

Abstract
Alzheimer’s disease (AD) is predominantly characterized by progressive neuronal loss in the brain. It has been recently found that adult neurogenesis in the hippocampal dentate gyrus of AD patients is significantly enhanced, while its functional significance is still unknown. By using an AD-like neurodegeneration mouse model, we show here that neurogenesis in the dentate gyrus was neurodegenerative stage-dependent. At early stages of neurodegeneration, neurogenesis was significantly enhanced and newly generated neurons migrated into the local neuronal network. Up to late stages of neurodegeneration, however, the survival of newly generated neurons was impaired so that the enhanced neurogenesis could not be detected any more. Most interestingly, these dynamic changes in neurogenesis were correlated with the severity of neuronal loss in the dentate gyrus, indicating that neurogenesis may work as a self-repairing mechanism to compensate for neurodegeneration. Therefore, to enhance endogenous neurogenesis at early stages of neurodegeneration may be a valuable strategy to delay neurodegenerative progress.
Keywords: Alzheimer’s disease, adult neurogenesis, neuronal loss, presenilin 1, neurodegeneration, conditional knockout, mice
Introduction
Alzheimer’s disease (AD) is clinically characterized by progressive loss of memory, and at late stages, by dementia (Heston, et al., 1981). Although senile plaques and neurofibrillary tangles (NFTs) in the brain are considered the pathological hallmarks, the presences of these hallmarks seem not correlated to the severity of dementia (Arriagada, et al., 1992, Samuel, et al., 1994, Gomez-Isla, et al., 1997). In contrast, neuronal loss in certain selected brain regions represents a direct pathological cause for dementia (Bondareff, et al., 1982, Whitehouse, et al., 1982, Samuel, et al., 1994, Gomez-Isla, et al., 1997, Bussiere, et al., 2003). Furthermore, the hippocampus is one of the most vulnerable brain regions to AD, and the degeneration in this brain structure may directly underlie memory deficit, the earliest symptom of AD (Barber, et al., 2001, Thompson, et al., 2004). Therefore, to attenuate or stop neuronal loss in the brain is a fundamental strategy that may eventually cure this disease.
During the past decade, it has been firmly established that new neurons are continually generated in the brain post-developmentally, a process that is called adult neurogenesis (Altman and Das, 1965, Gage, 2000). In adult mammalian brains, neurogenesis is restricted to two regions, the dentate gyrus and olfactory bulb (Eriksson, et al., 1998, Gould, et al., 1999, Lledo, et al., 2006). The other brain regions such as the cortex are considered non-neurogenic regions (Gage, 2000, Ming and Song, 2005, Lledo, et al., 2006). In the dentate gyrus, new neurons are produced via mitosis of neural progenitor cells, which are located in the subgranular zone (SGZ) (Gage, et al., 1998, Kokaia and Lindvall, 2003, Ming and Song, 2005, Lledo, et al., 2006). It is estimated that about 800–1,600 new neurons are generated everyday in the dentate gyrus of a mouse (Cameron and McKay, 2001, Hayes and Nowakowski, 2002). Although the functional significance is still not clear, accumulating evidence has indicated that neurogenesis may play an important role in the maintenance of normal hippocampal functions such as learning and memory (van Praag, et al., 2000, Shors, et al., 2001, Aimone, et al., 2006) and may be modulated by environmental factors (Kempermann, et al., 1997, Rampon, et al., 2000, van Praag, et al., 2000), stress (Mirescu and Gould, 2006, Warner-Schmidt and Duman, 2006) and antidepressants (Santarelli, et al., 2003, Duman, 2004). Moreover, certain acute brain injuries such as ischemia, hypoxia, seizures, and trauma increase neurogenesis (Kokaia and Lindvall, 2003, Rice, et al., 2003, Itoh, et al., 2005, Overstreet-Wadiche, et al., 2006, Qiu, et al., 2007), suggesting that neurogenesis may work as a protective mechanism for the brain. Interestingly, enhanced neurogenesis was recently found in brains of neurodegenerative diseases including Huntington’s disease (Curtis, et al., 2003), Parkinson’s disease (Hoglinger, et al., 2004), and AD (Jin, et al., 2004a). However, neither the course of neurogenesis during the neurodegenerative processes nor the functional significance of neurogenesis in neurodegeneration is well understood.
Here, we report that neurogenesis in the dentate gyrus is neurodegenerative stage-dependent. At early stages of neurodegeneration (ESND), neurogenesis was most evident. Moreover, the newly generated neurons migrated into the neuronal network such as the granule cell layer (GCL) and hilus region. At late stages of neurodegeneration (LSND), however, the survival of newly generated neurons was dramatically impaired so that the increased neurogenesis could not be found any more. Most interestingly, these dynamic changes were correlated with the severity of neurodegeneration in the dentate gyrus, which showed modest neuronal loss at ESND when there was robust neurogenesis while severe neuronal loss at LSND when there was no significant neurogenesis. These results indicate that to enhance endogenous neurogenesis at early stages of AD may be a valuable strategy to delay the disease course or to cure the disease.
Materials and Methods
Generation of presenilin 1 (PS1) and presenilin 2 (PS2) double knockout (PS1/PS2-KO) mice
In PS1/PS2-KO mice, PS1 was conditionally knocked out and PS2 was conventionally knocked out. The Cre/loxP recombinant system was used to delete PS1 as we previously published (Feng, et al., 2001). Briefly, the exon 4 of the PS1 gene was flanked by two loxP elements for the generation of PS1 floxed mice. A specific Ca2+-calmodulin kinase-II (CaMKII)-Cre transgenic mouse strain was used to delete the floxed exon 4. In these Cre transgenic mice, as the expression of Cre recombinase began in neurons of the whole forebrain 6 months postnatally, the knockout of PS1 in neurons in this region occurred thereafter. PS2 knockout mice were the same as previously published (Donoviel, et al., 1999), in which the whole exon 5 was deleted. Both PS1 floxed mice and PS2 knockout mice were backcrossed into B6/CBA F1 mice for about 12 generations to dilute the 129v background. PS1/PS2-KO mice were generated by three steps of breeding. First, floxed PS1/floxed PS1 mice were crossed into PS2−/− mice to produce double heterozygous mice (floxed PS1/+//PS2 +/−) and at the same time PS2 −/− mice were crossed into Cre transgenic mice to produce PS2 +/−//Cre/− mice (Cre/− indicates Cre hemizygous transgenic mice). Second, floxed PS1/+//PS2 +/− mice were crossed into PS2 +/−//Cre/− mice to produce floxed PS1/+//PS2 −/−//Cre/− mice and at the same time floxed PS1/+//PS2 +/− mice were crossed together to produce floxed PS1/+//PS2−/− mice. Finally, floxed PS1/+//PS2 −/−//Cre/− mice were crossed into floxed PS1/+//PS2−/− mice to produce floxed PS1/PS1//PS2−/−//Cre/− mice, which are PS1/PS2-KO mice. The genotypes were determined by PCR analyses of these three alleles. Primers of 5′-AGA TGT TCG CGA TTA TC-3′ and 5′AGC TAC ACC AGA GAC GG-3′ were used to amplify the Cre transgene (490 bp). Primers of 5′-CAG ACA TTA GCA CTG TCT GTA AGG AGT C-3′ and 5′-GTT CCT AAA CCT CTA AAC TTC CAT GAG C-3′ were used to amplify PS1 wild-type allele (645 bp) and PS1-floxed allele (703 bp). Primers of 5′-AAG TAT CGA TGC TAC AAG GTG AGG-3′ and 5′-CCC ACA TGA TAA AAG GAG GAC -3′ were used to amplify PS2 wild-type allele (640 bp) and PS2 knockout allele (380 bp). As PS1/PS2-KO mice carry two homozygous alleles (PS1 floxed and PS2 knockout) and one hemizygous allele (Cre transgene), it is impossible to obtain real wild-type mice from PS1/PS2-KO littermates as controls. We used PS2-KO or PS1-KO mice as controls. In order to confirm that there would be no significant difference between wild-type and these control mice, we characterized three control groups in our pilot experiments in terms of neurodegeneration, neurogenesis, and learning and memory. The results showed that there was no significant difference between wild-type and PS2−− mice or between wild-type and PS−/+ mice. Hence, littermates of PS1/PS2-KO mice with either PS1-KO heterozygous or PS2-KO homozygous mice were used as control mice. Male and female mice were mixed in all the experiments. All studies in animals were conducted in accordance with NIH guidelines for the use of animals. All procedures were reviewed and approved by the Institutional Animal Care and Use Committee in the University of Chicago.
Histological studies
Nissl staining
Animals were anesthetized with ketamine (100 mg/kg)/xylazine (20 mg/kg) mixture and were perfused transcardially with 0.9% saline, followed by 4% paraformaldehyde (PFA). Brains were post-fixed in 4% PFA overnight and then equilibrated with 30% sucrose in phosphate buffer overnight. Total of 16 coronal brain sections (20 μm) were collected from each brain with a Cryostat (Leica, CM1900) and every two sections were spaced by 5 (PS1/PS2-KO mice) and 6 (control mice) sections to ensure that the examined sections were at a similar level between control and PS1/PS2-KO mice. Both the cortex and hippocampus were examined. For the cortex, neuron numbers were counted in two fields in each side of the cortex and total four fields were counted in each section. The whole dentate gyrus in two sides was counted. In each animal, total of 8 sections were examined with a computerized imaging analysis system (Q-imaging). The results were expressed as density (number/mm2) and were calculated as percentage of control mice. Results were analyzed by Student’s t test.
Adult neurogenesis
Bromodeoxyuridine (BrdU) labeling
Thymidine analog BrdU (Sigma, St. Louis, MO) was used to label newly synthesized DNA. In order to detect different processes of neurogenesis, a single dose injection (intraperitoneally) of BrdU at the concentration of 100 mg/kg was used.
Immunohistochemistry
Tissue preparation was the same as in Nissl staining. A stereological method was used to collect brain sections (Chui, et al., 1999). Briefly, total of 16 coronal sections (40 μm) were collected from each brain with the Cryostat. Every two sections were spaced by 5 sections in PS1/PS2-KO mice and 6 sections in control mice to ensure that the examined sections were at a similar level between PS1/PS2-KO and control mice and that at least 200 μm apart between each two sections from the same brain. A floating staining protocol and immunofluorescent method was used. For BrdU staining, DNA was denatured by incubating the sections in 50% formamide in 2 × SSC at 65°C for 2 hrs. The sections were rinsed twice with 2 × SSC and incubated in 2N HCL at 37°C for 30 min. The sections were blocked with 3% blocking serum in TBS/0.1 Triton X-100 for 1 h and then were stained with single or double immunofluorescence. The primary antibodies and working concentration were as following: rat anti-BrdU at 1:800 (Abcam, Cambridge, UK), mouse anti-NeuN at 1:800 (Chemicon, Temecula, CA), and mouse anti-GFAP at 1:800 (Chemicon, Temecula, CA).
Proliferation
Proliferation is generally completed within 24 hrs. Accordingly, mice were killed 24 hrs after a single dose of BrdU injection. The tissue preparation was the same as described above. The total number of BrdU labeled cells was an index for cell proliferation, after which the cell fate includes neurogenesis and gliosis. All the sections were red-counter stained.
Neuronal differentiation
Neuronal differentiation indicates a choice of neuronal fate decision. Immunostaining with an immature neuronal marker, doublecortin (DCX), was used to evaluate neuronal differentiation. A goat anti-DCX antibody (Santa Cruz Biotechology, Santa Cruz, CA) at the concentration of 1:200 was used. After incubated overnight at 4°C, sections were washed and incubated with the second antibody, FITC conjugated donkey anti-goat IgG antibody (1:400, Jackson Immuno Research, West Grove, PA), then mounted onto glass slides. It should be noted that we did not use double staining of DCX with BrdU here, because DCX staining reveals the overall neuronal differentiation, while double staining of DCX with BrdU is limited by the effective time window associated with BrdU injection and this double staining can only detect the differentiated cells with BrdU incorporation, and thus may dramatically underestimate the capacity of total neuronal differentiation. However, in order to have a clear vision for neuron-specific staining, all these sections were double-stained with NeuN.
New neuron generation
Generally, newly proliferated cells are able to differentiate into neurons in about 4 days. In order to have a wider time window to detect new neuron generation, we used a two-week protocol, in which mice were killed at two weeks after the single BrdU injection. Double staining with BrdU and NeuN was used to determine new neuron generation and double staining with BrdU and GFAP for new glial cells.
New neuron survival
After four weeks of survival, the number of newly generated neurons is relatively stable and a functional adult neurogenesis must indicate those that can live over this time window. Thus, the number of new neurons four weeks after BrdU injection is an index for new neuron survival. Accordingly, mice were killed 4 weeks after the single dose of BrdU injection and double staining with BrdU and NeuN was used to identify neuronal phenotype while double staining with BrdU and GFAP for glial cell phenotype.
Cell counting and confocal microscopic analysis
All assessments were done by experimenters who were blind to the genotypes of each brain section. The number of BrdU immunoreactive nuclei in the SGZ, GCL and hilus region of the hippocampus were counted regardless of size or shape with a confocal laser microscope (Olympus Fluoview 200, Japan) equipped with laser emitting at 488 nm, 543 nm and 630 nm. All cell counting was conducted with a “real-time” fashion on a computer monitor. Briefly, each BrdU immunoreactive nucleus was defined by careful z-series analyses of stained section stacks. This was performed by examining sequential optical sections at intervals of 1 μm in z-axis. For proliferation analysis, only the green spots that were confirmed within the nucleus with z-axis analysis were counted as positive stains. Any green sports that were not localized within the nucleus were considered as artifacts. For new neuron generation and new glial cell generation, BrdU-positive cells that were co-localized with NeuN, for neuronal phenotype, or with GFAP, for glial phenotype, were determined. For these double staining counts, we first confirmed the nucleus-BrdU-staining and then with different channels to the other staining. All co-localization signals were assessed by this XYZ zoom analysis with z-axis analysis of stacks of single cells at 3 μm intervals.
Statistical analysis
One-way analysis of variance (ANOVA) followed by post hoc analysis and Student’s t test were used whereas it was appropriate. A difference was considered significant when p value is less than 0.05.
Results
Generation of PS1/PS2-KO mice
As knockout of PS1 from the first ontogenesis led to premature death of null mutant mice and PS2 could partially compensate for PS1 null mutation, we decided to combine both conventional and conditional knockout techniques in this study. In double knockout mice, PS2 deletion was universal and PS1 deletion was limited to neurons in the whole forebrain from postnatal 6 months. Before 6 months old, mice with PS1/PS2-KO genotype only showed PS2, but not PS1, knockout. Our in situ hybridization with 35S oligo probes has confirmed these knockout patterns (data not shown). PS1/PS2-KO mice could live up to 24 months, and the average lifetime was slightly but significantly shorter than their littermate control (thereafter control) mice (data not shown).
Differential neuronal loss in the dentate gyrus of PS1/PS2-KO mice at different neurodegenerative stages
In PS1/PS2-KO mice, AD-like neurodegeneration was observable within one month after PS1 deletion and was most evident in neuronal loss. No significant neurodegeneration was found in single knockout, either PS1 or PS2, mice. Surprisingly, although neurodegeneration in the cortex and hippocampus was both progressive, significant difference in the severity in neuronal loss was observed between these two regions. Fig. 1A-D show Nissl staining and Fig. 1E and F show NeuN staining in control (A, C, E) and PS1/PS2-KO mice (B, D, F) at LSND, when mice were 18–20 months old. At ESND, when mice were 7–9 months old, neuronal loss was about 24% in the cortex (p < 0.01) and about 8% in the dentate gyrus, compared to control mice, respectively (Fig. 1G). In contrast, at the LSND, neuronal loss was about 62% in the cortex and 36% in the dentate gyrus, compared to control mice (p < 0.001, respectively; Fig. 1H). It will be interest to elucidate the mechanism that leads to this differential neuronal loss in the brain.
Fig. 1. Brain regional-specific neuronal loss in PS1/PS2-KO mice.

A–D. Nissl staining of coronal sections of brains from control (A and C) and PS1/PS2-KO (B and D) mice at the age of 18 months old to show neuronal loss at lower magnification (A and B) and higher magnification (C and D), which shows the marked region from control and PS1/PS2-KO mice in A and B, respectively. E and F. NeuN staining of coronal sections of brains from control (E) and PS1/PS2-KO (F) mice at the age of 18 months old to show neuron density. G and H. Quantitative analyses of neuronal loss in the cortex and dentate gyrus of control (n = 5) and PS1/PS2-KO (n = 6) mice at the age of 7–9 months old (G) and 18–20 months old (H) (control mice, n = 6; PS1/PS2-KO mice n = 5). Data are expressed as percentage of control mice and error bars are SD. **, p < 0.01, ***, p < 0.001, Student’s t test. The magnification in A, B, E, and F is the same and the scale bars represent 200 μm. The magnification in C and D is 5-fold higher than in A and B. DG: dentate gyrus.
Cell proliferation in the dentate gyrus of PS1/PS2-KO mice at different neurodegenerative stages
In order to determine whether or not the neurodegeneration triggers progenitor cell proliferation, control and PS1/PS2-KO mice received the single dose of BrdU injection and were then sacrificed at 24 hrs after the injection. Both the cortex and dentate gyrus were examined. In the cortex, no accountable BrdU staining was found in control mice, while a very low level of BrdU staining was found in PS1/PS2-KO mice at both ESND and LSND (data not shown), indicating that neurodegeneration does not trigger robust cell proliferation in these non-neurogenic regions. In the dentate gyrus, however, robust BrdU signals were detected in both of control (Fig. 2A, PS2-KO) and knockout mice (Fig. 2B) at ESND. A quantitative analysis revealed a highly significant difference in total number of BrdU labeled cells between control and PS1/PS2-KO mice [F(3,17) = 12.66, p > 0.001] (Fig. 2E). Post hoc analysis confirmed the significant difference between PS1/PS2-KO mice and any group of these groups: wild-type, PS2-KO, and PS1-KO (p < 0.01, respectively), but not between any two of these three control groups, further validating the use of PS2-KO or PS1-KO mice (or the heterozygous mice) as controls. Following aging, the capacity of proliferation in the dentate gyrus was dramatically decreased in control (Fig. 2C) and PS1/PS2-KO (Fig. 2D) mice. Nevertheless, a significant difference was still observed between these two groups (p < 0.05; Student’s t test; Fig. 2F). These results indicate that the neurodegeneration is able to trigger robust cell proliferation in the dentate gyrus but not the cortex, and that although less robust, this effect continues into the LSND.
Fig. 2. Enhanced cell proliferation in the dentate gyrus of PS1/PS2-KO mice.

A–D. BrdU staining of coronal sections of brains from control (A and C) and PS1/PS2-KO (B and D) mice at the age of 9 months old (A and B) and 18 months old (C and D). This is single BrdU staining with red-counter staining to visualize the nucleus. E and F. Quantitative analyses of the number of BrdU stained cells in the dentate gyrus of wild-type (n = 4), PS2-KO (n = 6), PS1-KO (n = 5), and PS1/PS2-KO (n = 6) mice at the age of 7–9 months old (E) and control (n = 6) and PS1/PS2-KO (n = 5) mice at the age of 18–20 months old (F). Data is expressed as mean ± SD. ***, p < 0.001, one-way ANOVA followed by Dunnett’s post hoc analyses; *, p < 0.05, Student’s t test. The magnification is the same in all the photo-pictures and the scale bar in D equals 50 μm.
Neuronal differentiation in the dentate gyrus of PS1/PS2-KO mice at different neurodegenerative stages
The increased cell proliferation does not necessarily indicate that the proliferated cells must differentiate into neurons. In order to determine whether these proliferated cells commit to neuronal differentiation, double staining with DCX, an immature neuronal marker, and NeuN, a marker for mature neuron, was used. As shown in Fig. 3, significantly increased staining signals were observed in the dentate gyrus of PS1/PS2-KO mice at both ESND (Fig. 3D–F) and LSND (Fig. 3J–L), compared to those in control mice (Fig. 3A–C and G–I), respectively, although a much higher level was observed at ESND. Experiments were repeated in 4–6 mice in each group and the results were consistent, indicating that the proliferated cells triggered by PS1/PS2-KO-caused neurodegeneration are able to differentiate into neuronal phenotype.
Fig. 3. Enhanced neuronal differentiation in the dentate gyrus of PS1/PS2-KO mice.
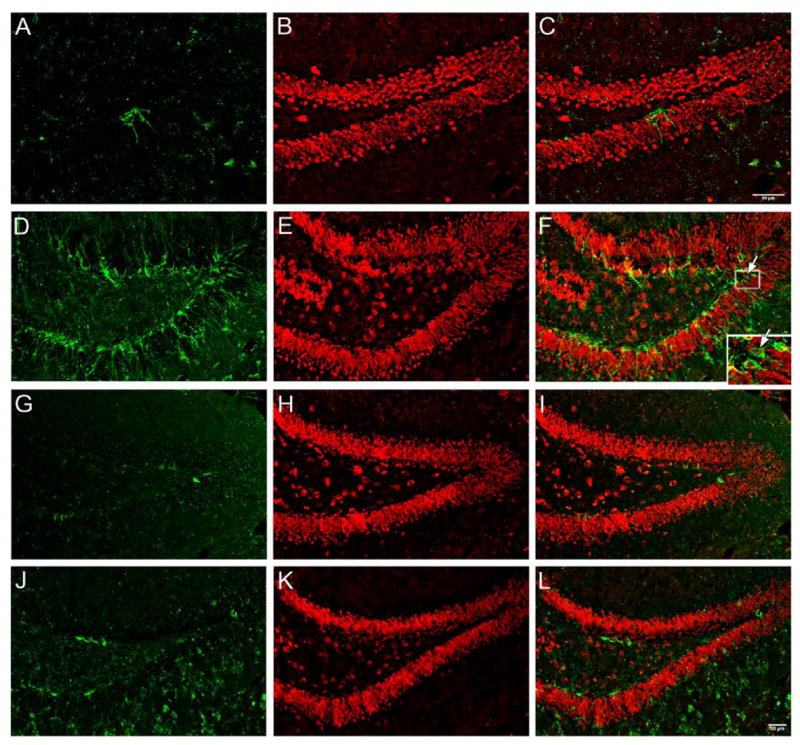
Representative confocal imaging of coronal brain sections from control (A-C and G-I) and PS1/PS2-KO mice (D-F and J-L) at the age of 9 (A to F) and 18 months old (G to L). A, D, G, and J show the DCX staining (green). B, E, H, and K show the NeuN staining (red). C, F, I, and L are DCX/NeuN merged imaging. The insect box in F shows the colocalization of DCX and NeuN. The magnification is the same in all the photo-pictures and the scale bar in L represents 50 μm.
New neuron generation in the dentate gyrus of PS1/PS2-KO mice at different neurodegenerative stages
It is well known that immature neurons may die before or during maturation (Cameron and McKay, 2001) and therefore the enhanced neuronal differentiation does not necessarily mean that neurogenesis must be enhanced. Accordingly, newly generated neurons were examined two weeks after a single dose of BrdU injection with a confocal imaging system, which detects double staining of BrdU with either NeuN (a mature neuron marker) or GFAP (a marker for glial cell). Fig. 4A–F are representative photos for BrdU/NeuN staining and Fig. 4G–L are for BrdU/GFAP staining at ESND. Statistical analyses of group data indicated that the number of total BrdU stained, BrdU/NeuN stained, and BrdU/GFAP stained cells were significantly increased in PS1/PS2-KO mice at either ESND (Fig. 4M) or LSND (Fig. 4N), compared to those in control mice, respectively. These results confirm the increased proliferation in the dentate gyrus of PS1/PS2-KO mice and further demonstrate that both new neuron generation and gliosis are dramatically enhanced in the hippocampus of PS1/PS2-KO mice at both ESND and LSND.
Fig. 4. Enhanced new neuron generation and gliosis in the dentate gyrus of PS1/PS2-KO mice.

A–L. Representative confocal imaging of coronal brain sections from control (A-C and G-I) and PS1/PS2-KO mice (D-F and J-L) at the age of 9 (A to F) and 18 months old (G to L). A, D, G, and J show the BrdU staining (green). B and E show NeuN staining (red) while H and K show GFAP staining (red). C and F are BrdU/NeuN merged imaging, while I and L are BrdU/GFAP merged imaging. The insects in C and F show colocalization of BrdU and NeuN with 3X (XYZ) analysis. The insects in I and L show colocalization of BrdU and GFAP with 3X (XYZ) analysis. The magnification is the same in all the sections and the scale bar in L equals 50 μm. M and N. Quantitative analyses of the number of double stained cells in the dentate gyrus of control (n = 5) and PS1/PS2-KO (n = 6) mice at the age of 7–9 months old (M) or control (n = 5) and PS1/PS2-KO (n = 7) mice at the age of 18–20 months old (N). Data is expressed as mean ± SD. ***, p < 0.001; **, p < 0.01; *, p < 0.05, Student’s t test. The magnification is the same in all the photo-pictures and the scale bar in L represents 50 μm.
New neuron survival and migration in the dentate gyrus of PS1/PS2-KO mice at different neurodegenerative stages
The survival time of newly generated neurons and some other functional properties such as migration are the most essential criteria to judge the significance of neurogenesis in the brain. These indexes were examined 4 weeks after a single dose of BrdU injection. Fig. 5A–C are representative photos showing new neuron migration in the hippocampus of PS1/PS2-KO mice at ESND. The black arrow is for a new neuron in the SGZ, red arrow for a new neuron in the hilus region, and white arrow for a new neuron in the GCL. As neither the hilus nor the GCL contains progenitor cells, the appearance of new neurons in these areas is indicative of migration from the SGZ (Kokaia and Lindvall, 2003, Lledo, et al., 2006). Quantitative analysis revealed that about 22–25% of new neurons migrated into the GCL and about 6–7% into the hilus region in PS1/PS2-KO and control mice with no significant difference between these two groups (Fig. 5D). As shown in Fig. 5E, at ESND, the total number of new neurons and glial cells in the dentate gyrus of PS1/PS2-KO mice was significantly higher than that in control mice, indicating that these new neurons survived over the maturation stage. Most surprisingly, no significant difference in total number of either new neurons or new glial cells was found between control and PS1/PS2-KO mice at LSND (Fig. 5F), indicating that newly generated neurons and glial cells in the dentate gyrus of PS1/PS2-KO mice cannot survive over the maturation stage at LSND.
Fig. 5. New neuron migration and survival in the dentate gyrus of PS1/PS2-KO mice.

A–C. Representative confocal imaging of coronal brain sections from a PS1/PS2-KO mouse at the age of 9 months old showing new neuron migration. Black arrow indicates a new neuron in the SGZ, red arrow for a new neuron in the hilus region, and white arrow for a new neuron in the GCL. Insect in C shows single XY image and XZ imaging demonstrating the co-localization of BrdU and NeuN in a new neuron from GCL. D. Quantitative analyses of the number of newly-born neuron migration in the hippocampal sub-regions of control (n = 5) and PS1/PS2-KO (n = 5) mice at the age of 7–9 months old. E. Quantitative analyses of the number of new neuron survival in the dentate gyrus of control (n = 5) and PS1/PS2-KO (n = 5) mice at the age of 7–9 months old. F. Quantitative analyses of the number of new neuron survival in the dentate gyrus of control (n = 6) and PS1/PS2-KO (n = 6) mice at the age of 18–20 months old. Data is expressed as mean ± SD. ***, p < 0.001, Student’s t test. The magnification is the same in all the photo-pictures and the scale bar in L represents 200 μm
Discussion
Recently, it has been found that knockout of PS1 and PS2 in the mouse is able to trigger robust neurodegeneration in the brain (Feng, et al., 2004, Saura, et al., 2004). We have provided further evidence to show that this neurodegeneration is featured by many characteristics of AD neurodegeneration, including the presences of NFTs, the increased tau hyperphosphorylation, and the presence of intracellular filamentous aggregates (submitted results). Especially, the neuronal loss in the brain represents the most comparable neurodegenerative status in terms of both the severity and selectivity in neuronal loss in AD. As shown in Fig. 1, at LSND, neuronal loss was about 62% in the cortex of PS1/PS2-KO mice, which is very similar to the extent at a late stage of AD, at which over 50% or even up to 70–90% of neurons are lost (Gomez-Isla, et al., 1997, Bussiere, et al., 2003). Most importantly, all these features show an age-dependent manner, which is phenotypically similar to AD. Therefore, we have reasons to claim that the neurodegeneration in the knockout mice is an AD-like type. It should be noted that, however, this model does not represent a comprehensive AD model, as no β-amyloid deposition could be found in the brain.
With this model, we have systematically characterized the involvement of neurogenesis in the neurodegenerative processes. One important finding in this study is that the neurodegeneration itself can trigger robust neurogenesis. It should be mentioned that in our previous study (Feng, et al., 2001) and the unpublished observation, we found that either conditional knockout of PS1 only or conventional knockout of PS2 only, but not double knockout, had no significant effect on neurogenesis. Since in these single knockout mouse strains, no observable neurodegeneration could be detected, the enhanced neurogenesis in PS1/PS2-KO mice therefore may be directly related to neurodegeneration, but not PS1/PS2 knockout per se. Virtually, in the conditional PS1 single knockout mice, we observed a significant deficit in neurogenesis in the mice that were subjected to environmental enrichment (Feng, et al., 2001), an experimental paradigm that triggers robust neurogenesis in the rodents (Kempermann, et al., 1997, Rampon, et al., 2000), further suggesting that it is very unlikely that the enhanced neurogenesis found in the current study was due to the loss of presenilin function per se. Therefore, our results indicate that neurodegeneration, when it comes to be the dominant event over the loss of PS1 function, could reverse PS1’s effect on inhibiting neurogenesis or trigger more robust neurogenesis in the brain. Many acute brain insults such as ischemia, hypoxia, seizures, and trauma enhance neurogenesis (fore review see Min and Song, 2005). Similarly, increased neurogenesis is also found in various neurodegenerative diseases (Curtis, et al., 2003, Hoglinger, et al., 2004, Jin, et al., 2004a), indicating that chronic insults can trigger neurogenesis too. Together with all those findings, our results suggest that neurogenesis may work as a universal protective mechanism endogenously for the brain to against different neuronal insults.
It should be mentioned that neurogenesis has been extensively investigated in many other AD models. In those models, FAD-linked APP or/and PS1 mutations were introduced into the mouse genome, either by transgenic expression or knock-in. Overall, the findings in those models were inconsistent. For example, in transgenic/knock-in mice expressing mutant PS1, mutant APP, or both, neurogenesis in the brain was impaired (Wang, et al., 2004, Wen, et al., 2004, Chevallier, et al., 2005, Donovan, et al., 2006, Zhang, et al., 2007) or enhanced (Jin, et al., 2004b, Greenberg and Jin, 2007). The reasons that led to this discrepancy among those different transgenic or knock-in mouse strains or between those studies and ours are not clear. It should be noted that one consistent finding in those models is that the deposition of Aβ is dramatically increased in the brain. There is evidence that the deposition of Aβ itself may disrupt neurogenesis (Haughey, et al., 2002, Zhang, et al., 2007). Therefore, it might be possible that the changes in neurogenesis were, at least partially, dependent on the amount of Aβ deposition. Most importantly, unlike in PS1/PS2-KO mice, all those transgenic/knock-in mouse models do not exhibit AD comparable neuronal loss, conforming that neurodegeneration is a dominant event that triggers neurogenesis in the brain.
Adult neurogenesis consists of four distinctive stages, proliferating from stem or progenitor cells (proliferation), differentiating into immature neurons (differentiation), developing into mature neurons (new neuron generation), and migrating into neuronal network, axon/dendrite targeting and synaptic integrating (survival or functional neurogenesis) (Gage, 2000, Dayer, et al., 2003). A well-designed BrdU injection paradigm is able to specifically address these distinctive neurogenic processes. By doing so, we have found that neurogenesis is dynamically involved in neurodegeneration. This dynamic involvement can be understood at two levels. First, the effect of neurodegeneration on neurogenesis is neurodegenerative stage-dependent. At ESND, the effects on all neurogenic stages including proliferation, differentiation, new neuron generation, and survival were robust. At LSND, however, these effects were dramatically decreased, indicating that the overall effect of the neurodegeneration on neurogenesis is decreased following neurodegenerative progression. As neurogenesis is down regulated by aging (Amrein, et al., 2004, Bondolfi, et al., 2004, Verret, et al., 2007), it is not clear whether this shift is or is not purely due to the effect of aging on neurogenesis. Second, the effect of neurodegeneration on neurogenesis is neurogenic stage-specific. At ESND, the effects on all of neurogenic stages were enhanced. At LSND, however, although the effects on proliferation, differentiation, and new neuron generation were still observable, the effect on survival was significantly impaired so that there was no significantly increased neurogenesis in the dentate gyrus of PS1/PS2-KO mice. The mechanism for this neurogenic stage-specific effect is still not clear. It might be possible that the changes in some microenvironment in neurons following the neurodegenerative progression are not favorable for the survival of newly generated neurons. This remains a particularly interesting topic for the further studies, such as to determine whether the expression of BDNF is involved in this neurodegenerative stage-dependent neurogenesis or other epigenetic events are involved.
It has been demonstrated that many newly generated neurons die within a couple of weeks after the birth and only about 20–40% of these new neurons can eventually survive over this time duration (Kempermann, et al., 2003). The number of new neurons that can survive over four weeks is relatively stable and thus this time scale has been considered as an essential criterion to judge whether new neurons have the potential to develop into functional units in the neuronal network. Indeed, there is evidence that the survival of new neurons over four weeks makes them express electrophysiological properties similar to other granule cells in the dentate gyrus (van Praag, et al., 2000, Dayer, et al., 2003, Overstreet-Wadiche, et al., 2006). Interestingly, we have found that neurodegeneration-triggered new neurons in PS1/PS2-KO mice survive over a period of four weeks at ESND, implying that this neurogenesis might be potentially functional. As no significantly increased neurogenesis was observed at LSND, this potential is lost following the neurodegenerative progression. Another important criterion to show the potentially functional neurogenesis is new neuron migration. In the dentate gyrus, new neurons are produced via mitosis of neural progenitor cells, which are located in the SGZ of the dentate gyrus (Gage, 2000, Ming and Song, 2005, Lledo, et al., 2006). No progenitor cells are found in the other places of the hippocampus. Thus, the appearance of new neurons in other hippocampal regions rather than the SGZ indicates that these newly generated neurons are able to migrate. The target areas for this migration include the GCL and hilus region, where the young neurons develop into mature neurons and integrate into local neuronal network through axon/dendrite targeting or synaptic formation. As shown in Fig. 5, newly generated neurons in the dentate gyrus of PS1/PS2-KO mice at ESND migrated into both the GCL and hilus region. However, there is no significant difference in the migration rate between control and PS1/PS2-KO mice (Fig. 5D), indicating that the neurodegeneration itself does not affect migration, once these new neurons have been generated. These results indicate that neurogenesis-triggered by neurodegeneration has the basic properties for functional neurogenesis.
The most important finding in this study is that the dynamic involvement of neurogenesis in the dentate gyrus is functionally correlated with the severity of neuronal loss in this brain region. At ESNG, a highly significant neuronal loss (24% neurons lost, p < 0.01) was found in the cortex (Fig. 1G), whereas no observable neurogenesis could be detected. At the same stage, however, a significant neurogenesis was found in the dentate gyrus, where only modest neuronal loss (8% neurons lost) could be detected in PS1/PS2-KO mice, in comparison with control mice. It should be mentioned that in the dentate gyrus, it is estimated that about 800–1,600 new neurons are generated everyday in a normal mouse (Cameron and McKay, 2001). Given the increased scale (2–3 fold) of newly generated neurons in our PS1/PS2-KO mice at ESND, it is reasonably to speculated that the increased neurogenesis in the dentate gyrus may efficiently, although not completely, compensate for the neuronal loss in this region. This speculation has been further supported by the observations of neurogenesis at LSND, at which a significant neuronal loss (36% neurons lost, p < 0.01) was found. At this stage, no significant neurogenesis could be detected. These results indicate that adult neurogenesis is not only dynamically involved in neurodegeneration, but also functionally correlated to the severity of neuronal loss in the dentate gyrus of the neurodegenerative animal model.
Conclusion
In this study, we have produced a robust neurodegeneration model in the mouse. Although this mouse strain is not a comprehensive AD model, it expresses many features of AD neurodegeneration. Especially, neuronal loss in the brain is comparable to AD brain. With this mouse model, we have demonstrated that neurodegeneration trigger robust adult neurogenesis in the dentate gyrus. This effect is dynamically associated with different neurodegenerative stages. Most importantly, the dynamic involvement of neurogenesis in the dentate gyrus is correlated to the degree of neuronal loss, providing the first evidence that the endogenous neurogenesis may functionally work as a self-repairing mechanism to compensate, at least partially, for the neuronal loss-induced by neurodegeneration. As the hippocampus is one of the most vulnerable regions in the brain to AD neurodegeneration and this vulnerability may contribute to the earliest symptom, learning and memory deficit, in AD, the demonstration of the dynamic changes of neurogenesis in this region suggests that to enhance the endogenous adult neurogenesis at ESND may be a valuable strategy to delay or stop the neurodegenerative progression (Becker, et al., 2007, Tchantchou, et al., 2007).
Acknowledgments
We greatly thank Dr. Vytautas P. Bindokas at the Department of Pharmacology, Physiology, and Neurobiology, and the University core facility for confocal microscope, the University of Chicago, in helping collecting data and data analyses. We also thank Dr. Elliot S. Gershon at the University of Chicago for the stimulating discussion and Dr. George Lagan and his staff in the animal facility for the excellent care of our animals. This study was supported by grants from NIMH/NIH (MH066243), Alzheimer’s Association (NIRG-02-4368), NSF (0213112), Brain Research Foundation (Chicago), Louis Block Foundation, and NARSAD, all to YPT.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aimone JB, Wiles J, Gage FH. Potential role for adult neurogenesis in the encoding of time in new memories. Nat Neurosci. 2006;9:723–727. doi: 10.1038/nn1707. [DOI] [PubMed] [Google Scholar]
- Altman J, Das GD. Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol. 1965;124:319–335. doi: 10.1002/cne.901240303. [DOI] [PubMed] [Google Scholar]
- Amrein I, Slomianka L, Poletaeva II, Bologova NV, Lipp HP. Marked species and age-dependent differences in cell proliferation and neurogenesis in the hippocampus of wild-living rodents. Hippocampus. 2004;14:1000–1010. doi: 10.1002/hipo.20018. [DOI] [PubMed] [Google Scholar]
- Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- Barber R, McKeith IG, Ballard C, Gholkar A, O’Brien JT. A comparison of medial and lateral temporal lobe atrophy in dementia with Lewy bodies and Alzheimer’s disease: magnetic resonance imaging volumetric study. Dement Geriatr Cogn Disord. 2001;12:198–205. doi: 10.1159/000051258. [DOI] [PubMed] [Google Scholar]
- Becker M, Lavie V, Solomon B. Stimulation of endogenous neurogenesis by anti-EFRH immunization in a transgenic mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2007;104:1691–1696. doi: 10.1073/pnas.0610180104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondareff W, Mountjoy CQ, Roth M. Loss of neurons of origin of the adrenergic projection to cerebral cortex (nucleus locus ceruleus) in senile dementia. Neurology. 1982;32:164–168. doi: 10.1212/wnl.32.2.164. [DOI] [PubMed] [Google Scholar]
- Bondolfi L, Ermini F, Long JM, Ingram DK, Jucker M. Impact of age and caloric restriction on neurogenesis in the dentate gyrus of C57BL/6 mice. Neurobiol Aging. 2004;25:333–340. doi: 10.1016/S0197-4580(03)00083-6. [DOI] [PubMed] [Google Scholar]
- Bussiere T, Giannakopoulos P, Bouras C, Perl DP, Morrison JH, Hof PR. Progressive degeneration of nonphosphorylated neurofilament protein-enriched pyramidal neurons predicts cognitive impairment in Alzheimer’s disease: stereologic analysis of prefrontal cortex area 9. J Comp Neurol. 2003;463:281–302. doi: 10.1002/cne.10760. [DOI] [PubMed] [Google Scholar]
- Cameron HA, McKay RD. Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. J Comp Neurol. 2001;435:406–417. doi: 10.1002/cne.1040. [DOI] [PubMed] [Google Scholar]
- Chevallier NL, Soriano S, Kang DE, Masliah E, Hu G, Koo EH. Perturbed neurogenesis in the adult hippocampus associated with presenilin-1 A246E mutation. Am J Pathol. 2005;167:151–159. doi: 10.1016/S0002-9440(10)62962-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chui DH, Tanahashi H, Ozawa K, Ikeda S, Checler F, Ueda O, Suzuki H, Araki W, Inoue H, Shirotani K, Takahashi K, Gallyas F, Tabira T. Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat Med. 1999;5:560–564. doi: 10.1038/8438. [DOI] [PubMed] [Google Scholar]
- Curtis MA, Penney EB, Pearson AG, van Roon-Mom WM, Butterworth NJ, Dragunow M, Connor B, Faull RL. Increased cell proliferation and neurogenesis in the adult human Huntington’s disease brain. Proc Natl Acad Sci U S A. 2003;100:9023–9027. doi: 10.1073/pnas.1532244100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayer AG, Ford AA, Cleaver KM, Yassaee M, Cameron HA. Short-term and long-term survival of new neurons in the rat dentate gyrus. J Comp Neurol. 2003;460:563–572. doi: 10.1002/cne.10675. [DOI] [PubMed] [Google Scholar]
- Donovan MH, Yazdani U, Norris RD, Games D, German DC, Eisch AJ. Decreased adult hippocampal neurogenesis in the PDAPP mouse model of Alzheimer’s disease. J Comp Neurol. 2006;495:70–83. doi: 10.1002/cne.20840. [DOI] [PubMed] [Google Scholar]
- Donoviel DB, Hadjantonakis AK, Ikeda M, Zheng H, Hyslop PS, Bernstein A. Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev. 1999;13:2801–2810. doi: 10.1101/gad.13.21.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman RS. Depression: a case of neuronal life and death? Biol Psychiatry. 2004;56:140–145. doi: 10.1016/j.biopsych.2004.02.033. [DOI] [PubMed] [Google Scholar]
- Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH. Neurogenesis in the adult human hippocampus. Nat Med. 1998;4:1313–1317. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- Feng R, Rampon C, Tang YP, Shrom D, Jin J, Kyin M, Sopher B, Miller MW, Ware CB, Martin GM, Kim SH, Langdon RB, Sisodia SS, Tsien JZ. Deficient neurogenesis in forebrain-specific presenilin-1 knockout mice is associated with reduced clearance of hippocampal memory traces. Neuron. 2001;32:911–926. doi: 10.1016/s0896-6273(01)00523-2. [DOI] [PubMed] [Google Scholar]
- Feng R, Wang H, Wang J, Shrom D, Zeng X, Tsien JZ. Forebrain degeneration and ventricle enlargement caused by double knockout of Alzheimer’s presenilin-1 and presenilin-2. Proc Natl Acad Sci U S A. 2004;101:8162–8167. doi: 10.1073/pnas.0402733101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage FH. Mammalian neural stem cells. Science. 2000;287:1433–1438. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- Gage FH, Kempermann G, Palmer TD, Peterson DA, Ray J. Multipotent progenitor cells in the adult dentate gyrus. J Neurobiol. 1998;36:249–266. doi: 10.1002/(sici)1097-4695(199808)36:2<249::aid-neu11>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- Gould E, Reeves AJ, Graziano MS, Gross CG. Neurogenesis in the neocortex of adult primates. Science. 1999;286:548–552. doi: 10.1126/science.286.5439.548. [DOI] [PubMed] [Google Scholar]
- Greenberg DA, Jin K. Regenerating the brain. Int Rev Neurobiol. 2007;77:1–29. doi: 10.1016/S0074-7742(06)77001-5. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Nath A, Chan SL, Borchard AC, Rao MS, Mattson MP. Disruption of neurogenesis by amyloid beta-peptide, and perturbed neural progenitor cell homeostasis, in models of Alzheimer’s disease. J Neurochem. 2002;83:1509–1524. doi: 10.1046/j.1471-4159.2002.01267.x. [DOI] [PubMed] [Google Scholar]
- Hayes NL, Nowakowski RS. Dynamics of cell proliferation in the adult dentate gyrus of two inbred strains of mice. Brain Res Dev Brain Res. 2002;134:77–85. doi: 10.1016/s0165-3806(01)00324-8. [DOI] [PubMed] [Google Scholar]
- Heston LL, Mastri AR, Anderson VE, White J. Dementia of the Alzheimer type. Clinical genetics, natural history, and associated conditions. Arch Gen Psychiatry. 1981;38:1085–1090. doi: 10.1001/archpsyc.1981.01780350019001. [DOI] [PubMed] [Google Scholar]
- Hoglinger GU, Rizk P, Muriel MP, Duyckaerts C, Oertel WH, Caille I, Hirsch EC. Dopamine depletion impairs precursor cell proliferation in Parkinson disease. Nat Neurosci. 2004;7:726–735. doi: 10.1038/nn1265. [DOI] [PubMed] [Google Scholar]
- Itoh T, Satou T, Hashimoto S, Ito H. Isolation of neural stem cells from damaged rat cerebral cortex after traumatic brain injury. Neuroreport. 2005;16:1687–1691. doi: 10.1097/01.wnr.0000183330.44112.ab. [DOI] [PubMed] [Google Scholar]
- Jin K, Galvan V, Xie L, Mao XO, Gorostiza OF, Bredesen DE, Greenberg DA. Enhanced neurogenesis in Alzheimer’s disease transgenic (PDGF-APPSw,Ind) mice. Proc Natl Acad Sci U S A. 2004a;101:13363–13367. doi: 10.1073/pnas.0403678101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC, Greenberg DA. Increased hippocampal neurogenesis in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2004b;101:343–347. doi: 10.1073/pnas.2634794100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempermann G, Gast D, Kronenberg G, Yamaguchi M, Gage FH. Early determination and long-term persistence of adult-generated new neurons in the hippocampus of mice. Development. 2003;130:391–399. doi: 10.1242/dev.00203. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Kuhn HG, Gage FH. More hippocampal neurons in adult mice living in an enriched environment. Nature. 1997;386:493–495. doi: 10.1038/386493a0. [DOI] [PubMed] [Google Scholar]
- Kokaia Z, Lindvall O. Neurogenesis after ischaemic brain insults. Curr Opin Neurobiol. 2003;13:127–132. doi: 10.1016/s0959-4388(03)00017-5. [DOI] [PubMed] [Google Scholar]
- Lledo PM, Alonso M, Grubb MS. Adult neurogenesis and functional plasticity in neuronal circuits. Nat Rev Neurosci. 2006;7:179–193. doi: 10.1038/nrn1867. [DOI] [PubMed] [Google Scholar]
- Ming GL, Song H. Adult neurogenesis in the mammalian central nervous system. Annu Rev Neurosci. 2005;28:223–250. doi: 10.1146/annurev.neuro.28.051804.101459. [DOI] [PubMed] [Google Scholar]
- Mirescu C, Gould E. Stress and adult neurogenesis. Hippocampus. 2006;16:233–238. doi: 10.1002/hipo.20155. [DOI] [PubMed] [Google Scholar]
- Overstreet-Wadiche LS, Bromberg DA, Bensen AL, Westbrook GL. Seizures accelerate functional integration of adult-generated granule cells. J Neurosci. 2006;26:4095–4103. doi: 10.1523/JNEUROSCI.5508-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu L, Zhu C, Wang X, Xu F, Eriksson PS, Nilsson M, Cooper-Kuhn CM, Kuhn HG, Blomgren K. Less neurogenesis and inflammation in the immature than in the juvenile brain after cerebral hypoxia-ischemia. J Cereb Blood Flow Metab. 2007;27:785–794. doi: 10.1038/sj.jcbfm.9600385. [DOI] [PubMed] [Google Scholar]
- Rampon C, Tang YP, Goodhouse J, Shimizu E, Kyin M, Tsien JZ. Enrichment induces structural changes and recovery from nonspatial memory deficits in CA1 NMDAR1-knockout mice. Nat Neurosci. 2000;3:238–244. doi: 10.1038/72945. [DOI] [PubMed] [Google Scholar]
- Rice AC, Khaldi A, Harvey HB, Salman NJ, White F, Fillmore H, Bullock MR. Proliferation and neuronal differentiation of mitotically active cells following traumatic brain injury. Exp Neurol. 2003;183:406–417. doi: 10.1016/s0014-4886(03)00241-3. [DOI] [PubMed] [Google Scholar]
- Samuel W, Terry RD, DeTeresa R, Butters N, Masliah E. Clinical correlates of cortical and nucleus basalis pathology in Alzheimer dementia. Arch Neurol. 1994;51:772–778. doi: 10.1001/archneur.1994.00540200048015. [DOI] [PubMed] [Google Scholar]
- Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, Weisstaub N, Lee J, Duman R, Arancio O, Belzung C, Hen R. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 2003;301:805–809. doi: 10.1126/science.1083328. [DOI] [PubMed] [Google Scholar]
- Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, Chattarji S, Kelleher RJ, 3rd, Kandel ER, Duff K, Kirkwood A, Shen J. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42:23–36. doi: 10.1016/s0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Miesegaes G, Beylin A, Zhao M, Rydel T, Gould E. Neurogenesis in the adult is involved in the formation of trace memories. Nature. 2001;410:372–376. doi: 10.1038/35066584. [DOI] [PubMed] [Google Scholar]
- Tchantchou F, Xu Y, Wu Y, Christen Y, Luo Y. EGb 761 enhances adult hippocampal neurogenesis and phosphorylation of CREB in transgenic mouse model of Alzheimer’s disease. Faseb J. 2007 doi: 10.1096/fj.06-7649com. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Hayashi KM, De Zubicaray GI, Janke AL, Rose SE, Semple J, Hong MS, Herman DH, Gravano D, Doddrell DM, Toga AW. Mapping hippocampal and ventricular change in Alzheimer disease. Neuroimage. 2004;22:1754–1766. doi: 10.1016/j.neuroimage.2004.03.040. [DOI] [PubMed] [Google Scholar]
- van Praag H, Kempermann G, Gage FH. Neural consequences of environmental enrichment. Nat Rev Neurosci. 2000;1:191–198. doi: 10.1038/35044558. [DOI] [PubMed] [Google Scholar]
- Verret L, Trouche S, Zerwas M, Rampon C. Hippocampal neurogenesis during normal and pathological aging. Psychoneuroendocrinology. 2007;27:6771–6780. doi: 10.1016/j.psyneuen.2007.04.014. [DOI] [PubMed] [Google Scholar]
- Wang R, Dineley KT, Sweatt JD, Zheng H. Presenilin 1 familial Alzheimer’s disease mutation leads to defective associative learning and impaired adult neurogenesis. Neuroscience. 2004;126:305–312. doi: 10.1016/j.neuroscience.2004.03.048. [DOI] [PubMed] [Google Scholar]
- Warner-Schmidt JL, Duman RS. Hippocampal neurogenesis: opposing effects of stress and antidepressant treatment. Hippocampus. 2006;16:239–249. doi: 10.1002/hipo.20156. [DOI] [PubMed] [Google Scholar]
- Wen PH, Hof PR, Chen X, Gluck K, Austin G, Younkin SG, Younkin LH, DeGasperi R, Gama Sosa MA, Robakis NK, Haroutunian V, Elder GA. The presenilin-1 familial Alzheimer disease mutant P117L impairs neurogenesis in the hippocampus of adult mice. Exp Neurol. 2004;188:224–237. doi: 10.1016/j.expneurol.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR. Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215:1237–1239. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- Zhang C, McNeil E, Dressler L, Siman R. Long-lasting impairment in hippocampal neurogenesis associated with amyloid deposition in a knock-in mouse model of familial Alzheimer’s disease. Exp Neurol. 2007;204:77–87. doi: 10.1016/j.expneurol.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]