

Abstract
Familial primary localized cutaneous amyloidosis (FPLCA) is an autosomal-dominant disorder associated with chronic skin itching and deposition of epidermal keratin filament-associated amyloid material in the dermis. FPLCA has been mapped to 5p13.1–q11.2, and by candidate gene analysis, we identified missense mutations in the OSMR gene, encoding oncostatin M-specific receptor β (OSMRβ), in three families. OSMRβ is a component of the oncostatin M (OSM) type II receptor and the interleukin (IL)-31 receptor, and cultured FPLCA keratinocytes showed reduced activation of Jak/STAT, MAPK, and PI3K/Akt pathways after OSM or IL-31 cytokine stimulation. The pathogenic amino acid substitutions are located within the extracellular fibronectin type III-like (FNIII) domains, regions critical for receptor dimerization and function. OSM and IL-31 signaling have been implicated in keratinocyte cell proliferation, differentiation, apoptosis, and inflammation, but our OSMR data in individuals with FPLCA represent the first human germline mutations in this cytokine receptor complex and provide new insight into mechanisms of skin itching.
Introduction
One of the most common and least well understood symptoms in dermatology is itchy skin (also known as pruritus).1 To date, relatively little is known about the key itch mediators, receptors, and pathways, and consequently effective therapies for alleviating symptoms of itch are limited. One particular pruritic skin disorder often seen by dermatologists is primary localized cutaneous amyloidosis (PLCA [MIM 105250]). This condition usually presents with itching (especially on the lower legs) and visible changes of skin hyperpigmentation and thickening (lichenification) that may be exacerbated by chronic scratching and rubbing.2 Despite its name, PLCA is not associated with other systemic forms of amyloidosis: the skin in PLCA shows fibrillary degeneration of basal keratinocytes with increased apoptosis, disruption of dermal unmyelinated nerve fibers, and accumulation of melanosomes in dermal macrophages and Schwann cells.3–5 The “amyloid” in PLCA probably reflects a combination of degenerate keratin filaments, serum amyloid P component, and secondary deposition of immunoglobulins. Nevertheless, PLCA has been reported to coexist with several other clinical disorders such as connective tissue diseases (e.g., systemic lupus erythematosus, rheumatoid arthritis, systemic sclerosis, scleroderma, primary biliary cirrhosis, and dermatomyositis) as well as multiple endocrine neoplasia type 2A (MEN2A [MIM 171400]).2 PLCA in some individuals with MEN2A has been associated with specific amino acid substitutions in the RET proto-oncogene (especially involving codon 634), but no RET gene pathology has been disclosed in cases of PLCA in the absence of MEN2A.6–9 Most clinical cases of PLCA are sporadic but the disorder is more common in certain parts of the world, including South America and Southeast Asia, where up to 10% of cases may be familial (autosomal dominant). Genetic heterogeneity is suspected, however, because recent genome-wide scans for familial PLCA (FPLCA) in Taiwan have suggested genetic linkage to 1q23 or 5p13.1-q11.2 or perhaps other undisclosed loci, with the most significant LOD scores for the disease locus on chromosome 5.10,11 Identifying the gene for FPLCA, therefore, might be expected to give significant new insight into certain pathophysiological mechanisms underlying skin itching, inflammation, and keratinocyte apoptosis.
In this study, we mapped a large Brazilian family with FPLCA to 5p13.1-q11.2 and performed candidate gene analysis by sequencing genomic DNA. We found a missense mutation in the OSMR gene, which encodes the oncostatin M receptor β (OSMRβ), in all the affected individuals of the Brazilian FPLCA family. Further investigation in two other white families with FPLCA (from the United Kingdom and South Africa) also disclosed mutations in OSMR. Abnormalities in OSMRβ signaling after stimulation with the ligands OSM and IL-31 were demonstrated, thus providing functional data to support OSMR gene mutations being responsible for FPLCA as well as generating intriguing new data on mechanisms of pruritus and keratinocyte apoptosis in human skin.
Material and Methods
DNA Samples, Microsatellite Analysis, and Sequencing
After Ethical Committee approval and in compliance with the Helsinki Accord, and after obtaining informed consent from all subjects, genomic DNA was extracted from peripheral blood samples obtained from three FPLCA families (I-1; II-3 to 6; III-1, 3 to 6, 8 to 17; IV-2 to 4, 6 to 9 of Family 1; three affected individuals from Family 2, and two affected individuals from Family 3). DNA was amplified with five primer sets for microsatellite markers located between 5p13.2 and 5q11.2 (Table 1). These primers were obtained from the ABI PRISM Linkage Mapping Set Version 2.5 (Applied Biosystems). The PCR products were analyzed on an ABI 310 DNA sequencer with Genescan 2.1 and Genotyper 2.0 software (Applied Biosystems). Two-point LOD scores were computed by the MLINK algorithm of LINKAGE version 5.1 under the assumptions of a mutant allele frequency of 0.00001 and 100% penetrance. Candidate gene analysis was then performed (see Results for rationale of gene selection). For sequencing, DNA samples were amplified with primers sited in introns flanking individual exons of the OSMR gene (details available on request) and IL31RA, IL6ST, and LIFR genes (primer details available on request). PCR products were sequenced with ABI BigDye Terminator reagents (Applied Biosystems) in an ABI 310 sequencer.
Table 1.
Results of Microsatellite Analysis on FPLCA Locus 5p13.1–q11.2
| Recombination Fractions |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Locus | Z Max | θ | 0 | 0.01 | 0.05 | 0.1 | 0.2 | 0.3 | 0.4 |
| D5S426 | 2.650448 | 0 | 2.65045 | 2.60028 | 2.396885 | 2.13661 | 1.597563 | 1.041051 | 0.488485 |
| D5S2021 | 3.475509 | 0.05 | −∞ | 3.087335 | 3.475509 | 3.371219 | 2.800568 | 2.015219 | 1.079287 |
| D5S418 | 4.801343 | 0 | 4.80134 | 4.720222 | 4.387974 | 3.954395 | 3.021799 | 1.999922 | 0.927309 |
| D5S1969 | 3.474038 | 0 | 3.47404 | 3.414591 | 3.171226 | 2.854256 | 2.178197 | 1.455041 | 0.714624 |
| D5S407 | 1.352818 | 0.09 | −∞ | 0.776918 | 1.288535 | 1.350272 | 1.156844 | 0.821459 | 0.426546 |
RT-PCR
mRNA was extracted from skin biopsy specimens from individual III-16 in Family 1 and one of the affected individuals from Families 2 and 3, respectively, as well as normal control skin samples, with RNeasy fibrous tissue kits (QIAGEN) with subsequent cDNA synthesis with Superscript II reverse transcriptase (Invitrogen). Primers targeting near the 3′-end of the cDNA were used to amplify IL-6-type cytokine receptors and the corresponding ligands (primer details available on request).
Keratinocyte Isolation and Culture
Primary keratinocyte cultures were isolated according to a standard procedure.12 In brief, after mechanical dissociation, skin biopsy fragments were immersed for 1 hr at 37°C in trypsin-EDTA solution. The solution was then filtered through a 100 μm pore cell strainer (VWR), and then medium supplemented with 10% fetal bovine serum (FBS) was added to neutralize the trypsin. Cells were isolated with a centrifuge (5 min, 1000 rpm), and the pellet was resuspended in normal keratinocyte medium. Finally, the cells were seeded in T25 flasks containing feeders. The keratinocytes were maintained in DMEM:Ham's F12 medium supplemented with 10% FBS, 5 μg/ml transferrin, 0.4 μg/ml hydrocortisone, 10−10 M cholera toxin, 10 ng/ml epidermal growth factor (EGF)1, 5 μg/ml insulin, and 2 × 10−11 M liothyronine. Fresh mitomycin C-treated NIH 3T3 feeder cells were added to primary keratinocytes twice a week. Early passage keratinocytes were immortalized with HPV16 (E6∧E7), as described previously.13
Cytokine Stimulation
Keratinocytes were seeded into 6-well plates and grown to confluency. Cells were maintained in normal keratinocyte media for 3–5 days after confluency. Prior to cytokine stimulation, cells were washed twice in phosphate-buffered saline (PBS) and incubated in keratinocyte-defined serum-free media (Invitrogen) for either 2 or 24 hr. Cultures were then stimulated with the relevant cytokine for 15 min prior to the preparation of lysates for immunoblotting. Cytokines used in this study were recombinant human OSM, IL-6, and IL-31 (R&D Systems) (see Results). The concentrations of cytokines used were 10, 50, and 100 ng/ml for OSM and 100 ng/ml for IL-6 and IL-31.
Immunoblotting
The cells were washed once with ice-cold PBS and lysed with chilled RIPA buffer containing anti-proteases. Cell lysates were resolved with 4%–12% NuPAGE Novex Bis-Tris Gels (Invitrogen). The fractionated proteins were transferred to Hybond-ECL nitrocellulose transfer membrane (Amersham Biosciences). The membrane was blocked with 5% nonfat milk-TTBS for 2 hr at room temperature and incubated overnight at 4°C with the relevant primary antibody. Antibody-antigen complexes were visualized by enhanced chemiluminescence (Amersham Biosciences), according to the manufacturer's instructions. The optical density of bands on the blot was calculated with Image J software. Primary antibodies used were STAT1(42H3)(9175), STAT3(124H6)(9139), STAT5(3H7)(9358), p44/42 MAP Kinase (Erk1/2)(9102), Akt (9272), phospho-STAT1-Tyr701(9171), phospho-STAT3-Tyr705(9131), phospho-STAT5-Tyr694(9351), phospho-p44/42 MAPK-Thr202/Tyr204 (phosphor-Erk1/2; 9106), phospho-Akt-Ser473(9271) (Cell Signaling Technology, Inc.). Secondary antibodies were polyclonal goat anti-mouse immunoglobulins/HRP and polyclonal swine anti-rabbit immunoglobulins/HRP (Dako Cytomation).
Results
Clinical and Histopathological Features of FPLCA
Sixteen subjects over four generations of the Brazilian pedigree were known to have FPLCA, with inheritance by autosomal-dominant transmission (Figure 1A). In affected individuals, symptoms typically started during childhood with severe pruritus on the lower legs, leading to focal skin lichenification (Figures 1B and 1C). Histopathology of lesional skin showed pigmentary incontinence and amorphous eosinophilic material in the papillary dermis (Figure 1D), which stained positively for thioflavin-T, consistent with amyloid deposition (Figure 1E). Clinico-pathological details of Family 2 (white British) and Family 3 (white South African) with autosomal-dominant FPLCA have been reported previously.14,15
Figure 1.
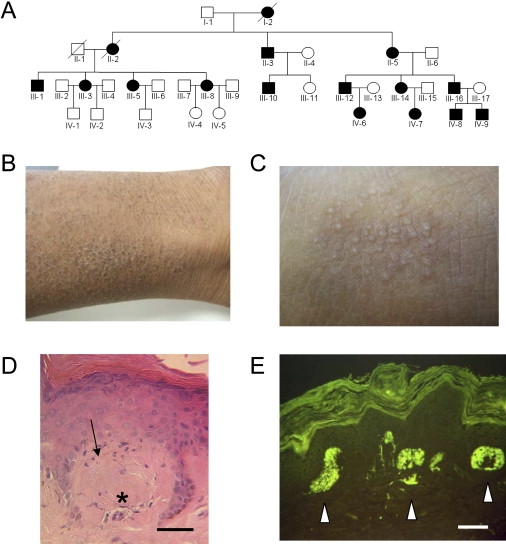
Clinical, Histological, and Genetic Aspects of FPLCA
(A) The pedigree of Family 1 showing autosomal-dominant inheritance; filled symbols represent affected individuals.
(B) Clinical features of FPLCA. Skin on the lower leg (individual III-16) appears dry, scaly, and thickened with some hyperpigmentation.
(C) At higher magnification, there is accentuation of the normal skin creases and focal skin papules (lichenification).
(D) Histopathological features of FPLCA. Skin biopsy of lesional skin shows pigmentary incontinence (arrow) and amorphous eosinophilic material (asterisk) in the superficial papillary dermis (hematoxylin and eosin) (scale bar represents 25 μm).
(E) These collections of material show bright fluorescent labeling with thioflavin-T (white arrows) (scale bar represents 25 μm).
Identification of OSMR as the FPLCA Gene
We performed linkage analysis in Family 1 with five microsatellite markers covering 5p13.1–q11.2 and confirmed this as the FPLCA locus. The disease-associated allele was flanked by D5S2021 and D5S407 with a maximum LOD score of 4.8 for the microsatellite marker D5S418 (Table 1). The boundary of this interval on the short arm of chromosome 5 was defined by recombination between D5S2021 and D5S418 in individual III-1, whereas the boundary on the long arm of chromosome 5 was defined by recombination between D5S1969 and D5S407 in individual IV-2 (on the assumption that the penetrance of the condition is complete). This ∼18 cM interval, which spans the centromere of chromosome 5, contains more than 150 genes and in silico predicted genes. This region spans the entire region of linkage on chromosome 5 previously reported,11 but widens the linkage interval by ∼2.8 cM beyond the boundary on the long arm. Within the interval, we searched for genes that might have pathophysiological relevance to skin itching. We focused on the genes coding interleukin (IL)-6-type receptors, given that overexpression of IL-31 in mice has recently been shown to cause itchy dermatitis and that oncostatin M (OSM) is a potent keratinocyte activator involved in skin inflammation.16,17 Within our linked region, there are four IL-6 type receptors: IL6ST, LIFR, IL31RA, and OSMR (which encode gp130, leukemia inhibitory factor receptor, IL-31 receptor A, and OSM-specific receptor β, respectively). Two of these genes, OSMR and LIFR, are also present within the Taiwanese linkage interval.11 Sequencing of genomic DNA in Family 1 disclosed a heterozygous point mutation in OSMR, c.2072T→C (NM_003999) (Figure 2A), which was present in all the affected individuals but not in any of the unaffected individuals. This mutation converts isoleucine to threonine at amino acid 691 (p.I691T). No mutations were found in any of the other putative candidate genes. We then sequenced the OSMR gene in Families 2 and 3 and found a heterozygous missense mutation c.1853G→C (p.G618A) common to the affected individuals in both pedigrees (Figure 2A). Subsequent microsatellite analyses around the OSMR gene indicated that Families 2 and 3 are likely to share a common British ancestor (data not shown). Neither of the missense mutations p.I691T or p.G618A was found in screening 210 ethnically matched control chromosomes. The mutations in the FPLCA families occur within the extracellular fibronectin type III-like (FNIII) domains of OSMRβ (Figure 2B), sites that have previously been shown to be necessary for correct receptor function and signal transduction.18 In addition, these particular mutated amino acids are well conserved throughout various evolutionary lineages (Figure 2C).
Figure 2.
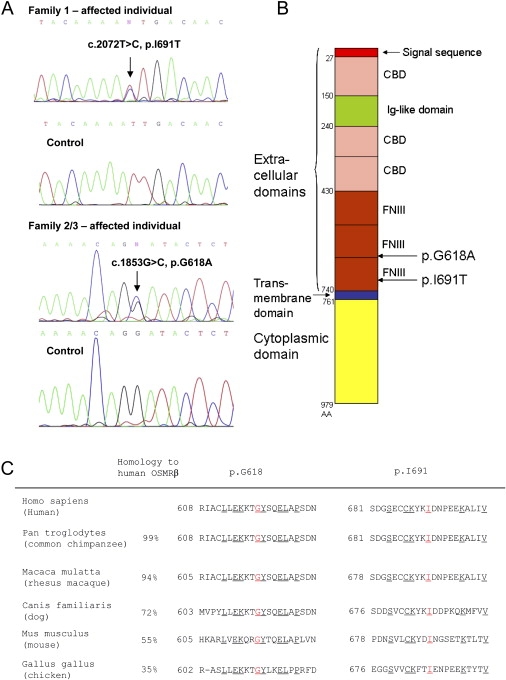
The Molecular Basis of FPLCA Involves Heterozygous Missense Mutations in the OSMR Gene
(A) DNA sequencing of OSMR. Sequencing of genomic DNA in individuals with FPLCA reveals heterozygous single-nucleotide substitutions in OSMR c.2072T→C in Family 1 and c.1853G→C in Families 2/3 (NM_003999). These convert an isoleucine to threonine and a glycine to alanine, designated p.I691T and p.G618A, respectively.
(B) Structural model of OSMRβ depicting the functional domains and the sites of the missense mutations. FNIII, fibronectin type III-like domain; CBD, cytokine binding domain; Ig-like domain, immunoglobulin-like domain. Amino acid (AA) numbers of the 979 amino acid OSMRβ protein are shown on the left of the figure. The missense mutations p.I691T and p.G618A are located within the extracellular FNIII domains.
(C) Protein homology and amino acid identity indicate that amino acids p.I691 and p.G618 are well conserved across several species. The numbers at the start of the depicted sequences indicate the amino acid numbers. Underlined amino acids are conserved in all species shown here; p.I691 and p.G618 are highlighted in red.
Expression and Function of OSMRβ in FPLCA
OSMRβ is a component of both the OSM type II receptor19,20 and the IL-31 receptor (Figure 3).7 One of its ligands, OSM, has known roles in cell proliferation, apoptosis, differentiation, and inflammation,17,21 and in keratinocytes, OSM has been shown to modulate expression of several genes involved in innate immunity, angiogenesis, adhesion, cell motility, tissue remodeling, cell-cycle regulation, and transcription.21 In the OSM type II receptor, OSMRβ couples with gp130, which is the most common receptor subunit of the IL-6-type cytokine receptors. OSM also binds to the OSM type I receptor (LIFR and gp130), although LIFR is barely expressed in keratinocytes.17 The other ligand for OSMRβ is IL-31, which has been shown to induce severe pruritus in an animal model.16 In the IL-31 receptor, OSMRβ couples with the IL-31 receptor A subunit. The IL-6 family of cytokine receptors typically invoke signaling via the Jak/STAT, MAPK, and PI3K/Akt pathways.19
Figure 3.
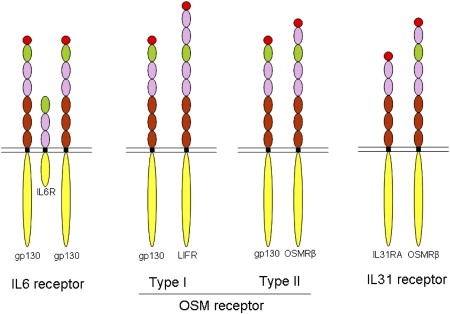
Schematic Model of IL-6 Family Cytokine Receptors
The IL-6 receptor comprises two gp130 and one IL-6R molecules. There are two types of OSM receptors: type I is composed of gp130 and LIFR, whereas type II comprises gp130 and OSMRβ. The IL-31 receptor is composed of OSMRβ and IL31RA. Each domain of the receptors is colored similarly to the schematic shown in Figure 2B.
To assess the functional consequences of the OSMR mutations, we first assessed mRNA expression levels of the relevant IL-6 type cytokines and their receptors in the affected subjects' and control skin samples and cultured keratinocytes by RT-PCR. All the IL-6-type cytokines and their receptors examined were detected and expressed at similar levels in all samples (not illustrated). Next, FPLCA cultured keratinocytes (from Family 2) were stimulated with OSM or IL-31, and the levels of phosphorylated STATs, Erk1/2, and Akt were observed by western blotting. Normal control keratinocytes responded to the OSM strongly and, to a lesser extent, to IL-31, as recently documented.22 In FPLCA, however, the level of pSTATs, pErk, and pAkt were reduced by ∼65%–95% (assessed by optical densitometry) after OSM stimulation, and no phosphorylation was observed after IL-31 stimulation (Figure 4, top and middle). Response to OSM was dose dependent in both FPLCA and control keratinocytes (Figure 4, top). Stimulation with IL-6, which does not include OSMRβ in its receptor complex, did not result in altered phosphorylation in the FPLCA keratinocytes (Figure 4, bottom).
Figure 4.
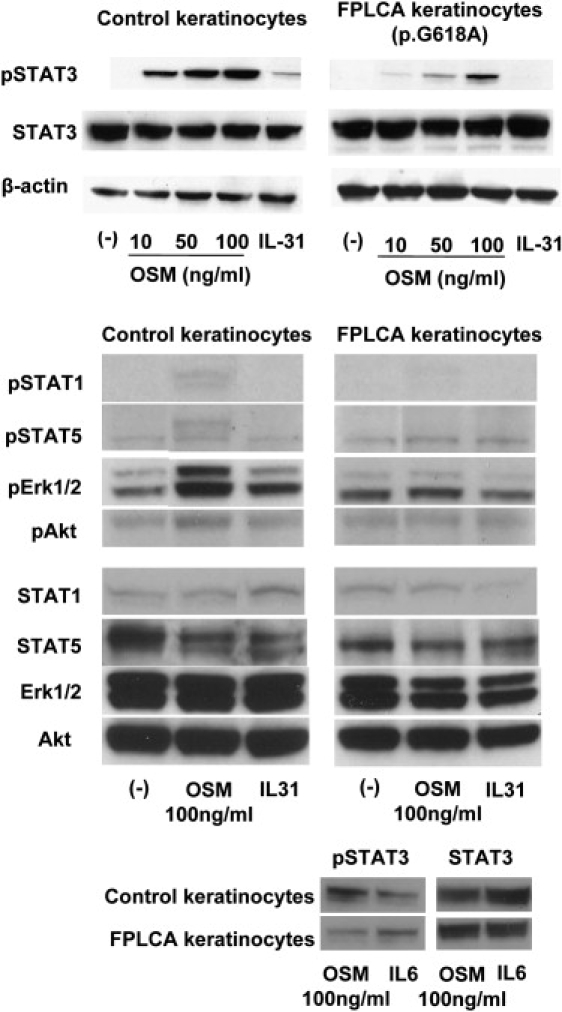
Western Blotting of Phosphorylated (p)-STATs, pErk1/2, and pAkt of Cultured Keratinocytes Stimulated with OSM or IL-31
Phosphorylation of STATs, Erk1/2, and Akt was observed after stimulation by OSM and IL-31 in normal human keratinocytes, but in FPLCA keratinocytes (here illustrated for Family 2), there was reduced phosphorylation of STATs, Erk1/2, and Akt by OSM and no activation by IL-31 (top and middle). The reduction of pSTAT3 was more clearly demonstrated at lower OSM concentrations (top). Stimulation by IL-6 did not result in altered phosphorylation in FPLCA keratinocytes (bottom).
Discussion
This study has identified that mutations in the OSMRβ receptor underscore the molecular basis of FPLCA and provide intriguing new insight into mechanisms of itch and apoptosis in human skin. The OSMRβ missense mutation p.G618A is located within the second FNIII domain, and p.I691T occurs within the first FNIII domain adjacent to the transmembranous domain (Figure 2B). Previous in vitro studies of FNIII deletions or single amino acid substitutions in gp130 have disclosed key roles for these domains in receptor dimerization for both gp130 homodimers or gp130-LIFR heterodimers.18,23 Given that OSMRβ has a similar domain organization to gp130, we predict that the amino acid substitutions p.G618A and p.I691T in the FNIII regions of this receptor may interfere with the normal receptor coupling between OSMRβ and gp130 as well as OSMRβ with IL31RA. This then leads to the decrease in signal activation by OSM and IL-31 in FPLCA keratinocytes (Figure 4 and illustrated schematically in Figure 5).
Figure 5.
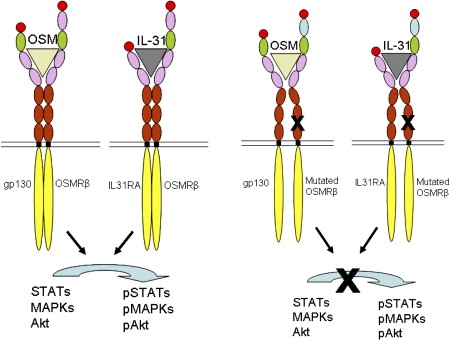
Structural Representation of OSMR Type II and IL-31 Receptor Signaling in Normal and FPLCA Keratinocytes
Each domain of the receptors is colored similarly to the schematic shown in Figure 2B. After binding of OSM or IL-31 to these receptors, the FNIII domains of both receptor subunits interact and signal transduction occurs. The missense mutations seen in our cases occur within the FNIII domains (black crosses), regions critical for receptor dimerization and subsequent signaling. Failure to form conformationally functional receptor complexes in FPLCA leads to reduced receptor signaling after stimulation with either OSM or IL-31.
OSMRβ is widely expressed in many cells and tissues20 and a key question therefore is why do these mutations result in what is predominantly a skin disease? Part of the explanation for why heterozygous human missense mutations result in FPLCA, however, could relate to lack of the OSM-type I receptor in keratinocytes which, when present in other tissues, may compensate for any dysfunctional OSM-type II receptor. Another possibility might relate to recent findings of coexpression of OSMRβ and IL-31RA in skin cells (specific cell types yet to be determined) as well as in nociceptive neurons in dorsal root ganglia that project into the dermis in skin.24,25 Thus, it is plausible that the clinicopathological features of FPLCA may only manifest at sites where both types of receptor that contain OSMRβ are expressed.
The precise cellular mechanisms through which the OSMRβ mutations result in reduced OSM and IL-31 signaling, such as impaired signaling via a mutant receptor complex or premature degradation of the mutant OSMRβ leading to reduced amounts of OSMRβ available to form dimers with gp130 or IL-31R upon ligand activation, are not yet known. Nevertheless, our new data are able to provide an explanation for the histological finding of increased keratinocyte apoptosis in FPLCA. We found decreased activation of Jak/STAT, Erk1/2, and PI3K/Akt signaling, pathways that have been reported to have antiapoptotic effects in several tumor cell lines.26,27 Keratinocytes with mutated OSMRβ, therefore, may be more susceptible to apoptosis, resulting in cell death and accumulation of degenerate keratinous material in the superficial dermis. It is not currently known whether similar apoptosis occurs in nociceptive neurons in FPLCA, although degenerative changes in some nerves in the dermis have been noted on transmission electron microscopy.5
How mutations in OSMRβ lead to increased skin itching, however, requires further study. Our current data show decreased signaling in FPLCA keratinocytes after stimulation with OSM or IL-31, in contrast to other reports indicating that itchy dermatitis in mice results from overexpression of IL-31.16 The role of the other OSMRβ-associated ligand, OSM, in nociceptive neurons has not been fully elucidated, although it has been suggested that OSM may sustain pain during inflammation.28 Precisely how OSMRβ gene mutations might affect the function of nociceptive neurons, however, is not clear, but if a link can be shown, this mechanism might have relevance to explaining the concept of neurological itching in skin (“neurodermatitis”), which is often a clinical consideration in many patients with chronic skin itching. With regard to FPLCA, therefore, this disorder could represent the first example of a true neurodermatitis rather than a primary disorder of keratinocytes.
Demonstration of pathogenic mutations in OSMRβ in FPLCA now provides an opportunity to explore whether abnormalities in signaling via this cytokine receptor are also present in sporadic cases of PLCA as well as perhaps in acquired itchy dermatoses with lichenoid patterns of skin inflammation, such as lichen planus or graft-versus-host disease. Moreover, given the association of PLCA with various connective tissue and autoimmune pathologies,2 our findings may also help focus assessment of specific IL-6 signaling responses in these common disorders. Such investigations may lead to new therapeutic targets for these conditions as well as for the most common of all dermatological symptoms, itch.
Web Resources
The URLs for data presented herein are as follows:
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for OSMR)
ImageJ, http://rsb.info.nih.gov/ij/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for PLCA)
Acknowledgments
We are grateful to Irwin McLean (University of Dundee) and David Goudie (Tayside University Hospitals NHS Trust) for their assistance with the genetic linkage analysis; Lu Liu (Guy's and St Thomas' NHS Foundation Trust), Tracy Wong (King's College London), and Vesarat Wessagowit (Institute of Dermatology, Bangkok) for technical assistance; and to Kurt Q. Lu (Case Western University) for helpful discussions. This work was kindly supported by a project grant from Action Medical Research (UK).
References
- 1.Paus R., Schmelz M., Biro T., Steinhoff M. Frontiers in pruritus research: scratching the brain for more effective itch therapy. J. Clin. Invest. 2006;116:1174–1186. doi: 10.1172/JCI28553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Breathnach S.M. Amyloid and the amyloidosis of the skin. In: Burns T., Breathnach S., Cox C., Griffiths C., editors. Rook's Textbook of Dermatology. Volume 3. Blackwell Scientific; Oxford: 2004. pp. 57.36–57.51. [Google Scholar]
- 3.Kumakiri M., Hashimoto K. Histogenesis of primary localized cutaneous amyloidosis: sequential change of epidermal keratinocytes to amyloid via filamentous degeneration. J. Invest. Dermatol. 1979;73:150–162. doi: 10.1111/1523-1747.ep12581609. [DOI] [PubMed] [Google Scholar]
- 4.Kobayashi H., Hashimoto K. Amyloidogenesis in organ-limited cutaneous amyloidosis: an antigenic identity between epidermal keratin and skin amyloid. J. Invest. Dermatol. 1983;80:66–72. doi: 10.1111/1523-1747.ep12531130. [DOI] [PubMed] [Google Scholar]
- 5.Schepis C., Siragusa M., Gagliardi M.E., Torre V., Cicciarello R., Albiero F., Cavallari V. Primary macular amyloidosis: an ultrastructural approach to diagnosis. Ultrastruct. Pathol. 1999;23:279–284. doi: 10.1080/019131299281428. [DOI] [PubMed] [Google Scholar]
- 6.Verga U., Fugazzola L., Cambiaghi S., Pritelli C., Alessi E., Cortelazzi D., Gangi E., Beck-Peccoz P. Frequent association between MEN 2A and cutaneous lichen amyloidosis. Clin. Endocrinol. (Oxf.) 2003;59:156–161. doi: 10.1046/j.1365-2265.2003.01782.x. [DOI] [PubMed] [Google Scholar]
- 7.Hofstra R.M., Sijmons R.H., Stelwagen T., Stulp R.P., Kousseff B.G., Lips C.J., Steijlen P.M., Van Woorst Vader P.C., Buys C.H. RET mutation screening in familial cutaneous lichen amyloidosis and in skin amyloidosis associated with multiple endocrine neoplasia. J. Invest. Dermatol. 1996;107:215–218. doi: 10.1111/1523-1747.ep12329651. [DOI] [PubMed] [Google Scholar]
- 8.Lee D.D., Huang J.Y., Wong C.K., Gagel R.F., Tsai S.F. Genetic heterogeneity of familial primary cutaneous amyloidosis: lack of evidence for linkage with the chromosome 10 pericentromeric region in Chinese families. J. Invest. Dermatol. 1996;107:30–33. doi: 10.1111/1523-1747.ep12297840. [DOI] [PubMed] [Google Scholar]
- 9.Seri M., Celli I., Betsos N., Claudiani F., Camera G., Romeo G. A Cys634Gly substitution of the RET proto-oncogene in a family with recurrence of multiple endocrine neoplasia type 2A and cutaneous lichen amyloidosis. Clin. Genet. 1997;51:86–90. doi: 10.1111/j.1399-0004.1997.tb02425.x. [DOI] [PubMed] [Google Scholar]
- 10.Lin M.W., Lee D.D., Lin C.H., Huang C.Y., Wong C.K., Chang Y.T., Liu H.N., Hsiao K.T., Tsai S.F. Suggestive linkage of familial primary cutaneous amyloidosis to a locus on chromosome 1q23. Br. J. Dermatol. 2005;152:29–36. doi: 10.1111/j.1365-2133.2004.06254.x. [DOI] [PubMed] [Google Scholar]
- 11.Lee D.D., Lin M.W., Chen I.C., Huang C.Y., Liu M.T., Wang C.R., Chang Y.T., Liu H.N., Liu T.T., Wong C.K., Tsai S.F. Genome-wide scan identifies a susceptibility locus for familial primary cutaneous amyloidosis on chromosome 5p13.1-q11.2. Br. J. Dermatol. 2006;155:1201–1208. doi: 10.1111/j.1365-2133.2006.07524.x. [DOI] [PubMed] [Google Scholar]
- 12.Rheinwald J.G. Human epidermal keratinocyte cell culture and xenograft systems: applications in the detection of potential chemical carcinogens and the study of epidermal transformation. Prog. Clin. Biol. Res. 1989;298:113–125. [PubMed] [Google Scholar]
- 13.Halbert C.L., Demers G.W., Galloway D.A. The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J. Virol. 1991;65:473–478. doi: 10.1128/jvi.65.1.473-478.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Newton J.A., Jagjivan A., Bhogal B., McKee P.H., McGibbon D.H. Familial primary cutaneous amyloidosis. Br. J. Dermatol. 1985;112:201–208. doi: 10.1111/j.1365-2133.1985.tb00084.x. [DOI] [PubMed] [Google Scholar]
- 15.Hartshorne S.T. Familial primary cutaneous amyloidosis in a South African family. Clin. Exp. Dermatol. 1999;24:438–442. doi: 10.1046/j.1365-2230.1999.00526.x. [DOI] [PubMed] [Google Scholar]
- 16.Dillon S.R., Sprecher C., Hammond A., Bilsborough J., Rosenfeld-Franklin M., Presnell S.R., Haugen H.S., Maurer M., Harder B., Johnston J. Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat. Immunol. 2004;5:752–760. doi: 10.1038/ni1084. [DOI] [PubMed] [Google Scholar]
- 17.Boniface K., Diveu C., Morel F., Pedretti N., Froger J., Ravon E., Garcia M., Venereau E., Preisser L., Guignouard E. Oncostatin M secreted by skin infiltrating T lymphocytes is a potent keratinocyte activator involved in skin inflammation. J. Immunol. 2007;178:4615–4622. doi: 10.4049/jimmunol.178.7.4615. [DOI] [PubMed] [Google Scholar]
- 18.Kurth I., Horsten U., Pflanz S., Timmermann A., Kuster A., Dahmen H., Tacken I., Heinrich P.C., Muller-Newen G. Importance of the membrane-proximal extracellular domains for activation of the signal transducer glycoprotein 130. J. Immunol. 2000;164:273–282. doi: 10.4049/jimmunol.164.1.273. [DOI] [PubMed] [Google Scholar]
- 19.Heinrich P.C., Behrmann I., Haan S., Hermanns H.M., Muller-Newen G., Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mosley B., De Imus C., Friend D., Boiani N., Thoma B., Park L.S., Cosman D. Dual oncostatin M (OSM) receptors. Cloning and characterization of an alternative signaling subunit conferring OSM-specific receptor activation. J. Biol. Chem. 1996;271:32635–32643. doi: 10.1074/jbc.271.51.32635. [DOI] [PubMed] [Google Scholar]
- 21.Finelt N., Gazel A., Gorelick S., Blumenberg M. Transcriptional responses of human epidermal keratinocytes to Oncostatin-M. Cytokine. 2005;31:305–313. doi: 10.1016/j.cyto.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 22.Chattopadhyay S., Tracy E., Liang P., Robledo O., Rose-John S., Baumann H. Interleukin-31 and oncostatin-M mediate distinct signaling reactions and response patterns in lung epithelial cells. J. Biol. Chem. 2007;282:3014–3026. doi: 10.1074/jbc.M609655200. [DOI] [PubMed] [Google Scholar]
- 23.Timmermann A., Küster A., Kurth I., Heinrich P.C., Müller-Newen G. A functional role of the membrane-proximal extracellular domains of the signal transducer gp130 in heterodimerization with the leukemia inhibitory factor receptor. Eur. J. Biochem. 2002;269:2716–2726. doi: 10.1046/j.1432-1033.2002.02941.x. [DOI] [PubMed] [Google Scholar]
- 24.Bando T., Morikawa Y., Komori T., Senba E. Complete overlap of interleukin-31 receptor A and oncostatin M receptor beta in the adult dorsal root ganglia with distinct developmental expresion patterns. Neuroscience. 2006;142:1263–1271. doi: 10.1016/j.neuroscience.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 25.Sonkoly E., Muller A., Lauerma A.I., Pivarcsi A., Soto H., Kemeny L., Alenius H., Dieu-Nosjean M.C., Meller S., Rieker J. IL-31: a new link between T cells and pruritus in atopic skin inflammation. J. Allergy Clin. Immunol. 2006;117:411–417. doi: 10.1016/j.jaci.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 26.Chen R.H., Chang M.C., Su Y.H., Tsai Y.T., Kuo M.L. Interleukin-6 inhibits transforming growth factor-beta-induced apoptosis through the phosphatidylinositol 3-kinase/Akt and signal transducers and activators of transcription 3 pathways. J. Biol. Chem. 1999;274:23013–23019. doi: 10.1074/jbc.274.33.23013. [DOI] [PubMed] [Google Scholar]
- 27.Mirmohammadsadegh A., Mota R., Gustrau A., Hassan M., Nambiar S., Marini A., Bojar H., Tannapfel A., Hengge U.R. ERK1/2 is highly phosphorylated in melanoma metastases and protects melanoma cells from cisplatin-mediated apoptosis. J. Invest. Dermatol. 2007;127:2207–2215. doi: 10.1038/sj.jid.5700870. [DOI] [PubMed] [Google Scholar]
- 28.Tamura S., Morikawa Y., Miyajima A., Senba E. Expression of oncostatin M receptor β in a specific subset of nociceptive sensory neurons. Eur. J. Neurosci. 2003;17:2287–2298. doi: 10.1046/j.1460-9568.2003.02681.x. [DOI] [PubMed] [Google Scholar]