

Abstract
Vascular endothelial growth factor (VEGF) is a potent mitogen with a unique specificity for endothelial cells and a key mediator of aberrant endothelial cell proliferation and vascular permeability in a variety of human pathological situations, such as tumor angiogenesis, diabetic retinopathy, rheumatoid arthritis, or psoriasis. VEGF is a symmetric homodimeric molecule with two receptor binding interfaces lying on each pole of the molecule. Herein we report on the construction and recombinant expression of an asymmetric heterodimeric VEGF variant with an intact receptor binding interface at one pole and a mutant receptor binding interface at the second pole of the dimer. This VEGF variant binds to VEGF receptors but fails to induce receptor activation. In competition experiments, the heterodimeric VEGF variant antagonizes VEGF-stimulated receptor autophosphorylation and proliferation of endothelial cells. A 15-fold excess of the heterodimer was sufficient to inhibit VEGF-stimulated endothelial cell proliferation by 50%, and a 100-fold excess resulted in an almost complete inhibition. By using a rational approach that is based on the structure of VEGF, we have shown the feasibility to construct a VEGF variant that acts as an VEGF antagonist.
Vascular endothelial growth factor (VEGF), also known as vascular permeability factor, is a potent mitogen in vasculogenesis and angiogenesis with a unique specificity for endothelial cells (for recent reviews, see refs. 1–4). The pivotal role of VEGF in vasculogenesis was exemplified in recent studies on targeted inactivation of the VEGF gene in mice that resulted, even when heterozygous, in fatal deficiencies in vascularization (5, 6). In a variety of human pathological situations that are associated with aberrant endothelial proliferation and aberrant neovascularization, such as tumor angiogenesis (7), diabetic retinopathy (8), rheumatoid arthritis (9), or psoriasis (10), elevated expression of VEGF has been observed. Two homologous receptor tyrosine kinases, fms-like tyrosine kinase 1 (Flt-1) and kinase domain receptor (KDR), bind VEGF with high affinity (11, 12). Interference with the VEGF/VEGF-receptor system by using neutralizing antibodies, dominant negative receptor, or recombinant soluble receptor domains has been shown to inhibit tumor growth (13, 14) and retinal neovascularization (15).
VEGF is a homodimeric glycosylated protein that exists in five isoforms of 121, 145, 165, 189, and 206 amino acids. VEGF, placental growth factor (16), VEGF-B (17), and VEGF-C (18) form a family of related growth factors with structural homology to platelet-derived growth factor (PDGF). In particular, the cysteines building up the structural fold of these dimeric proteins (three intramolecular disulfide bridges and two intermolecular disulfide bridges cross-linking the polypeptide chains, the cystine knot motif) are conserved for these growth factors. Modeling of the VEGF sequence onto the crystal structure of PDGF-B (19) predicts three surface loops or turns that cluster at each end of the dimeric molecule with loop/turn II of one VEGF chain in close proximity to loop/turn III of the other VEGF chain. This model and the overall structural similarity of VEGF and PDGF was confirmed by the very recently solved crystal structure of VEGF (20). Receptor binding has been associated with loop/turn II and loop/turn III (21). Because of the symmetry, the VEGF dimer contains two receptor binding interfaces lying on each pole of the molecule. Each of the two binding interfaces should be able to contact one receptor monomer thereby inducing receptor dimerization and activation. On the basis of this model, an asymmetric VEGF variant that contains only one receptor binding interface at one pole of the dimer should act as an VEGF antagonist.
Herein we show the construction of an antagonistic variant of a dimeric growth factor. A heterodimeric VEGF molecule with an intact receptor binding interface at one pole and a mutated receptor binding interface at the second pole of the dimer was constructed by combination of a loop/turn II mutant VEGF chain and of a loop/turn III mutant VEGF chain. The heterodimeric molecule binds to VEGF receptors but fails to induce receptor activation. Furthermore, it antagonizes VEGF-stimulated receptor autophosphorylation and proliferation of endothelial cells.
EXPERIMENTAL PROCEDURES
Construction and Bacterial Expression of VEGF Mutant Proteins.
VEGF variants were generated by PCR amplification of the human VEGF121 cDNA or VEGF165 cDNA, respectively, cloned in pBluescript vector (22), and using the following primers. For VEGF121-L2 and VEGF165-L2, respectively, the 5′ primer L2B (5′-GGCTGCTGCAATACCTCGTCCGTGGAGTGTGTG-3′) and the 3′ primer 252c (5′-CCCGCATCGCATCAGGGGCAC-3′) were used. VEGF121-L3 was generated by a first round of mutagenesis using the 5′ primer Q87 (5′-CAAACCTCACCGAAAGAAGCACATAGGAG-3′) and the 3′ primer 326c (5′-ATCCGCATAATCTGCATGGTG-3′) followed by a second round of mutagenesis using the 5′ primer R82 (5′-CATGCAGATTATGGAGATCGAACCTCACCGAAAG-3′) and the 3′ primer 308c (5′-GTGATGTTGGACTCCTCAGTG-3′) with the recloned product of the first round of mutagenesis as template. VEGF121-C61S was generated by using the 5′ primer C61S (5′-GGCTGCTCCAATGACGAGGGC-3′) and the 3′ primer 252c. The resulting PCR fragments containing mutant VEGF sequences and vector sequence were phosphorylated by T4 polynucelotide kinase (Pharmacia), gel-purified, religated, and transformed into Escherichia coli XL1-blue. The mutant VEGF cDNAs were cloned into the His-pET vector (23) via NcoI and BamHI sites and transformed into E. coli BL21DE3 (24). All constructs were verified by DNA sequencing. Solubilization of VEGF proteins from inclusion bodies, refolding, and purification was performed essentially as described for wild-type VEGF121 (23).
For generation of the heterodimeric VEGF165-L2/VEGF121-L3 variant urea-solubilized VEGF165-L2 and VEGF121-L3 were mixed in an equimolar proportion before refolding and purification procedures. The protein solution was dialyzed against 50 mM Mes, pH 5.0/100 mM NaCl and subsequently loaded onto a 5-ml TSK heparin column (TosoHaas, Stuttgart, FRG). The proteins were eluted in 500-μl fractions with a 20-ml gradient of 1,050 mM NaCl to 1,120 mM NaCl in 50 mM Mes (pH 5.0) buffer by using an FPLC liquid chromatography system (Pharmacia).
Immunoprecipitation and Immunoblotting.
Human umbilical vein artery cells were prepared from fresh umbilical cords by mild digestion of arteries with dispase II (0.6 unit/ml; Boehringer Mannheim) in PBS for 40 min at 37°C. Cell suspension was expanded for two passages in endothelial growth medium (Promocell, Heidelberg, FRG) containing 5% fetal calf serum (FCS). For detection of KDR phosphorylation, 1.5 × 105 human umbilical artery endothelial cells per well were expanded in six-well plates for 48 h in endothelial growth medium/5% FCS, starved for 24 h in endothelial basic medium (Promocell) containing 1.3% FCS, and either left untreated or treated with VEGF variants, as indicated, for 1 h on ice. The cells were washed twice with ice-cold PBS containing 100 μM sodium orthovanadate, lysed within 300 μl of lysis buffer [20 mM Tris⋅HCl, pH 7.8/137 mM NaCl/50 mM NaF/10 mM sodium pyrophosphate/2 mM EDTA/2 mM sodium orthovanadate/2 mM Pefablock/10% glycerol/1% Triton X-100/aprotinin (13 μg/ml)/leupeptin (13 μg/ml)], and frozen at −70°C. The lysate was thawed, transferred to Eppendorf tubes, vortex-mixed, incubated on ice for 10 min, and centrifuged for 10 min at 14,000 rpm and 4°C in a tabletop centrifuge (Heraeus HFA 22.2 rotor). KDR was precipitated by incubation of the supernatant with 3 μg of 3G2 monoclonal antibody directed against the extracellular domain of KDR (P.R. and D.M., unpublished result) and 35 μg of anti-mouse IgG-agarose (Sigma) for 2 h at 4°C. Precipitates were collected by centrifugation, electrophoresed on SDS/7% polyacrylamide gels, electrotransferred to poly(vinylidene difluoride) membranes, and probed with a polyclonal anti-phosphotyrosine antibody (Transduction Laboratories/Dianova, Hamburg, FRG) or with monoclonal anti-KDR antibody 19H7 (P.R. and D.M., unpublished result). Detection was performed by using an enhanced chemiluminescence detection kit (Amersham).
Binding Assays.
Recombinant extracellular domain of human VEGF receptor Flt-1 and KDR, respectively, which were expressed as fusion proteins with the Fc region of human immune IgG1 at its C terminus (G.M.-B., B.B., F. Totzke, D.M. and G.S., unpublished result), were coated onto Maxisorb plates (1 μg per well) and were incubated with biotinylated VEGF165 (10 ng/ml for Flt-1 or 30 ng/ml for KDR) in the presence of increased concentrations of VEGF variant proteins as described (25).
Thymidine Incorporation Assays.
Quiescent human umbilical vein endothelial (HUVE) cells (Promocell) were stimulated with increased concentrations of VEGF variant proteins. After 18 h of VEGF-incubation, [3H]thymidine (0.5 μCi; 1 Ci = 37 GBq) was added and the incubation was continued for additional 6 h. The cells were washed and the incorporation of radioactivity was determined by scintillation counting.
RESULTS
Construction of Receptor-Selective VEGF Variants.
Analysis of the crystal structure of PDGF-B revealed that domains that are involved in receptor binding are located within loops or turns exposed on the dimeric protein (19). By using the Swiss-Model facilities (26), a partial structural model of VEGF was derived (data not shown), which was based on the PDGF-B crystal structure. We used the model to predict loop/turn structures within the VEGF protein probably involved in receptor recognition. Within these areas, the VEGF amino acids were substituted with the corresponding amino acids from PDGF that were selected to avoid disturbance of the overall structure of the VEGF mutant proteins. Point mutations were introduced into VEGF121 cDNA by using PCR techniques, and mutant proteins were refolded and purified from an E. coli expression system as described for wild-type VEGF121 (23). All of the mutant VEGF proteins analyzed were in the dimeric conformation (data not shown) with the exception of VEGF121-C61S, a monomeric VEGF mutant with Cys61 replaced with Ser that did not bind to endothelial cells and showed no significant biological activity (27), which was used as a control. The proteins were characterized for their binding affinity to VEGF-receptors Flt-1 and KDR, respectively, and for biological activity by their ability to stimulate DNA synthesis of HUVE cells. Substitution of amino acids Asp63, Glu64, Gly65, and Leu66 from loop/turn II by the corresponding sequence from PDGF-A (Thr-Ser-Ser-Val) abolished binding of the VEGF variant (VEGF121-L2) to Flt-1 (Fig. 1A) and decreased binding affinity for KDR 4- to 5-fold (Fig. 1B). Protein concentration necessary for half-maximal induction of DNA synthesis in HUVE cells increased 4- to 5-fold (Fig. 1C). Substitution of amino acids Arg82 with Glu, Gln87 with Arg, Gly88 with Lys, and Gln89 with Lys, which were derived from the corresponding sequence of PDGF-A/B, and of Lys84, which is conserved between VEGF and PDGF-A and -B, with Glu in loop/turn III had no effect on binding affinity of the VEGF variant (VEGF121-L3) to Flt-1 (Fig. 1A). In contrast, binding affinity for KDR was decreased more than 40-fold (Fig. 1B) and protein concentration necessary for half-maximal induction of DNA synthesis in HUVE cells increased more than 100-fold (Fig. 1C) as compared with wild-type VEGF121. These experiments show that loop/turn II of VEGF is involved in recognition of VEGF receptor Flt-1 and that loop/turn III is essential for recognition of VEGF-receptor KDR and resulted in the identification of receptor-selective VEGF variants. Our findings are in total agreement with the results of Keyt et al. (21) who identified VEGF determinants for binding to Flt-1 and KDR by performing alanine-scanning mutagenesis of charged amino acids of VEGF165. Projection of these results on the very recently solved crystal structure of VEGF (20), which confirmed the structural similarity of VEGF and PDGF and in addition confirmed the model that was used as a basis for our mutagenic analysis of VEGF, reveals that one binding site for each VEGF receptor is located at each pole of the VEGF dimer. Loop/turn II of one VEGF chain and loop/turn III of the other VEGF chain are in close proximity on each pole of the VEGF dimer thereby forming a receptor binding interface for both receptors at each pole (Fig. 2). Each of the two binding interfaces should be able to contact one receptor monomer to induce receptor dimerization.
Figure 1.

Competition binding of VEGF variants to VEGF receptors and stimulation of endothelial cells. Recombinant extracellular domains of VEGF receptors Flt-1 (A) and KDR (B), respectively, coated onto Maxisorb plates were incubated with biotinylated VEGF165 (10 ng/ml for Flt-1 or 30 ng/ml for KDR) in the presence of increased concentrations of various VEGF121 variants expressed in E. coli. (C) Quiescent HUVE cells were stimulated with increased concentrations of various VEGF121 variants, and growth factor-stimulated DNA synthesis was measured as [3H]thymidine incorporation. VEGF variants used were wild-type VEGF121 (•), VEGF121-L2 (□), VEGF121-L3 (◊), and VEGF121-C61S (▴).
Figure 2.
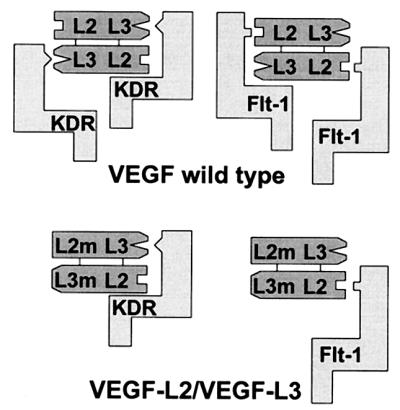
Scheme of receptor recognition by wild-type VEGF and by the heterodimeric VEGF165-L2/VEGF121-L3 variant. Major determinants for recognition of Flt-1 and KDR are located on loop/turn II (L2) and loop/turn III (L3). A heterodimeric VEGF molecule consisting of a loop/turn II mutant (L2 m) chain and of a loop/turn III mutant (L3 m) chain displays only one intact receptor binding interface.
Construction of a Heterodimeric VEGF Variant.
Construction of a heterodimeric VEGF molecule consisting of a loop/turn II mutant chain and of a loop/turn III mutant chain would have only one intact receptor binding interface (Fig. 2). Such a heterodimeric variant should bind to receptor monomers thereby antagonizing wild-type VEGF-induced receptor activation. On the basis of the characterization of receptor-selective VEGF variants, a heterodimeric VEGF variant was constructed. To distinguish between homodimeric and heterodimeric VEGF variants simply by size, the heterodimeric variant was constructed by combining a loop/turn II mutant VEGF165 with a loop/turn III mutant VEGF121. With respect to receptor binding and stimulation of HUVE cell proliferation homodimeric VEGF165-L2, which contained an amino acid substitution identical to VEGF121-L2, behaved as VEGF121-L2 did (data not shown). For construction of the heterodimeric VEGF variant, solubilized inclusion bodies containing VEGF165-L2 and VEGF121-L3 proteins, respectively, were mixed in an approximately equimolar ratio, and the refolding and purification procedure was performed on the mixture as described for wild-type VEGF121. After the refolding and Ni2+ affinity purification procedures, the solution was expected to contain VEGF165-L2 and VEGF121-L3 homodimers and VEGF165-L2/VEGF121-L3 heterodimers. For purification of the heterodimer, the mixture was applied to a heparin column and material was eluted with a NaCl gradient, which resulted in the separation of the VEGF variants in individual fractions. For analysis, aliquots of the mixture before heparin chromatography (Fig. 3, lanes 1 and 5) and aliquots of pooled heterodimer containing fractions (lanes 3 and 7) were subjected to SDS/PAGE along with VEGF121-L3 (lanes 2 and 6) and VEGF165-L2 (lanes 4 and 8) homodimers. Electrophoresis under reducing conditions (lanes 1–4) showed that VEGF121-L3 was most prominent in the mixture before heparin chromatography (lane 1), whereas the purified heterodimer fraction (lane 3) contained VEGF121-L3 and VEGF165-L2 in an approximately equimolar proportion. Electrophoresis under nonreducing conditions (lanes 5–8) showed the formation of the heterodimeric variant in the mixture (lane 5) besides the prominent VEGF121-L3 homodimer and only a very faint band corresponding to VEGF165-L2 homodimer. The differences in formation of the dimeric VEGF proteins are most likely due to preferential precipitation of VEGF165-containing variants during the refolding procedure. As described (23), VEGF121 variants migrated under nonreducing conditions as a single band (lane 6), whereas for VEGF165 variants two bands were observed (lane 8) that might be due to heterogenicity in refolding of VEGF165. The purified heterodimer (lane 7) contained an approximately 20% contamination with VEGF121-L3 homodimer as determined by videodensitometer evaluation of Coomassie-stained gels. The results show the feasibility to produce VEGF121/VEGF165-heterodimeric variants by a simultaneous refolding produce followed by separation of the dimeric proteins by heparin affinity chromatography resulting in a considerable purity of the heterodimer.
Figure 3.

SDS/PAGE analysis of the heterodimeric VEGF165-L2/VEGF121-L3 variant. Aliquots of the refolded mixture of VEGF165-L2 and VEGF121-L3 before heparin chromatography (lanes 1 and 5) and of the pooled VEGF165-L2/VEGF121-L3 heterodimer containing fractions after heparin chromatography (lanes 3 and 7) were electrophoresed on a SDS/15% gel along with VEGF121-L3 (lanes 2 and 6) and VEGF165-L2 (lanes 4 and 8) homodimers under reducing (lanes 1–4) and under nonreducing (lanes 5–8) conditions.
The Heterodimeric VEGF165-L2/VEGF121-L3 Variant Binds to VEGF Receptors.
The heterodimeric VEGF variant was analyzed for binding to recombinant extracellular domain of VEGF receptor Flt-1 and KDR, respectively, by solid-phase competition binding assays. Competition of the heterodimer with biotinylated wild-type VEGF165 for binding to Flt-1 was within the range of wild-type VEGF121 (Fig. 4A), whereas the competition experiments on KDR (Fig. 4B) showed a slightly lower affinity of the heterodimer compared with wild-type VEGF.
Figure 4.

Competition binding of the VEGF165-L2/VEGF121-L3 heterodimer to VEGF receptors. Recombinant extracellular domains of VEGF receptors Flt-1 (A) and KDR (B), respectively, coated onto Maxisorb plates were incubated with biotinylated VEGF165 (10 ng/ml for Flt-1 or 30 ng/ml for KDR) in the presence of increased concentrations of the VEGF165-L2/VEGF121-L3 heterodimer (⧫) and of VEGF121 (•), respectively.
The Heterodimeric VEGF165-L2/VEGF121-L3 Variant Is a VEGF Antagonist.
The heterodimeric VEGF165-L2/VEGF121-L3 variant was tested for its ability to antagonize VEGF-stimulated KDR autophosphorylation and proliferation of human endothelial cells. Analysis of autophosphorylation of KDR after VEGF stimulation in the absence or presence of the VEGF165-L2/VEGF121-L3 heterodimer showed a strong inhibition of ligand-induced autophosphorylation with increasing concentrations of the heterodimer (Fig. 5). No detectable KDR autophosphorylation was observed upon incubation of the cells with 30 nM of the heterodimer in the absence of wild-type VEGF (data not shown). The heterodimer failed to stimulate proliferation of HUVE cells significantly, although a slight stimulation, which decreased at higher concentrations, was fully reproducible in several experiments with various preparations of the heterodimer. In competition experiments, HUVE cells were stimulated with VEGF121 at 0.36 nM (10 ng/ml), which is sufficient to support 85% of maximal VEGF-induced cell proliferation, and increasing amounts of the heterodimer VEGF165-L2/VEGF121-L3 or of the homodimers VEGF165-L2 and VEGF121-L3, respectively, were added. Whereas the homodimeric VEGF variants were unable to suppress VEGF121-stimulated proliferation of HUVE cells, the heterodimer diminished VEGF121-stimulated proliferation in a concentration-dependent manner (Fig. 6). A 50% inhibition of HUVE cell proliferation stimulated by 0.36 nM of VEGF121 was achieved with an approximately 15-fold molar excess of the heterodimer. An approximately 100-fold molar excess of the heterodimer resulted in an almost complete inhibition of VEGF-stimulated proliferation of HUVE cells. Competition experiments using the VEGF165 isoform to stimulate HUVE cells gave similar results (data not shown).
Figure 5.

Inhibition of VEGF-stimulated KDR autophosphorylation. (A) Quiescent human umbilical artery endothelial cells (lane 1) were stimulated with VEGF121 (10 ng/ml; lane 2) in the presence of increased concentrations of the VEGF165-L2/VEGF121-L3 heterodimer (HD; lanes 3–6). KDR was immunoprecipitated and the blot was probed with an anti-phosphotyrosine antibody (αpY), and with an anti-KDR antibody (αKDR). (B) KDR phosphorylation was calculated by videodensitometry and normalized on KDR contents.
Figure 6.

Competition of VEGF121-stimulated proliferation of endothelial cells. Quiescent HUVE cells were stimulated either with increased concentrations of VEGF121 (•) and of the VEGF165-L2/VEGF121-L3 heterodimer (⧫), respectively, or with of VEGF121 (10 ng/ml) and increased concentrations of VEGF165-L2 homodimer (▪), of VEGF121-L3 homodimer (□), and of VEGF165-L2/VEGF121-L3 heterodimer (◊), respectively, were added. Growth factor-stimulated DNA synthesis was measured as [3H]thymidine incorporation. The dotted line represents level of DNA synthesis achieved by stimulation with VEGF121 (10 ng/ml).
DISCUSSION
By using a rational approach which is based on the structure of VEGF, we have shown the feasibility to construct a VEGF variant that acts as an VEGF antagonist. The strategy used herein for construction of the VEGF antagonist should be applicable to other disease-associated dimeric growth factors, e.g., PDGF, which is involved in several human pathological settings including atherosclerosis and carcinomas (28). Our approach differs substantially from experiments in which dominant negative mutant chains of PDGF-A were coexpressed in cells expressing wild-type PDGF-A or -B (29–31). The dominant negative approach resulted in the formation of low-affinity receptor-binding heterodimers of the mutant PDGF-A chain with with either wild-type PDGF-A or -B. These heterodimers showed reduced receptor-agonistic activity as compared with wild-type PDGF that was sufficient for disruption of autocrine loops in sis-transformed cells. No receptor-antagonistic effects were observed with these PDGF variants. Another PDGF-A mutant (29) and the VEGF165 mutant C101-S (32) were not secreted from transfected cells but suppressed expression of the respective wild-type growth factor when coexpressed with the wild-type form in the same cell. The mechanism involved in this case most likely is the formation of unstable heterodimers.
Antagonistic variants of monomeric cytokines had been studied in detail. For example, a variant of human growth hormone, G120R, in which binding site 2 for human growth hormone receptor had been mutated, functioned as a potent antagonist (33). A 20-fold excess of G120R was required for a 50% inhibition of growth hormone-stimulated cell proliferation. Introduction of additional mutations in variant G120R within binding site 1 resulted in tighter binding to the receptor via site 1 and in a 10 times higher antagonistic activity.
Because a 15-fold molar excess of the heterodimeric VEGF165-L2/VEGF121-L3 variant is required to inhibit VEGF-stimulated endothelial cell proliferation by 50%, this variant is already a very potent VEGF antagonist in vitro. Ongoing experiments should show its efficiency as an useful antiangiogenic reagent in vivo. For this purpose, improvement of the refolding and purification procedures will be necessary. Although the VEGF165-L2/VEGF121-L3 preparations used herein contained up to 20% contamination with VEGF121-L3 homodimer, they showed an efficient inhibition of VEGF-stimulated endothelial cell proliferation. VEGF121-L3 has a strongly decreased affinity to KDR, the VEGF receptor that transduces a proliferative signal (21, 34). Therefore, a contamination of the heterodimeric variant by VEGF121-L3 homodimer should not compromise the antagonistic action in a proliferation assay significantly. In contrast, VEGF121-L3 showed high affinity to Flt-1. This might be a reason for the apparently high-affinity Flt-1 binding of the heterodimer preparations. Although activation of Flt-1 was shown to mediate biological responses, such as endothelial and monocyte cell migration and tissue factor induction (35, 36), but obviously no proliferative signals (34), its in vivo function remains elusive. The recently solved crystal structure of VEGF (20) should facilitate an optimization of the mutations to be introduced in VEGF to achieve a higher affinity of the antagonistic variant to receptor monomers and, at the same time, a lower residual agonistic activity of the variant.
Acknowledgments
We thank Steffi Koidl and Katja Mohrs for excellent technical assistance. This work was supported by “Kirstins Weg.”
ABBREVIATIONS
- Flt-1
fms-like tyrosine kinase-1
- KDR
kinase domain receptor
- HUVE
human umbilical vein endothelial
- PDGF
platelet-derived growth factor
- VEGF
vascular endothelial growth factor
References
- 1.Martiny-Baron G, Marmé D. Curr Opin Biotechnol. 1995;6:675–680. doi: 10.1016/0958-1669(95)80111-1. [DOI] [PubMed] [Google Scholar]
- 2.Thomas K A. J Biol Chem. 1996;271:603–606. doi: 10.1074/jbc.271.2.603. [DOI] [PubMed] [Google Scholar]
- 3.Breier G, Risau W. Trends Cell Biol. 1996;6:454–456. doi: 10.1016/0962-8924(96)84935-x. [DOI] [PubMed] [Google Scholar]
- 4.Risau W. Nature (London) 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- 5.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, et al. Nature (London) 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 6.Ferrara N, Carver-Moore, Chen H, Dowd M, Lu L, O’Shea K S, Powell-Braxton L, Hillan K J, Moore M W. Nature (London) 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 7.Plate K H, Breier G, Weich H A, Risau W. Nature (London) 1992;359:845–848. doi: 10.1038/359845a0. [DOI] [PubMed] [Google Scholar]
- 8.Aiello L P, Avery R L, Arrigg P G, Keyt B A, Jampel H D, Shah S T, Pasquale L R, Thieme H, Iwamoto M A, Park J E, et al. N Engl J Med. 1994;331:1480–1487. doi: 10.1056/NEJM199412013312203. [DOI] [PubMed] [Google Scholar]
- 9.Fava R A, Olsen N J, Spencer-Green G, Yeo K T, Yeo T K, Berse B, Jackman R W, Senger D R, Dvorak H F, Brown L F. J Exp Med. 1994;180:341–346. doi: 10.1084/jem.180.1.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Detmar M, Brown L F, Claffey K P, Yeo K T, Kocher O, Jackman R W, Berse B, Dvorak H F. J Exp Med. 1994;180:1141–1146. doi: 10.1084/jem.180.3.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Vries C, Escobedo J A, Ueno H, Houck K, Ferrara N, Williams L T. Science. 1992;255:989–991. doi: 10.1126/science.1312256. [DOI] [PubMed] [Google Scholar]
- 12.Terman B I, Dougher-Vermazen M, Carrion M E, Dimitrov D, Armellino D C, Gospodarowicz D, Bohlen P. Biochem Biophys Res Commun. 1992;187:1579–1586. doi: 10.1016/0006-291x(92)90483-2. [DOI] [PubMed] [Google Scholar]
- 13.Kim K J, Li B, Winer J, Armanini M, Gillett N, Phillips H S, Ferrara N. Nature (London) 1993;362:841–844. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- 14.Millauer B, Shawver L K, Plate K H, Risau W, Ullrich A. Nature (London) 1994;367:576–579. doi: 10.1038/367576a0. [DOI] [PubMed] [Google Scholar]
- 15.Aiello L P, Pierce E A, Foley E D, Takagi H, Chen H, Riddle L, Ferrara N, King G L, Smith L E H. Proc Natl Acad Sci USA. 1995;92:10457–10461. doi: 10.1073/pnas.92.23.10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maglione D, Guerriero V, Vigliette G, Delli-Bovi P, Persico M G. Proc Natl Acad Sci USA. 1991;88:9267–9271. doi: 10.1073/pnas.88.20.9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olofsson B, Pajusola K, Kaipainen A, Von Euler G, Joukov V, Saksela O, Orpana A, Pettersson R F, Alitalo K, Eriksson U. Proc Natl Acad Sci USA. 1996;93:2576–2581. doi: 10.1073/pnas.93.6.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joukov V, Pajusola K, Kaipainen A, Chilov D, Lahtinen I, Kukk E, Saksela O, Kalkkinen N, Alitalo K. EMBO J. 1996;15:290–298. [PMC free article] [PubMed] [Google Scholar]
- 19.Oefner C, D’Arcy A, Winkler F K, Eggimann B, Hosang M. EMBO J. 1992;11:3921–3926. doi: 10.1002/j.1460-2075.1992.tb05485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muller Y A, Li B, Christinger H W, Wells J A, Cunningham B C, de Vos A A. Proc Natl Acad Sci USA. 1997;94:7192–7197. doi: 10.1073/pnas.94.14.7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keyt B A, Nguyen H V, Berleau L T, Duarte C M, Park J, Chen H, Ferrara N. J Biol Chem. 1996;271:5638–5646. doi: 10.1074/jbc.271.10.5638. [DOI] [PubMed] [Google Scholar]
- 22.Weindel K, Marmé D, Weich H A. Biochem Biophys Res Commun. 1992;183:1167–1174. doi: 10.1016/s0006-291x(05)80313-4. [DOI] [PubMed] [Google Scholar]
- 23.Siemeister G, Schnurr B, Mohrs K, Schächtele C, Marmé D, Martiny-Baron G. Biochem Biophys Res Commun. 1996;222:249–255. doi: 10.1006/bbrc.1996.0730. [DOI] [PubMed] [Google Scholar]
- 24.Studier F W, Rosenberg A H, Dunn J J, Dubendorf J W. Meth Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 25.Barleon B, Totzke F, Herzog C, Blanke S, Kremmer E, Siemeister G, Marmé D, Martiny-Baron G. J Biol Chem. 1997;272:10382–10388. doi: 10.1074/jbc.272.16.10382. [DOI] [PubMed] [Google Scholar]
- 26.Peitsch M C. Bio/Technology. 1995;13:658–660. [Google Scholar]
- 27.Pötgens A J G, Lubsen N H, van Altena M C, Vermeulen R, Bakker A, Schoemakers J G G, Ruiter D J, de Waal R M W. J Biol Chem. 1994;269:32879–32885. [PubMed] [Google Scholar]
- 28.Heldin C H. EMBO J. 1992;11:4251–4259. doi: 10.1002/j.1460-2075.1992.tb05523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mercola M, Deininger P L, Shamah S M, Porter J, Wang C, Stiles C D. Genes Dev. 1990;4:2333–2341. doi: 10.1101/gad.4.12b.2333. [DOI] [PubMed] [Google Scholar]
- 30.Vassbotn F S, Andersson M, Westermark B, Heldin C H, Östman A. Mol Cell Biol. 1993;13:4066–4076. doi: 10.1128/mcb.13.7.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shamah S M, Stiles C D, Guha A. Mol Cell Biol. 1993;13:7203–7212. doi: 10.1128/mcb.13.12.7203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Claffey K P, Senger D R, Spiegelman B M. Biochim Biophys Acta. 1995;1246:1–9. doi: 10.1016/0167-4838(94)00144-6. [DOI] [PubMed] [Google Scholar]
- 33.Fuh G, Cunningham B C, Fukunaga R, Nagata S, Goeddel D V, Wells J A. Science. 1992;256:1677–1680. doi: 10.1126/science.256.5064.1677. [DOI] [PubMed] [Google Scholar]
- 34.Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin C H. J Biol Chem. 1994;269:26988–26995. [PubMed] [Google Scholar]
- 35.Barleon B, Sozzani S, Zhou D, Weich H A, Mantovani A, Marmé D. Blood. 1996;87:3336–3343. [PubMed] [Google Scholar]
- 36.Clauss M, Weich H, Breier G, Knies U, Röckl W, Waltenberger J, Risau W. J Biol Chem. 1996;271:17629–17634. doi: 10.1074/jbc.271.30.17629. [DOI] [PubMed] [Google Scholar]