

Abstract
Background/Aim:
The kd/kd mouse spontaneously develops severe and progressive nephritis leading to renal failure, characterized by cellular infiltration, tubular destruction and glomerular sclerosis. Recent identification of the mutant gene and the observation that podocytes are affected, led to the hypothesis that there are primary renal epithelial cell defects in this strain.
Methods:
Clinical and pathological signs of disease in a large cohort of kd/kd mice were studied by light microscopy, electron microscopy, and biochemical analyses of serum and urine at early stages of disease. Special attention was paid to mice under 140 days of age that had normal blood urea nitrogen (BUN) levels, but had developed albuminuria.
Results:
Although overt glomerular abnormalities are commonly observed either coincident with or after tubulointerstitial nephritis, we now report that albuminuria and visceral epithelial abnormalities, including hyperplasia and podocyte effacement may occur before the onset of either elevated BUN levels or severe interstitial nephritis, and this is accompanied by biochemical perturbations in serum typical of the nephrotic syndrome.
Conclusions:
The results suggest that the defect in kd/kd mice primarily affects both the tubular and glomerular visceral epithelium. The tubular epithelial defect triggers autoimmune interstitial nephritis, whereas a defect in podocytes leads to proteinuria and glomerulosclerosis. Thus, a single mitochondrial abnormality may result in differences in disease expression that vary with the type of epithelial cells. It is likely that the mitochrondrial perturbations in glomerular and tubular epithelia act in concert, through activation of different pathologic pathways, to accelerate disease progression leading to renal failure.
Keywords: Albuminuria, Gene, Glomerulus, Nephritis, Podocyte
Introduction
The kd/kd mouse is homozygous for the ‘kidney disease’ mutation, which occurred spontaneously in a CBA/CaH colony. Mutant homozygotes are apparently healthy for the first 8 weeks of life, but eventually die of end-stage renal disease [1]. Histological examination of kidneys at earlier time points reveals a mononuclear cell infiltrate and tubular dilatation with proteinaceous casts in cortical areas, which with time expands throughout the entire kidney and leads to renal failure [2-4]. The disease can be transferred from kd/kd mice by bone marrow cells to lethally irradiated recipients [2], and there is evidence for an MHC-restricted effector T cell response directed against a tubular basement membrane (TBM) antigen [3]. Nevertheless, multiple autoimmune pathways are involved, as doubly mutant mice with both the Rag-1−/− and kd/kd genotypes develop nephritis as readily and severely as kd/kd controls [5], indicating that neither functional B nor T cells are required for this phenotype. Glomerular abnormalities typically occur as kd/kd mice age, and this has been attributed to secondary consequences of renal failure and severe interstitial disease [2].
Recent elucidation that the kd allele has a missense mutation in a gene encoding a prenyltransferase-like mitochrondrial protein provided further pathogenic clues, as both renal tubular epithelial cells and hepatocytes were shown to have mitochrondrial abnormalities [6]. These findings raised the possibility that the autoimmune response was secondary to a primary epithelial defect and that other epithelia could be involved. In this regard, podocyte abnormalities have been observed late in disease [7], though it was uncertain whether this was due to either a primary glomerular epithelial defect or secondary to the pathologic events resulting from hyperfiltration and advancing renal failure. We now report that mutant mice develop visceral epithelial abnormalities, characterized by hyperplasia and effacement, associated with proteinuria. Furthermore, in some animals the pathology is observed prior to either elevation of the BUN levels or histologic evidence of severe interstitial disease. The results indicate that the podocytes are primarily affected by the kd/kd genotype independently of the interstitial nephritis, and that they contribute to pathogenesis of nephritis and disease progression.
Materials and Methods
Mice
All experiments were conducted according to NIH and institutional guidelines. The CBA/CaH-kd line no longer exists, so a new line was derived by transferring the kd allele to the C57BL/6J (B6) background with selection for closely linked markers [6, 8]. These mice develop severe interstitial nephritis and die from renal failure according to a pattern that appears indistinguishable from that of their CBA/CaH-kd/kd counterparts. Homozygous mice obtained after 12 generations of backcrossing are designated B6.CBACaH(CAST)-kd/Upa (B6.kd/kd). The B6.kd/kd,Rag-1−/− double mutants were previously described [5].
Mice were maintained in a 12-hour light-dark cycle at 22°C, provided water ad libitum, and were fed a standard rodent chow (LabDiet, Richmond, Ind., USA, 5001; 4.5% fat, 49.9% carbohydrate, 23.4% protein; 4 kcal/g).
Histology
Mutant and control mice were sacrificed, and their kidneys were evaluated for glomerular and tubular pathology. The frequencies of global and segmental glomerulosclerosis and of glomerular visceral and parietal epithelial hyperplasia were evaluated along with tubulointerstitial atrophy on a 0–4+ scale by a single observer (J.E.T.) who was blinded to the origin of the specimens. For this purpose, 50 glomeruli from each animal were evaluated and the frequency of global and segmental glomerulosclerosis, glomerular epithelial hyperplasia, tubular atrophy and tubular inflammation were determined.
For electron microscopy, tissue samples were fixed and processed as previously described [6]. All sections were examined in a JEOL 1010 electron microscope, and digital images were recorded with a Hamamatsu camera system.
Chemistries
Serum triglyceride, cholesterol and nonesterified fatty acid (NEFA) were measured using colorimetric assays (Wako Chemicals, Richmond, Va., USA; Stanbio Laboratories, Boerne, Tex., USA), as described [9]. Pooled serum samples from wild-type and kd/kd mice were subjected to fast-performance liquid chromatography (FPLC) analysis [10]. Urine was collected from mice maintained for 24 h in metabolic cages, and albuminuria was measured by quantitative ELISA (Bethyl Laboratories, Inc., Montgomery, Tex., USA).
Statistics
The lipid values showed a normal distribution, and were analyzed by the t test. As the urine albumin measurements were not normally distributed, they were analyzed using a nonparametric method (the two-tailed Mann Whitney U test).
Results
kd/kd Mice Develop Biochemical Features Typical of the Nephrotic Syndrome in Late Stages of Disease
As shown in figure 1, albuminuria rose dramatically in the B6.kd/kd mice as the mice aged (fig. 1a). There was no significant difference between B6 and B6.kd/kd in the urine albumin before 90 days of age (p = 0.125), but in the age group of 90–140 days, the difference was significant (p < 0.05), and it became even more significant over 140 days of age (p < 0.01). A decrease in serum albumin levels occurred in the B6.kd/kd mice by 150 days (fig. 1b; p = 0.014). In mice with more advanced disease, lipid abnormalities, which are typical of the nephrotic syndrome in mice, were observed. In the mice shown in figure 2 (152 ± 16 days of age), serum triglyceride and cholesterol levels were significantly higher in B6.kd/kd mice than in B6 controls.
Fig. 1.

a Urine albumin in mg/24 h from B6 vs. B6.kd/kd. b Serum albumin from B6 vs. B6.kd/kd.
Fig. 2.
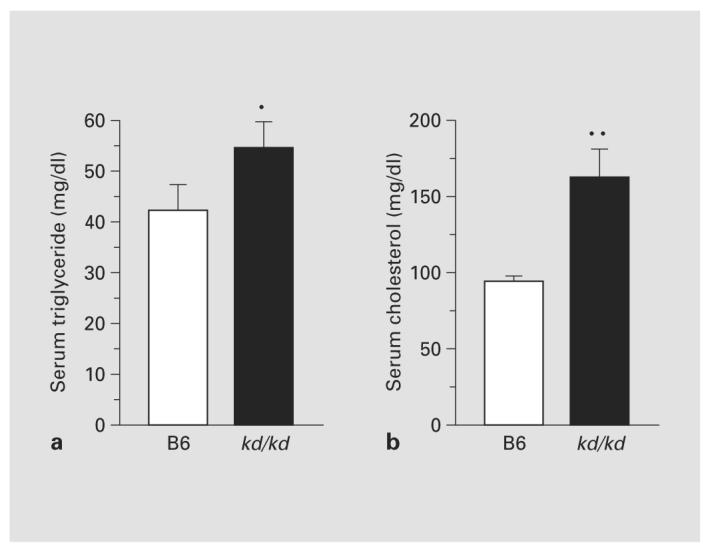
Serum triglyceride a and cholesterol b levels in chow-fed mice. Data are mean ± SEM, n = 5. * p < 0.05; ** p < 0.001.
The rise in cholesterol was confirmed in FPLC, showing increased very low density lipoprotein (VLDL), intermediate density lipoprotein (IDL) and high density lipoprotein (HDL) levels in kd/kd mice (fig. 3). The greatest increase is in the HDL-bound fraction, typical of nephrotic mice. This is in contrast to what would be observed in humans, as the lipid profile of mice is quite different from that of humans, since cholesterol circulates mostly as HDL cholesterol in mice. Nonesterified fatty acids were not different between wild-type and kd/kd mice (data not shown).
Fig. 3.
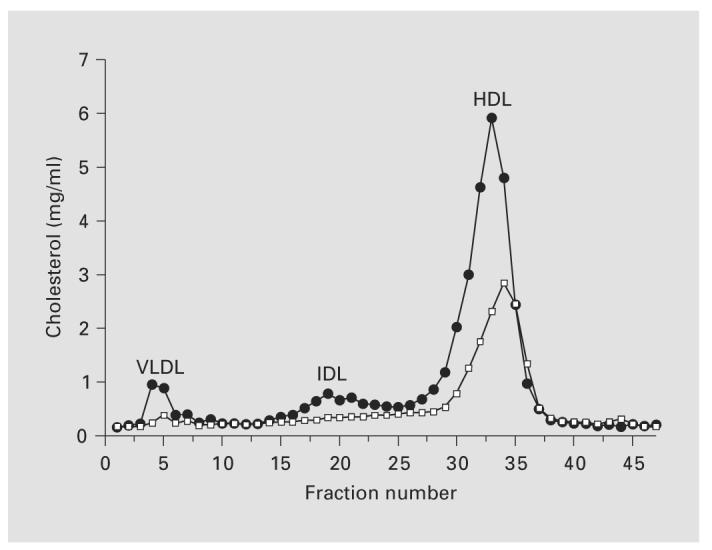
FPLC analysis of lipoproteins in plasma from B6 and B6.kd/kd mice maintained on a normal chow. Black circles, B6.kd/kd; open squares, B6.
Proteinuria and Podocyte Effacement Occur in Some kd/kd Mice prior to Elevated BUN Levels
As kd/kd mice age there is severe interstitial nephritis, and this is associated with elevated BUN levels greater than 100 mg/dl (data not shown). However, it appeared that some mice had glomerular pathology prior to the onset of severe interstitial disease, so we investigated a cohort of animals in the age range in which this appeared to occur. In table 1 are the results from mice of four different genotypes, all of which were within 90–140 days of age. All of the kd/kd mice had BUN levels that were within the normal range observed among the B6 controls (less than 35 mg/dl). However, both groups of mice with homozygous mutant genotypes had significant albuminuria in comparison to the B6 controls (p = 0.004 for B6.kd/kd; p = 0.009 for B6.kd/kd,Rag-1−/−; two-tailed Mann-Whitney U test). Of particular interest, all of the B6.kd/kd,Rag-1−/− developed proteinuria, and 4 of the 5 B6.kd/+,Rag-1−/− heterozygote mice had proteinuria outside of the normal range, despite the absence of TCR-αβ+ T cells.
Table 1.
Serum triglycerides, cholesterol, BUN, and histological signs at 90–140 days of age
| Mouse | Age days |
Triglyceride mg/dl |
Cholesterol mg/dl |
BUN mg/dl |
Urine albumin mg/24 h |
GGSa % |
SGSb % |
V/P Epithelial hyperplasiac, % |
Interstitial fibrosisd |
Inflammatione |
|---|---|---|---|---|---|---|---|---|---|---|
| B6 | ||||||||||
| 1 | 91 | 34 | 138 | 12 | 0.2 | 0 | 0 | 0 | 0 | 0 |
| 2 | 91 | 37 | 120 | 15 | 0.1 | 0 | 0 | 0 | 0 | 0 |
| 3 | 110 | 50 | 120 | 32 | 0.2 | 0 | 0 | 0 | 0 | 0 |
| 4 | 110 | 29 | 117 | 21 | 0.4 | 0 | 0 | 0 | 0 | 0 |
| 5 | 114 | 40 | 112 | 25 | 0.1 | 0 | 0 | 0 | 0 | 0 |
| B6.kd/kd | ||||||||||
| 6 | 96 | 41 | 94 | 23 | 3.8 | 2 | 4 | 4 | 2 | 2 |
| 7 | 102 | 74 | 135 | 26 | 3.5 | 0 | 0 | 6 | 2 | 2 |
| 8 | 102 | 61 | 95 | 21 | 3.4 | 0 | 2 | 4 | 2 | 2 |
| 9 | 116 | 45 | 106 | 15 | 2.0 | 0 | 0 | 2 | 1 | 1 |
| 10 | 116 | 40 | 102 | 29 | 3.8 | 0 | 0 | 2 | 1 | 1 |
| 11 | 125 | 39 | 99 | 29 | 4.0 | 0 | 0 | 6 | 2 | 1 |
| 12 | 132 | 45 | 121 | 26 | 21.0 | 0 | 0 | 2 | 1 | 1 |
| B6.kd/+,Rag-1−/− | ||||||||||
| 13 | 131 | 37 | 81 | 22 | 1.9 | 0 | 0 | 6 | 1 | 1 |
| 14 | 133 | 47 | 97 | 17 | 0.2 | 0 | 0 | 0 | 0 | 0 |
| 15 | 131 | 44 | 92 | 21 | 9.8 | 0 | 0 | 2 | 1 | 1 |
| 16 | 131 | 49 | 94 | 19 | 5.4 | 0 | 0 | 0 | 0 | 0 |
| 17 | 131 | 46 | 94 | 19 | 3.7 | 0 | 0 | 0 | 0 | 0 |
| B6.kd/kd,Rag-1−/− | ||||||||||
| 18 | 122 | 35 | 96 | 19 | 12.7 | 0 | 0 | 4 | 2 | 2 |
| 19 | 122 | 34 | 95 | 23 | 2.7 | 0 | 0 | 8 | 2 | 2 |
| 20 | 130 | 36 | 112 | 18 | 13.5 | 0 | 0 | 0 | 2 | 2 |
| 21 | 133 | 65 | 118 | 21 | 8.3 | 0 | 0 | 14 | 2 | 2 |
| 22 | 133 | 46 | 176 | 22 | 19.5 | 0 | 0 | 2 | 2 | 2 |
Global glomerulosclerosis.
Segmental glomerulosclerosis.
Visceral/parietal epithelial hyperplasia.
Amount of interstitial fibrosis and tubular atrophy as % of cortex: Score legend: 0 = no feature, 1 = <2%, 2 = 2–10%, 3 = 11–50%, 4 = >50%.
Amount of inflammation as % of cortex; same scale as above.
In this subgroup, there was glomerular epithelial pathology without extensive interstitial nephritis. By light microscopy, glomerular visceral and parietal epithelial hyperplasias were observed, most frequently at the corticomedullary junction. All of the B6.kd/kd mice had visceral/parietal (V/P) epithelial hyperplasia as well as interstitial fibrosis and cortical inflammation; all 5 of the B6.kd/kd,Rag-1−/− mice had interstitial fibrosis and cortical inflammation, and 4 also had V/P epithelial hyperplasia; 2 of the B6.kd/+,Rag-1−/− mice had V/P epithelial hyperplasia, interstitial fibrosis, and cortical inflammation (table 1). Furthermore, there was diffuse effacement of foot processes along with visceral epithelial hyperplasia, despite the paucity of interstitial infiltrates. A representative section is shown in figure 4.
Fig. 4.
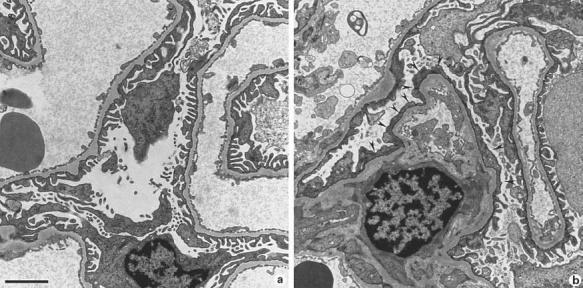
a Normal visceral epithelial cells from a 110 day old B6 mouse. b Diffuse effacement (arrowheads) of visceral epithelial cells from a 102-day-old B6.kd/kd mouse with proteinuria and normal BUN. × 10,000. Scale bar = 2 μm.
Discussion
The current results demonstrate that the genetic defect in kd/kd mice, previously shown to affect renal tubular epithelium [6], also involves glomerular epithelial cells, with mutant mice developing signs of the nephrotic syndrome [11]. Support for a primary glomerular epithelial defect is provided by the observation that in relatively early stages of disease (90–140 days of age), when all mice have BUN levels within the normal range, some kd/kd mice have heavy albuminuria, which is a sign of glomerular epithelial injury [12]. Furthermore, in this subgroup, there were visceral epithelial abnormalities, which were present prior to the occurrence of severe interstitial disease.
With advancing disease, the podocytes in kd/kd kidneys show de novo expression of cyclin D1, Ki-67 and desmin, with loss of synaptopodin and WT-1 expression, and this is associated with typical pathologic features of focal segmental glomerulosclerosis [7]. Nevertheless, in the context of the present observations, it is highly likely that mitochondrial abnormalities in kd/kd podocytes per se accelerate disease progression, even in mice in which interstitial nephritis may have been the predominant initial feature. In this situation, glomerular proteinuria would also be expected to contribute to ongoing interstitial inflammation, further accelerating disease progression.
The lack of glomerular inflammation early in the course of disease is consistent with the initial pathology in podocytes. This resulted in epithelial hyperplasia, effacement of foot processes and proteinuria. Presumably immune cells cannot traverse the glomerular capillary wall at this stage, since its integrity is intact. Nevertheless, with more advanced nephritis and disruption of glomerular and capsular basement membranes, there was cellular infiltration, with overt glomerulonephritis. With overwhelming interstitial inflammation and heavy proteinuria, cells either migrate in from the inflamed interstitium and/or are recruited from the bloodstream into the more severely injured glomeruli.
The immunological contribution to this disease was further dissected by placing the kd/kd genotype on the Rag-1−/− background. Although these mice have no functional CD4+ or CD8+ T cells, they nevertheless develop the phenotype characteristic of kd/kd mice [5]. Particularly relevant to the present conclusions, all of the B6.kd/kd,Rag-1−/− mice developed albuminuria and visceral epithelial abnormalities. In fact, albuminuria, V/P epithelial hyperplasia, interstitial fibrosis and cortical inflammation were even observed in most of the B6.kd/+, Rag-1−/− heterozygotes (table 1). The observation that kd/kd mice, and even most kd/+ heterozygotes, with a Rag-1−/− background develop nephritis without a functional system of adaptive immunity supports the importance of an initial epithelial defect, unrelated to the immune system.
The development of severe hyperlipidemia most likely resulted from the nephrotic syndrome, since it was only observed in animals with proteinuria. The lipid profile of these animals is typical of that observed in rodents with the nephrotic syndrome, for example, in nephrotoxic nephritis [13]. Nevertheless, we cannot exclude a contribution from the prenylation defect per se, and this will require future study to sort out such a potential contribution.
How a mutation in the PLMP gene leads to this cascade of events is not clear. Defects in the mitochondria of renal tubular epithelium [6] as well as the podocytes [7] have been demonstrated, so it is tempting to speculate that these defects lead to apoptosis and/or necrosis, which, in turn, stimulate either interstitial inflammation or proteinuria. Differences in pathology in these compartments may be related to access of inflammatory cells and functional properties of the region within the kidney. For example, early in disease, apoptotic tubular cells may more readily recruit inflammatory cells, whereas overt glomerulonephritis may not occur until there is either disruption of the glomerular capillary wall or cells migrate across Bowman's capsule from the interstitium. However, this has not been established experimentally, and targeted deletion studies will be needed to resolve this issue.
Apoptotic cell death does not normally induce an inflammatory response in contrast to necrosis [14], but apoptosis and necrosis are at opposite ends of a continuum, and mitochondrial permeability transition can be involved in both apoptosis and necrosis. Massive induction of permeability transition can lead to primary necrosis before apoptogenic proteases can come into action [15]. If bioenergetic catastrophe occurred in renal epithelial cells of kd/kd mice under appropriate conditions, cells that became necrotic could induce an inflammatory response. By contrast, renal epithelial cells not confined by special constraints could become detached from the basement membrane and lost in the urine, with the reduction of podocytes leading to proteinuria [16].
Acknowledgements
This work was supported by National Institutes of Health grants RO1-DK55852 (D.L.G.), P01-DK49210 (R.S.A.). Expertise for lipid chemistry was provided by the Penn DERC Mouse Phenotyping Core (P30–19525). We thank the members of the Morphology Core for Molecular Studies in Digestive and Liver Diseases (P30-DK50306) for histologic preparations.
References
- 1.Lyon MF, Hulse EV. An inherited kidney disease of mice resembling human nephronophthisis. J Med Genet. 1971;8:41–48. doi: 10.1136/jmg.8.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neilson EG, McCafferty E, Feldman A, Clayman MD, Zakheim B, Korngold R. Spontaneous interstitial nephritis in kdkd mice. I. An experimental model of autoimmune renal disease. J Immunol. 1984;133:2560–2565. [PubMed] [Google Scholar]
- 3.Kelly CJ, Korngold R, Mann R, Clayman M, Haverty T, Neilson EG. Spontaneous interstitial nephritis in kdkd mice. II. Characterization of a tubular antigen-specific, H-2K-restricted Lyt-2+ effector T cell that mediates destructive tubulointerstitial injury. J Immunol. 1986;136:526–531. [PubMed] [Google Scholar]
- 4.Sibalic V, Fan X, Wuthrich RP. Characterization of cellular infiltration and adhesion molecule expression in CBA/CaH-kdkd mice with tubulointerstitial renal disease. Histochem Cell Biol. 1997;108:235–242. doi: 10.1007/s004180050163. [DOI] [PubMed] [Google Scholar]
- 5.Hancock WW, Tsai T-L, Madaio MP, Gasser DL. Cutting edge: multiple autoimmune pathways in kd/kd mice. J Immunol. 2003;171:2778–2781. doi: 10.4049/jimmunol.171.6.2778. [DOI] [PubMed] [Google Scholar]
- 6.Peng M, Jarett L, Meade R, Madaio MP, Hancock WW, George AL, Jr, Neilson EG, Gasser DL. Mutant prenyltransferase-like mitochondrial protein (PLMP) and mitochondrial abnormalities in kd/kd mice. Kidney Int. 2004;66:20–28. doi: 10.1111/j.1523-1755.2004.00702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barisoni L, Madaio MP, Eraso M, Gasser DL, Nelson PJ. The kd/kd mouse is a model of collapsing glomerulopathy. J Am Soc Nephrol. 2005;16:2847–2851. doi: 10.1681/ASN.2005050494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dell KM, Li Y-X, Peng M, Neilson EG, Gasser DL. Localization of the mouse kidney disease (kd) gene to a YAC/BAC contig on chromosome 10. Mammalian Genome. 2000;11:967–971. doi: 10.1007/s003350010188. [DOI] [PubMed] [Google Scholar]
- 9.Takahashi N, Patel HR, Qi Y, Dushay J, Ahima RS. Divergent effects of leptin in mice susceptible or resistant to obesity. Horm Metab Res. 2002;34:691–697. doi: 10.1055/s-2002-38251. [DOI] [PubMed] [Google Scholar]
- 10.Maugeais C, Tietge UJ, Tsukamoto K, Glick JM, Rader DJ. Hepatic apolipoprotein E expression promotes very low density lipoprotein-apolipoprotein B production in vivo in mice. J Lipid Res. 2000;41:1673–1679. [PubMed] [Google Scholar]
- 11.Massry SG, Glassock RJ, editors. Massry and Glassock's Textbook of Nephrology. ed 4 Williams & Wilkins; Philadelphia, Lippincott: 2001. pp. 655–660. [Google Scholar]
- 12.Mathieson PW. The cellular basis of albuminuria. Clin Sci. 2004;107:533–538. doi: 10.1042/CS20040168. [DOI] [PubMed] [Google Scholar]
- 13.Zalou M, Azrolan N, Hayek T, Wang H, Wu L, Haghpassand M, Cizman B, Madaio MP, Millbrandt J, Marsh JB, Breslow JL, Fisher EA. The full induction of human Apoprotein A-I gene expression by the experimental nephrotic syndrome in transgenic mice depends on cis-acting elements in the proximal 256 base-pair promoter region and the trans-acting factor early growth response factor 1. J Clin Invest. 1998;101:1699–1707. doi: 10.1172/JCI2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 15.Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Ann Rev Physiol. 1998;60:619–642. doi: 10.1146/annurev.physiol.60.1.619. [DOI] [PubMed] [Google Scholar]
- 16.Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, Saunders TL, Dysko RC, Kohno K, Holzman LB, Wiggins RC. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol. 2005;16:2941–2952. doi: 10.1681/ASN.2005010055. [DOI] [PubMed] [Google Scholar]