-3′ | 5′-TACCATAA(TCATCGCTATCCCCACC)-3′ | 5′-(TCATCGACCTCCCCACC)CCATCCAAC-3′ | II | 7965 |
| 7 | 7414–15609 | 5′-CCCTACCACAC-3′ | 5′-GCCCCCCA(CCCTACCACAC)-3′ | 5′-(CCCTAA_CAAAC)TAGGAGGCG-3′ | II | 8195 |
| 8 | 7212–14261 | no repeat | 5′-TATCCGGAATGC-3′ | 5′-TCCTCCCGAA-3′ | III | 7049 |
| 9 | 8601–15731 | 5′-CTCCTCATTCTA-3′ | 5′-GCCGCAGTA(CTGATCATTCTA)-3′ | 5′-(CTCCTCATTCTA)ACCTGAATCG-3′ | II | 7151 |
| 10 | 7458–11845 | 5′-AAGC-3′ | 5′-AAAAGGAAGG(AATC)-3′ | 5′-(AAGC)CTCGCTAACC-3′ | II | 4390 |
| 11 | 8656–14811 | 5′-CACCCCA-3′ | 5′-GACTAATCA(CCACCCA)-3′ | 5′-(CACCCCA)TCCAACATCT-3′ | II | 6163 |
| 12 | 7819–16071 | 5′-CCCATC-3′ | 5′-TCATCG(CCATC)-3′ | 5′-(CCCATC)AACAACCGCT-3′ | II | 8252 |
This table displays all 30 mtDNA deletions detected in single substantia nigra neurons with primers to include amplification of the 16,070 bp deletion hotspot. Repeat sequences are shown in brackets, and where the deleted repeat is known, it is shown in italics; imperfect bases within repeats are underlined, and missing bases are indicated by an underscore.
Of the deletions that we investigated, 71 out of 89 involved a direct repeat of at least 3 bp. Thirty-seven out of eighty-nine of the breakpoints involved a perfect repeat, 34 out of 89 had an imperfect repeat, and 18 out of 89 had no direct repeat (example shown in Figure 2). Where there is an imperfect repeat, this allows identification of whether it is the 5′ or the 3′ end of the mtDNA molecule that remains after formation of the deletion. Of the imperfect repeats, 15 out of 34 involved deletion of the 5′ end, and in 16 out of 34, the 3′ end was deleted. Of interest, 2 out of 34 of the imperfect repeats were split in the middle so that the first half of the breakpoint corresponded to the 5′ end and the second half with the 3′ end, and for one of the imperfect repeats, the deleted repeat could not be distinguished. An example of one such imperfect repeats is shown in Figure 2E. The sequence proceeds through the repeat at the 5′ end until base 7406 (a cytosine), and until this point, the 3′ sequence is deleted (the 3′ sequence reads 5′-CCCCCCATCC-3′); then, after this base, it is the 5′ sequence that is deleted, and the sequence proceeds from base 12395 (a thymine) (the 5′ sequence reads 5′-T_ACCAC-3′).
Figure 2.
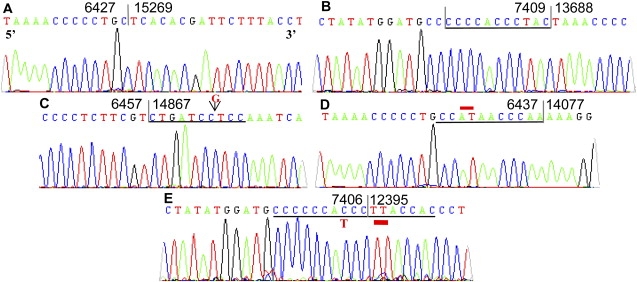
Sequence Chromatograms Displaying Examples of the Different Types of mtDNA Deletion Breakpoints Detected
This figure highlights the spectrum of different mtDNA deletion breakpoints detected in the neurons studied. Deletion A is from an aged patient and has no repeat sequence, and deletion B has an 11 bp direct repeat and was found twice in the neurons of a PD patient. Deletions C and D have imperfect repeat sequences. Deletion C is from the patient with multiple mtDNA deletions, and it is an example of a deletion where the imperfect 5′ repeat is deleted; in the 5′ sequence, there is an insertion of a g at the position indicated. Deletion D is from an aged control and is an example of where the 3′ repeat is deleted; in the 3′ sequence, the two bases highlighted are 5′-CT-3′. Finally, deletion E shows a split repeat sequence where part of both the 5′ and 3′ repeat is deleted.
The positions of the 5′ and 3′ breakpoints of the detected deletions with repeats were plotted against the frequency of the position (Figure 3). Although the 5′ breakpoints have a slightly more complicated distribution, it is noted that the 3′ break points seem to cluster in three distinct regions of the genome (at nt 11400–12400, nt 13400–14400, and nt 14800–16100) (Figure 3). In addition, there was no difference in the distribution of those deletions without repeat sequences compared to those with repeat sequences.
Figure 3.
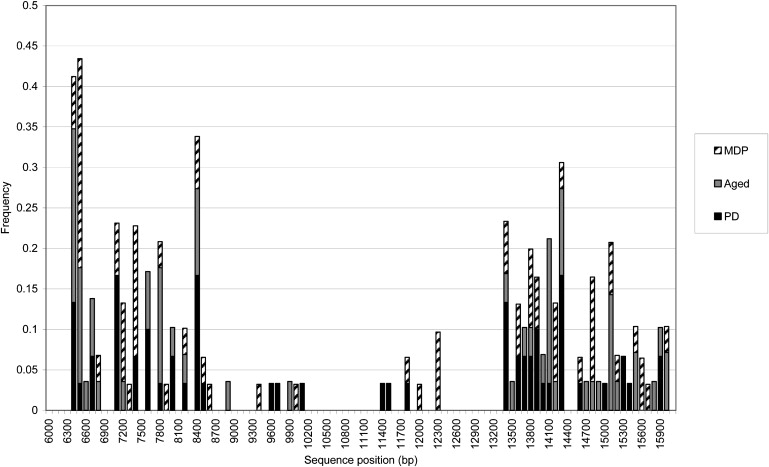
Distribution of the mtDNA Deletion Breakpoints Detected from Substantia Nigra Neurons
This graph shows the distribution of the detected mtDNA deletion breakpoints within the genome. It is clear that although the 5′ breakpoints have a complicated distribution, the 3′ breakpoints form three clusters. The breakpoint locations of the deletions from the PD patients are shown in black, those from the aged neurons are in gray, and those from the patient with multiple mtDNA deletions are hatched. MDP indicates a multiple-deletion patient.
We compared a number of different parameters between the deletions observed in the neurons from patients with PD, from aged control subjects, and from the patient with multiple deletions (Figure 4). We used a 2 × 3 Chi-square test to compare the parameter repeats versus no repeats, and there was no significant difference between the groups (p = 0.7683) (Figure 4A). When we compared the next parameter, perfect versus imperfect repeats, a 2 × 3 Chi-square test was also used, and again there was no significant difference (p = 0.1673) (Figure 4B). Finally, we looked at the length of the repeat, and again there was no significant difference between the three groups (p > 0.10, analysis of variance [ANOVA]) (Figure 4C). Therefore, for all parameters studied, there was no difference found in the types of deletion breakpoint detected between the three groups.
Figure 4.
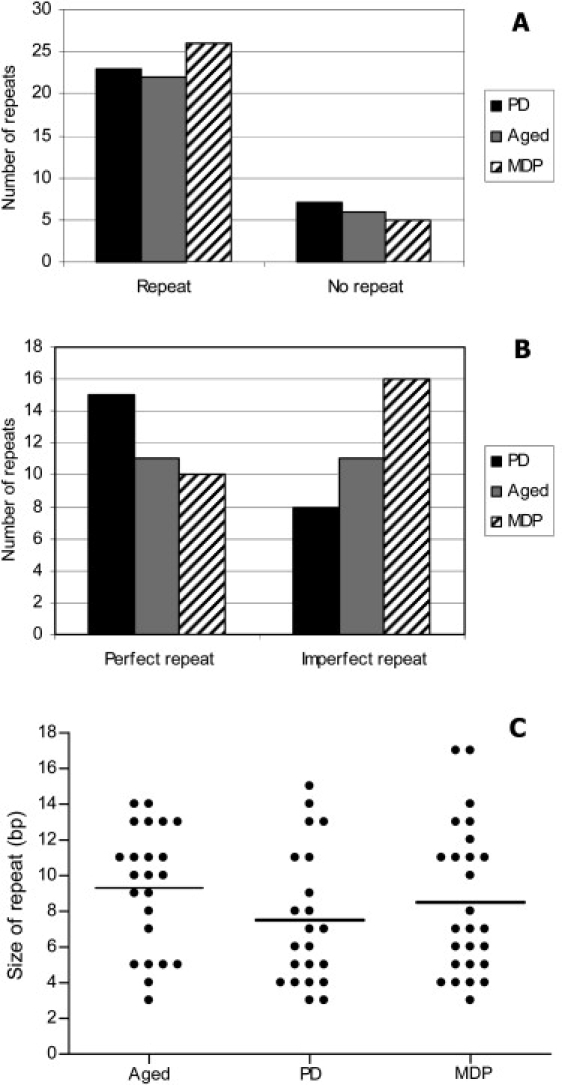
Graphs Displaying the Characteristics of the mtDNA Deletion Breakpoints within the Three Studied Groups
These graphs highlight the characteristics of the detected mtDNA deletion breakpoints. (A) shows repeat versus no repeat. A Chi-square test performed on these data showed the results were not significantly different (p = 0.7683). (B) displays perfect repeat versus imperfect repeat. A Chi-square test performed on these data showed that the results were not significantly different (p = 0.1673). (C) shows the size of all of the repeat sequences detected, whether imperfect or perfect. An ANOVA test performed on these data showed that the results were not significantly different (p > 0.10). For (A) and (B), mtDNA deletions from PD patients are in black, those from aged patients are in gray, and those from the patient with multiple mtDNA deletions are hatched. MDP indicates a multiple-deletion patient.
Our recent observations and those of several other groups have highlighted that mtDNA deletions are frequently observed in aging postmitotic tissues. These mtDNA deletions must arise somatically because there are different deletions in individual cells, an entirely different situation to those patients with single, large-scale deletion disorders in which the deletion is present in the oocyte and identical in all cells in which it can be detected. These studies have confirmed that there are high levels of clonally expanded mtDNA deletions in individual neurons from the substantia nigra in aging and patients with PD. In this study, we have observed similarities in the types of mtDNA deletion breakpoints occurring in aging subjects, patients with PD, and a patient with multiple mtDNA deletions. The majority of deletion breakpoints observed (∼79%) in this study are flanked by short (3–17 bp) direct repeats. Of these, approximately 41% are perfect, homologous repeats (class I deletions) and approximately 38% are imperfect (class II deletions). Only approximately 20% of deletions involve no direct repeat (class III deletions). In a study analyzing 263 pathogenic human mtDNA deletions, the authors found that approximately 60% were perfect repeats, approximately 30% were imperfect repeats, and approximately 10% had no direct repeat.8 These results are similar to those within our study, suggesting a common mechanism for the generation of mtDNA deletions in humans. Although a study identifying the deletion breakpoint characteristics of patients with POLG1 or PEO1 mutations did not find evidence of perfect repeats of at least 10 bp, almost all deletion breakpoints involved imperfect repeats of at least 2 bp, with homopolymeric runs being a common factor associated with the breakpoints.7
The distribution of 5′ end deletion breakpoints is spread out with some peaks around the nt 6500, 7800, and 8400 regions. For the 3′ deletion breakpoints ends, there are three clusters of where the breakpoints occur. There is no difference in the distribution of deletion breakpoints with no direct repeats. This suggests that the same fundamental mechanism exists to generate mtDNA deletions involving both direct repeats and those without breakpoint sequence homology.
The mechanism of mtDNA deletion formation remains uncertain. It has been postulated on the basis of the Clayton strand-displacement model of mtDNA replication that deletions occur on the basis of slipped-strand replication.9 However, this model of replication has recently been challenged with mtDNA replication occurring by a more conventional replication mechanism in which leading-lagging-strand DNA replication is coordinated.10 Recent studies have suggested these models are now rather similar, and even in the proposed leading-lagging-strand model, there is extensive involvement of RNA, suggesting potential mechanisms of deletion formation during replication.11
Analysis of the breakpoints with imperfect repeats (n = 34) showed that in 47% of cases, the 3′ end is deleted, compared with 44% of cases, in which the 5′ end is deleted. The latter result is interesting because it has been previously suggested that the 5′ repeat sequence is rarely deleted.8 This previous bias suggested exclusion of intramolecular recombination as a method of mtDNA deletion generation. However, in our data, the chance of the 5′ repeat sequence being deleted is almost equal; therefore, on the basis of this data, recombination might be a possible explanation.
Previous studies have shown that the substantia nigra is particularly prone to oxidative damage, and thus potentially there will be high levels of oxidative damage to the mitochondrial genome. This might result in either impaired replication or, alternatively, repair of damaged mtDNA, which could lead to deletion formation. Double-strand breaks have previously been shown to be a possible cause of deletion formation, as shown by the elegant experiments by Moraes and colleagues in mouse after the Pst1 restriction endonuclease was targeted to muscle.12
In conclusion, we have shown that the types of mtDNA deletions that have clonally expanded in substantia nigra neurons from patients with PD and age-matched control subjects are similar to those from a patient with multiple-deletion disorder and those reported in the literature from patients with single mtDNA deletions. The breakpoints of the deletions show similar characteristics and distribution to those from patients with single mtDNA deletions. These results indicate that the mechanism for mtDNA deletions is the same, regardless of the clinical scenario. At present, it is unknown what mechanism is generating mtDNA deletions; however, because we found high levels in substantia nigra neurons, cells shown to be under considerable oxidative stress,13 it seems likely that oxidative damage is involved. These current data confirm the previously reported high levels of mtDNA deletions in substantia nigra neurons. Previous studies have shown that higher levels of clonally expanded deletions are seen in the COX-deficient neurons, suggesting a functional effect of these deletions, similar to that observed in muscle.1,2,14
Acknowledgments
This work was funded by the MRC, Alzheimer's Research Trust, and the Wellcome Trust. We are grateful for the helpful comments from two anonymous referees.
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Bender A., Krishnan K.J., Morris C.M., Taylor G.A., Reeve A.K., Perry R.H., Jaros E., Hersheson J.S., Betts J., Klopstock T. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 2.Kraytsberg Y., Kudryavtseva E., McKee A.C., Geula C., Kowall N.W., Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006;38:518–520. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- 3.Itoh K., Weis S., Mehraein P., Muller-Hocker J. Cytochrome c oxidase defects of the human substantia nigra in normal aging. Neurobiol. Aging. 1996;17:843–848. doi: 10.1016/s0197-4580(96)00168-6. [DOI] [PubMed] [Google Scholar]
- 4.Sciacco M., Bonilla E., Schon E.A., DiMauro S., Moraes C.T. Distribution of wild-type and common deletion forms of mtDNA in normal and respiration-deficient muscle fibers from patients with mitochondrial myopathy. Hum. Mol. Genet. 1994;3:13–19. doi: 10.1093/hmg/3.1.13. [DOI] [PubMed] [Google Scholar]
- 5.Cottrell D.A., Blakely E.L., Johnson M.A., Ince P.G., Turnbull D.M. Mitochondrial enzyme-deficient hippocampal neurons and choroidal cells in AD. Neurology. 2001;57:260–264. doi: 10.1212/wnl.57.2.260. [DOI] [PubMed] [Google Scholar]
- 6.Zeviani M., Servidei S., Gellera C., Bertini E., DiMauro S., DiDonato S. An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the D-loop region. Nature. 1989;339:309–311. doi: 10.1038/339309a0. [DOI] [PubMed] [Google Scholar]
- 7.Wanrooij S., Luoma P., van Goethem G., van Broeckhoven C., Suomalainen A., Spelbrink J.N. Twinkle and POLG defects enhance age-dependent accumulation of mutations in the control region of mtDNA. Nucleic Acids Res. 2004;32:3053–3064. doi: 10.1093/nar/gkh634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Samuels D.C., Schon E.A., Chinnery P.F. Two direct repeats cause most human mtDNA deletions. Trends Genet. 2004;20:393–398. doi: 10.1016/j.tig.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Clayton D.A. Replication of animal mitochondrial DNA. Cell. 1982;28:693–705. doi: 10.1016/0092-8674(82)90049-6. [DOI] [PubMed] [Google Scholar]
- 10.Holt I.J., Lorimer H.E., Jacobs H.T. Coupled leading- and lagging-strand synthesis of mammalian mitochondrial DNA. Cell. 2000;100:515–524. doi: 10.1016/s0092-8674(00)80688-1. [DOI] [PubMed] [Google Scholar]
- 11.Yasukawa T., Reyes A., Cluett T.J., Yang M.Y., Bowmaker M., Jacobs H.T., Holt I.J. Replication of vertebrate mitochondrial DNA entails transient ribonucleotide incorporation throughout the lagging strand. EMBO J. 2006;25:5358–5371. doi: 10.1038/sj.emboj.7601392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Srivastava S., Moraes C.T. Double-strand breaks of mouse muscle mtDNA promote large deletions similar to multiple mtDNA deletions in humans. Hum. Mol. Genet. 2005;14:893–902. doi: 10.1093/hmg/ddi082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Halliwell B. Reactive oxygen species and the central nervous system. J. Neurochem. 1992;59:1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- 14.Bua E., Johnson J., Herbst A., Delong B., McKenzie D., Salamat S., Aiken J.M. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am. J. Hum. Genet. 2006;79:469–480. doi: 10.1086/507132. [DOI] [PMC free article] [PubMed] [Google Scholar]