

Abstract
Background
Mice that are homozygous for the kidney disease (kd) mutation are apparently healthy for the first 8 weeks of life, but spontaneously develop a severe form of interstitial nephritis that progresses to end-stage renal disease (ESRD) by 4 to 8 months of age. By testing for linkage to microsatellite markers, we previously localized the kd gene to a YAC/BAC contig.
Methods
The sequence of the entire critical region was examined, and candidate genes were identified. These candidate genes were sequenced in both mutant (kd/kd) mice and normal controls. The phenotype was further characterized by immunohistochemistry and electron microscopy. Transgenic mice were constructed that carried the wild-type allele of the prime candidate gene, and this transgene was transferred to a kd/kd background by breeding.
Results
We have obtained evidence that kd is a mutant allele of a novel gene for a prenyltransferase-like mitochondrial protein (PLMP). This gene is alternatively spliced, with the larger gene product having one domain that resembles transprenyltransferase and another that is similar to geranylgeranyl pyrophosphate synthase. The smaller gene product includes only the first domain. An antiserum to PLMP localizes to mitochondria, and ultrastructural defects are present in the mitochondria of renal tubular epithelial cells, and to a lesser extent, hepatocytes and heart cells from kd/kd mice. In a line of kd/kd mice that carried the wild-type PLMP allele as a transgene, only 1 out of 13 animals expressed the disease by 120 days of age.
Conclusion
The kd allele codes for a novel protein that localizes to the mitochondria, and the kd/kd mouse has dysmorphic mitochondria in the renal tubular epithelial cells. This mouse is therefore a unique animal model for studying mechanisms that lead to tubulointerstitial nephritis.
Keywords: interstitial nephritis, kd gene, mitochondria
A spontaneous mutation designated kidney disease (kd) occurred in a CBA/CaH colony, and in the homozygous condition produces interstitial nephritis leading to end-stage renal failure [1]. Examination of kidneys at early time-points reveals a mononuclear cell infiltrate and tubular dilatation in cortical areas, which with time expands throughout the entire kidney [2-4]. Histologic features of this disease resemble human nephronophthisis in some respects, but none of the mapped genes for this entity are in positions that correspond to the region of mouse chromosome 10 in which kd maps [1, 5]. These include NPHP1 on 2q13 [6], NPHP2 on 9q22–31 [7], NPHP3 on 3q22 [8], and NPHP4 on 1p36 [9].
The evidence for an autoimmune component in the phenotype of kd/kd mice is strong. An inflammatory reaction can be observed in histologic sections of the diseased kidneys [1-3]; the disease can be transferred by bone marrow cells to lethally irradiated recipients; and effector T cells can induce nephritis in this model [2]. However, we have recently shown that mice that are doubly homozygous for kd and Rag-1− developed nephritis as readily and as severely as the B6.kd/kd controls [10]. Mice with the Rag-1−/− genotype do not have functional T or B cells, and the leukocytes infiltrating the kidneys of the kd/kd,Rag-1−/− mice consisted of approximately 50% macrophages and 50% natural killer (NK) cells. These results suggested that the genetic defect in kd/kd mice is intrinsic to the kidney, and that an immune response involving either effector T cells or NK cells is a secondary consequence of this initial defect.
We used a positional cloning approach to identify the mutation. After analyzing the complete sequence of a previously reported contig [5], and using reverse transcription-polymerase chain reaction (RT-PCR) to compare mutant and control samples, we identified a missense mutation in one of the candidate genes. The sequence of the candidate gene suggested that the mutant enzyme could affect coenzyme Q in the respiratory pathway. Tissues from kd/kd mice, examined by electron microscopy, revealed ultrastructural abnormalities in the mitochondria of kidney, and to a lesser extent, liver and heart cells of kd/kd mice. The phenotype of kd/kd mice was rescued to a large extent by a BAC transgene that included the genomic DNA for the first of two alternatively spliced products of this prenyltransferase-like mitochondrial protein (PLMP) gene.
METHODS
Mouse genetics
All animal experiments were done in compliance with federal and university regulations. The B6.CBACaH (CAST)-kd/Upa (B6.kd/kd) strain was derived by mating a mouse carrying a recombinant kd haplotype [5] with B6, and backcrossing to the B6 line [10]. Transgenic lines carrying BAC inserts were generated by standard procedures, by injecting purified DNA from the 256E1 BAC (Research Genetics, Huntsville, AL, USA) into (B6 X SJL)F1 eggs fertilized by (B6 X SJL)F1 males. Pups identified to be positive for the transgene were grown up and then mated with B6.kd/kd partners. Transgene-positive progeny from that mating were mated with B6.kd/kd partners to produce a second backcross generation with a transgene-positive, kd/kd genotype. In order to evaluate the expression of the transgene, PCR products of the relevant region were sequenced after RNA extraction from kd/kd, transgene-positive mice. Among the transgenic lines that were generated, only one (line G) expressed the wild-type transgene at a high level. These mice were mated with one another, and the kd/kd, transgene-positive mice were evaluated histologically after the age of 120 days. As a control, a similar line on the same background that was vector-positive but did not express the transgene (line E) was also examined.
Protein expression and antiserum production
The normal PLMP gene product was expressed in vitro by use of the PCR T7 TOPO TA Expression Kit (Invitrogen, Carlsbad, CA, USA), using the PCR T7/NT-TOPO vector, and then purified with the ProBond purification system (Invitrogen). The HisG and Xpress epitopes were removed by enterokinase. This product was injected into Sprague-Dawley rats, and the antibody generated was used for Western blotting and immunohistology. The Western blotting was done by standard procedures using tissue extracts separated by sodium dodecyl sulfate (SDS) electrophoresis, transferring to Hybond-N+ membranes (Amersham Pharmacia Biotech, Buckinghamshire, UK), and incubation with antiserum or control serum.
Sequencing and RT-PCR
The mouse sequence database was searched with BAC end sequences that were known to map within the critical region for the kd gene [5]. RT-PCR was used to amplify coding regions from kd/kd mice with respect to numerous previously defined genes and expressed sequence tags (ESTs) that were identified within this region, and the sequences were compared with those in the database. The forward primer that was used to identify both the shorter and longer gene sequences described in this report was ATGAGCCTCCGGCAGCTGCTG. The reverse primer for the shorter product was TTACTTCATGTTGTCACTGCC, and for the longer product, TTCAAGAAAATCTGGTCACAGC. Both RT-PCR products were sequenced, and shown to be unique sequences that mapped in the expected location of the genome.
Histology and immunoperoxidase staining
The mice were sacrificed and examined histologically at various ages. Representative kidneys from normal C57BL/6 mice and B6.kd/kd mice were examined by immunoperoxidase using the anti-PLMP antibody or control IgG, as described [10]. B6.kd/kd kidneys were collected prior to onset of interstitial disease (<50 days), around the time of onset (75 to 100 days) and during advanced disease (>150 days).
Immunoelectron microscopy
Kidney, liver, and skeletal muscle were taken from control and diseased mice and fixed in 4% paraformaldehyde with 0.1% glutaraldehyde in 0.1 mol/L sodium cacodylate buffer, pH 7.4, overnight at 4°C. Two separate protocols were employed for each tissue. For a review of electron microscopic immunoelectron microscopic protocols, see Smith and Jarett [11]. Briefly, one group of tissue was permeablized with 0.2% saponin in 4% paraformaldehyde for 1 hour. After 3 sodium cacodylate buffer washes, samples were treated for 1 hour with blocking buffer [1% ovalbumin, 0.2% fish gelatin, in 0.1 mol/L phosphate-buffered saline (PBS), pH 7.4] before incubation with primary antibody (1:400 in PBS) overnight at 4°C. After three additional buffer washes, samples were dehydrated in a graded ethanol series to 90% ethanol, infiltrated, and embedded in LR White resin. A second group of tissues was processed to LR White resin by similar techniques, omitting the permeablization with saponin, and the incubation with primary antibody. After polymerization of both groups, thin sections were taken and picked up on nickel grids. All further treatment of thin sections was performed by floating the grids, section side down, on 20 uL drops of reagent displayed on a clean parafilm surface. For samples previously incubated with primary antibody, sections were treated with blocking buffer for 30 minutes at room temperature, and incubated with antirat IgG conjugated to 10 nm colloidal gold particles (1:20 in PBS) overnight at 4°C. Sections were then washed twice in PBS, rinsed in dH2O, and counterstained with 1% uranyl acetate. Samples not previously exposed to primary antibody were treated with blocking buffer for 1 hour at room temperature prior to incubation with primary antibody (1:500 in PBS) for 2 hours at room temperature. After three Tris-buffered saline (TBS) washes, sections were incubated with antirat IgG conjugated to 10 nm colloidal gold particles (1:40 in PBS) for 1 hour at room temperature. After several buffer washes, sections were rinsed in dH2O and counterstained with uranyl acetate. All sections were examined in a JEOL 100CX electron microscope and digital images recorded with a Hamamatsu camera system.
Ultrastructural analysis
Kidney, liver, heart, and muscle tissue samples, taken from control and diseased animals, were processed for electron microscopic ultrastructural analyses. They were fixed in 2.5% glutaraldehyde and 2.0% paraformaldehyde in 0.1 mol/L sodium cacodylate buffer, pH 7.4, overnight at 4°C. After three sodium cacodylate buffer washes, the samples were post-fixed with 2.0% osmium tetroxide in 0.1 mol/L sodium cacodylate buffer for 1 hour at 4°C. After two additional sodium cacodylate washes and one wash in dH2O, samples were stained en bloc with 2% aqueous uranyl acetate for 30 minutes at room temperature. Samples were rinsed again in dH2O subsequent to dehydration in a graded ethanol series, and infiltration and embedding in Embed-812 (Electron Microscopy Sciences, Fort Washington, PA, USA). All sections were examined in a JEOL 100CX electron microscope and digital images recorded with a Hamamatsu camera system.
Evaluation of nephritis
Transgenic mice were evaluated for nephritis as previously reported [10]. Hematoxylin and eosin sections were graded on a scale of 0 to 4+ by one observer (M. Madaio) who was blinded with respect to the origin of the specimens. Because mild symptoms are occasionally seen in nonmutant controls, a mouse was considered positive only if there was a score of 2+ with regard to the interstitial compartment. This excludes mice in which there were small pockets of 5 to 10 mononuclear cells.
RESULTS
The phenotype of the B6.kd/kd strain
The initial studies of this mutant made use of the original CBA/CaH-kd line [1-3]. To our knowledge this strain no longer exists, so we have utililized a congenic line which was derived by transferring the kd allele, along with closely linked microsatellite markers, to the B6 background [10]. Figure 1 demonstrates a histologic section from the kidney of a B6.kd/kd mouse in comparison to that of a B6 control. In Figure 1B, many of the tubules are dilated so extensively that the magnification appears to be greater than in Figure 1A, although they are both 100×. The section shown in Figure 1B, which is representative of what we have observed in B6.kd/kd mice, does not differ significantly in the degree of interstitial nephritis from those in previous reports that utilized the CBA/CaH-kd line.
Fig. 1.
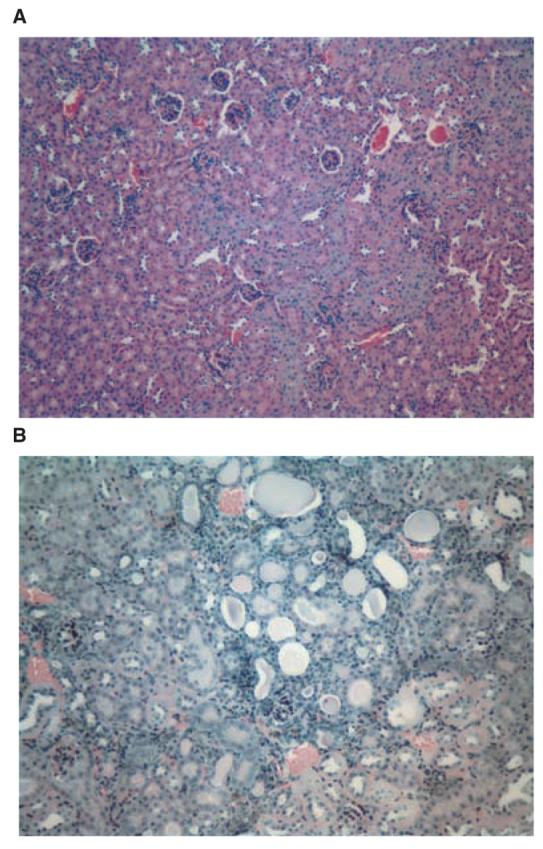
Histologic sections (both hematoxylin and eosin 100×) from a normal B6 mouse at 158 days of age (A) and a B6.kd/kd mouse at the age of 116 days (B) showing moderately severe tubulointerstitial nephritis (3+) with interstitial mononuclear cell infiltration and tubular dilatation.
Identification of a mutant candidate gene
When the various candidate genes in the critical region [5] were examined by amplifying the coding regions using RT-PCR and sequencing the products from mutant and control mice, a missense mutation was identified in one of the candidates (GenBank # AK017329) in the DNA from the original CBA/CaH-kd line. This mutation involved a CG to TG substitution on the antisense strand, which is consistent with the activity of a mutation hotspot (Fig. 2). The genomic DNA of the candidate gene consists of 8 exons, which are alternatively spliced to produce two possible products. The shorter product has a domain that is similar to transprenyltransferases from a number of species, and the mutated residue is especially well conserved (Table 1). The longer product includes most of the residues in the first message, including the mutated residue, in addition to a domain that is very similar to geranylgeranyl pyrophosphate synthase (Fig. 3). All of exon 3 is included in the short product, with a stop codon in exon 3b. Only part of exon 3 (3a) is in the longer product.
Fig. 2.

Sequence of the mutated portion of the candidate gene, showing the mutation at codon 117.
Table 1.
The mutation occurred in a highly conserved region: sequence of positions 116–119 in related proteins from various species
| Species and enzyme | Sequence |
|---|---|
| Mouse kd mutant protein | VMLL |
| Mouse wild-type prenyltransferase-like mitochondrial protein (PLMP) | VVLL |
| Mouse transprenyltransferase | VVLL |
| Human transprenyltransferase | VVLL |
| Zebrafish transprenyltransferase | IVIL |
| Drosophila transhexaprenyltransferase | IVLL |
| Cyanophora paradoxa prenyltransferase | IVLL |
| Cyanidium caldarium prenyltransferase | IVLL |
| Synechocystis prenyltransferase | IVLL |
| Porphyra purpura (Red alga) prenyltransferase | IVLL |
| Arabidopsis thaliana prenyltransferase | LVFL |
Fig. 3. Exons of the prenyltransferase-like mitochondrial protein (PLMP) gene (A) and protein sequences of the two alternatively spliced products of the kd allele (B and C).

The substituted residue, encoded by exon 2, is shown in bold and underlined.
Identification of PLMP in kidney sections
A cDNA for the shorter candidate gene product was used to produce the wild-type protein in a bacterial expression system, and an antiserum to this product was generated in Sprague-Dawley rats. When this antiserum was used for Western blotting, it was apparent that both alternatively spliced gene products are expressed in the kidneys of normal and kd/kd mice. One protein of approximately 92 kD was derived from the 1174 bp message, and a smaller protein of approximately 65 kD was derived from the message of 795 bp (Fig. 4). In both cases these proteins are somewhat larger than what would be expected from the amino acid sequence, so there apparently have been some posttranslational modifications. Control Western blots were run using normal rat serum and rat anti-PLMP serum incubated with the PLMP product obtained from a bacterial expression system, and both were negative (not shown). A subset of renal tubular epithelial cells in the B6.kdkd but not B6 mouse showed more strongly positive staining with the rat antibody (Fig. 5).
Fig. 4. Results of various evaluation techniques.
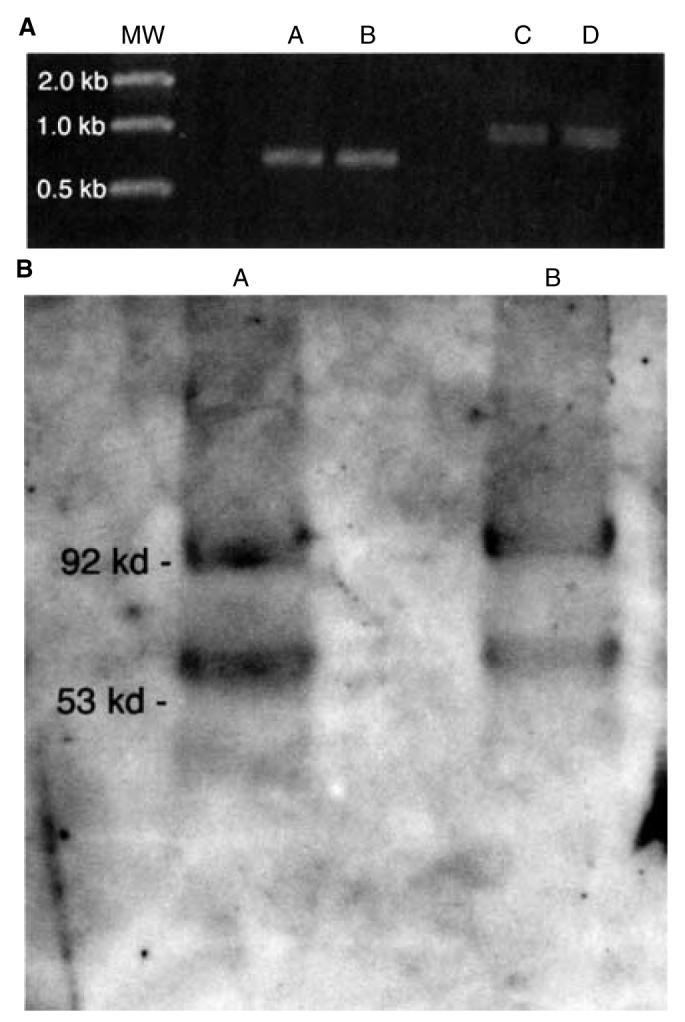
(A) Reverse transcription-polymerase chain reaction (RT-PCR) using primers for the shorter (lanes A and B) and longer (lanes C and D) alternatively spliced gene products. Lanes A and C, B6; lanes B and D, B6.kdkd. RNA was extracted from kidneys. (B) Western blot using kidney extracts and the rat antiserum against the shorter gene product. Lane A, B6; lane B, B6.kd/kd.
Fig. 5. Immunoperoxidase staining of renal tubular epithelium in B6.kd/kd kidneys using an anti-prenyltransferase-like mitochondrial protein (PLMP) antibody.
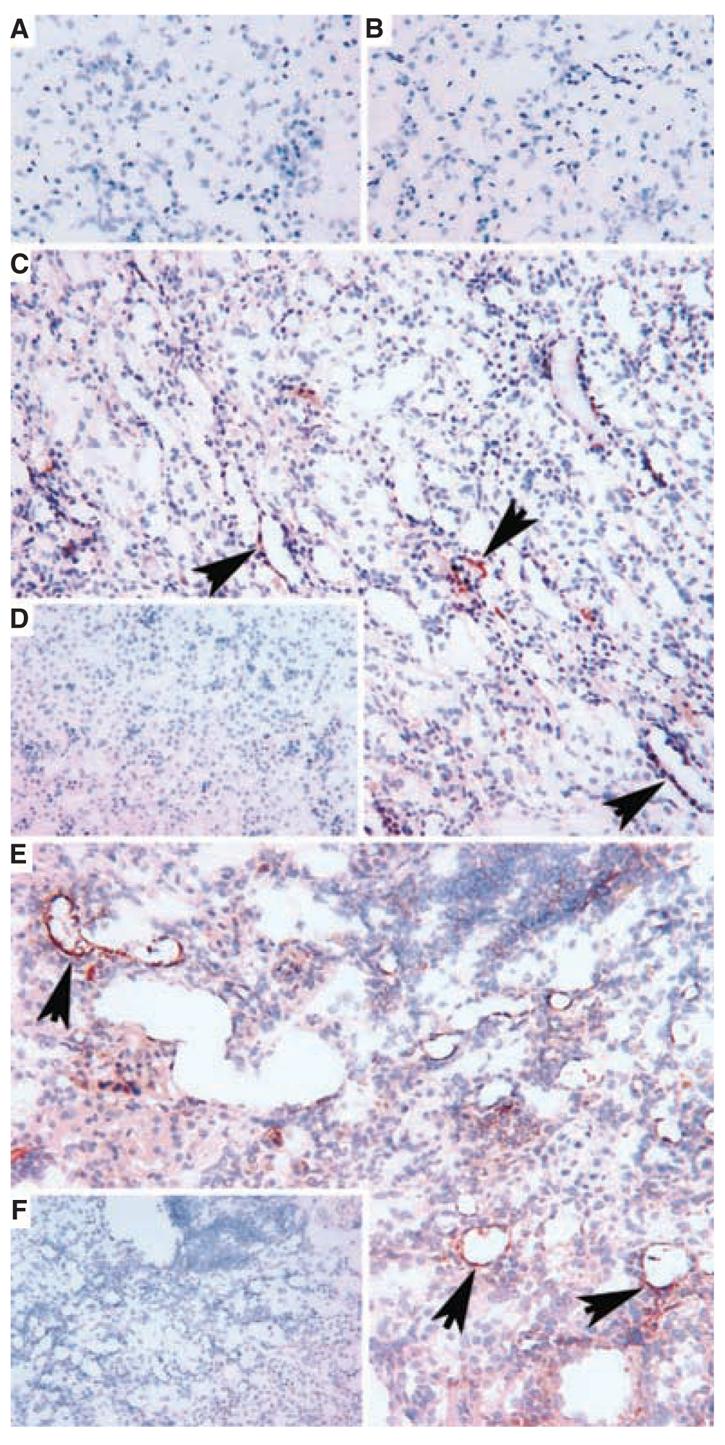
(A) Lack of staining of normal C57BL/6 kidney, at 286 days of age. (B) Lack of staining of B6.kd/kd mouse kidney at 49 days of life. (C) Focal renal tubular staining for PLMP (arrows) in a kidney from B6.kd/kd mouse at 91 days (around time of onset of interstitial nephritis). (D) Lack of staining of kidney from (C) with control IgG. (E) Extensive lack of staining using control IgG, rabbit antihorseradish peroxidase (HRP). (F) Lack of staining of kidney tubular staining for PLMP in the kidney of a B6.kd/kd mouse at 177 days (cryostat sections, hematoxylin counterstain, original magnifications ×250).
Localization of PLMP by immunoelectron microscopy
The same anti-PLMP antibody was then used for electron microscopy studies, with pre- and post-fixed immunogold techniques as detailed in the Methods section [11]. Samples of kidney, liver, and skeletal muscle were taken from B6 and B6.kd/kd mice and examined by both techniques. Using these immunogold techniques, the liver and skeletal muscle samples from B6 mice were indistinguishable from sections of the same tissue from B6.kd/kd mice. Both tissues showed only an occasional gold particle, indicating the presence of the kd gene product, mostly found on mitochondria (data not shown). The kidney from B6 mice (Fig. 6A) appeared normal with an occasional gold particle located mostly over mitochondria. In contrast, the B6.kd/kd kidney had markedly increased gold particles on mitochondria (Fig. 6B). These findings demonstrate that PLMP was over-expressed in the mitochondria of kidney from the B6.kd/kd mice, and provide further evidence that this defective gene product is responsible for the kidney disease.
Fig. 6. Immunoelectron microscopy labeling of prenyltransferase-like mitochondrial protein (PLMP) in B6 and B6.kd/kd mouse kidney samples.
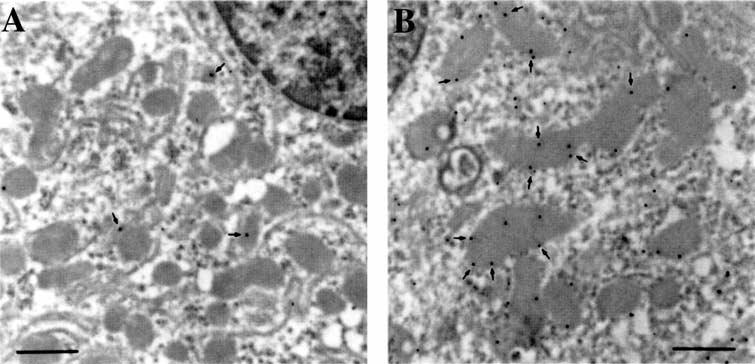
The B6 mouse was 308 days old, and the B6.kd/kd mouse was 267 days of age. (A) Immunogold labeling of B6 mouse kidney, demonstrating very few 10 nm gold particles (arrows), which are generally associated with mitochondria. (B) Immunogold labeling of B6.kd/kd mouse kidney demonstrating a greatly increased number of gold particles associated with mitochondria. The increase in the number of gold particles indicates an increased level of PLMP present in the B6.kd/kd kidney mitochondria (scale bars, 500 nm).
Ultrastructural defects in mitochondria of kd/kd mice
Tissues used for immunoelectron microscopy do not always demonstrate the level of preservation needed for fine structural analysis, as the fixation procedures are designed for preservation of antigenicity rather than fine structure. Therefore, tissue samples of kidney, liver, heart, and skeletal muscle were taken from control and experimental animals and were prepared expressly for fine structural analysis. The results of these examinations demonstrate clear alterations in the ultrastructure of kidney mitochondria from B6.kd/kd as compared to B6 cells The major difference between the B6 and B6.kd/kd kidney samples (Fig. 7A vs. Fig. 7B) is the complete absence of mitochondrial matrix granules in the latter tissue, compared to their prevalent inclusions in mitochondria of B6 kidney tissue. Furthermore, cristae of the B6 kidney tissue are more dilated than in the B6.kd/kd kidney tissue, where they are compressed. The mitochondrial matrix appears less dense and more heterogeneous in the B6.kd/kd tissue and in general the B6.kd/kd kidney mitochondria are smaller and have a more angular appearance.
Fig. 7. Ultrastructural analyses of mitochondria from B6 and B6.kd/kd mouse kidney and liver.
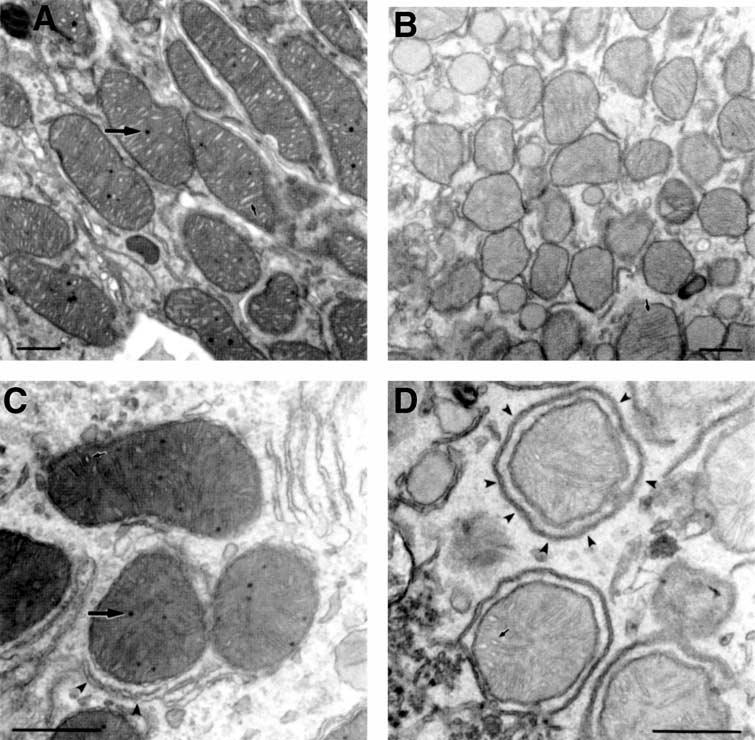
(A Electron micrograph of mitochondria from B6 mouse kidney. Many normal mitochondrial matrix granules are seen (large arrow), as well as well-defined cristae, which are moderately dilated (small arrow). The matrix is homogenous and the overall morphology is smooth and generally oblong. (B) Electron micrograph of mitochondria from B6.kd/kd mouse kidney demonstrating a lack of normal mitochondrial inclusions. In addition, the cristae (small arrow) are compressed, and the matrix is pale and granular. The overall morphology of the B6.kd/kd mitochondria appears smaller and more angular than mitochondria from B6 mice. (C) Electron micrograph for mitochondria from B6 mouse liver demonstrating normal matrix granules (large arrow), and clearly defined, moderately dilated cristae (small arrow). Endoplasmic reticulum (arrow heads) is often closely associated with the mitochondria. The mitochondrial matrix is homogenous and moderately dense. d, Electron micrograph of mitochondria from B6.kd/kd liver demonstrating characteristics similar to those of B6.kd/kd kidney mitochondria. There is an absence of normal matrix granules, and the matrix is paler and more granular than that of B6 mouse liver mitochondria. The cristae (small arrow) seem less affected and appear near normal. The overall mitochondrial morphology again appears generally more angular, but perhaps less so than in B6.kd/kd mouse kidney samples. The endoplasmic reticulum (arrow heads) often appears to completely surround the mitochondria (scale bars, 500 nm).
Some of the ultrastructural differences between the mitochondria of B6 and B6.kd/kd kidney tissue are also seen with liver mitochondria (Fig. 7C and D), including the lack of matrix granules, less dense mitochondrial matrix, and overall angular morphology. An especially interesting difference is that the endoplasmic reticulum (arrow heads in Fig. 7D) is often closely associated with the mitochondria. Although the liver mitochondria of the diseased mice have fine structural alterations similar to those in the kidney, they do not have an increased quantity of the gene product in their mitochondria. Some of the same abnormalities that were seen in hepatocytes were also seen in heart from kd/kd mice (not shown). In contrast to the results from the mitochondria from kidney and liver tissue, samples of skeletal muscle from B6 and B6.kd/kd mice were indistinguishable from each other and appeared normal (not shown).
Phenotype rescue in BAC transgenic mice
To determine whether the correct candidate gene had been identified, we attempted to rescue the phenotype by generating BAC transgenics that carried the wild-type candidate gene, PLMP. The BAC clone 256E1 includes the entire genomic DNA of the first alternatively spliced gene product, but not the second. All 15 of 15 control mice of a transgenic line that included the vector sequence but did not express the wild-type PLMP gene (line E), developed histologic evidence of disease by 120 days. In contrast, only one of 13 of the kd/kd transgenic mice with high expression of the wild-type transgene (line G), developed disease (Table 2).
Table 2.
Phenotypes of control and kd/kd mice at >120 days of age with or without expression of a wild-type transgene
Line E and line G both have a mixed genetic background from B6 and 129/J. Both have the kd/kd genotype, and both are positive for the vector used to construct the transgene for the candidate gene. Line G expresses the transgenic wild-type allele of the candidate gene, but line E does not.
DISCUSSION
The phenotype of the kd/kd mouse has been an interesting focus of investigation for many years. Although it was originally thought to resemble nephronophthisis most closely [1], none of the mapped genes for that disease [6-9] are in positions that correspond to the region of mouse chromosome 10 in which kd maps [1, 5]. The immune response that occurs in these mice has been studied in a number of laboratories [2-4], but the extent to which the pathology can be attributed to immunologic damage is still unclear. We have recently demonstrated that mice with the B6 background that are homozygous for both Rag-1− and kd get an inflammatory interstitial nephritis as readily and as severely as B6.kd/kd controls, although it differs in its cellular composition. The infiltrating leukocytes in this case are macrophages and NK cells rather than macrophages and T cells [10], but the pathologic mechanisms that are most relevant are yet to be determined.
In order to define the primary event in the initiation of this disease, we have used a positional cloning approach. We have demonstrated that the kd allele has a missense mutation in a novel gene that has one domain that resembles transprenyltransferase, and another that is similar to geranylgeranyl pyrophosphate synthase. However, genes for transprenyltransferase and geranylgeranyl pyrophosphate synthase have previously been mapped to mouse chromosomes 2 and 13, respectively, so the gene products described in this report are different from those that have been previously reported. Its greatest DNA sequence homology is with a human gene that is known in the databases as “candidate tumor suppressor protein” (NM 020381.2), which to our knowledge has not been described in the literature. When the protein sequences are compared, its greatest homology is with geranylgeranyl pyrophosphate synthases of various species, in a cluster of orthologous groups (COG) of highly conserved enzymes (COG0142). This gene has extensive similarity (85.7% alignment) with the IspA gene of Escherichia coli, which has farnesyl diphosphate (FPP) synthase activity. Hexaprenylpyrophosphate synthase isolated from mitochondria of yeast is a member of the same COG, and a strain with a mutation in this gene is deficient in coenzyme Q [12].
A BAC which included the genomic DNA for the first splice product was used to construct a transgenic line, and this transgene rescued the phenotype in 12 out of 13kd/kd mice. In mice that had the same genetic background but did not express the transgene, 15/15 developed nephritis (Table 1). The difference is highly significant (X2 = 24; P < 0.0001). That the phenotype was rescued in only 92% of the animals could be explained by the fact that these mice had two copies of the mutant kd gene and only one copy of the wild-type allele. A second possibility is that expression of both of the alternatively-spliced gene products may be needed for an entirely normal phenotype.
The sequence of this PLMP, as described in this report, suggests that it is one of the enzymes involved in prenylation [13]. How many target molecules are affected by this enzyme is not known, but by comparison to the yeast mutant [12], we suggest that coenzyme Q may be one of them. Because of that possibility, and the fact that antibodies to PLMP localized to mitochondria (Fig. 6), we examined mitochondria from kd/kd mice by electron microscopy. Sections optimized for ultrastructural detail demonstrated apparently defective mitochondria in both kidney and liver of B6.kdkd mice (Fig. 7) and, to some extent, in heart (not shown). There is an apparent correlation between the mitochondrial changes and increased gene product in kidney tissue with kidney disease, which could represent compensation for a defective enzyme. This increase, however, may be highly localized, as there were no obvious increases in the amount of gene product produced when whole kidney extracts were studied by RT-PCR or Western blotting (Fig. 4). The occurrence of an immune response in kd/kd mice that is directed to a kidney antigen is clearly relevant [4]. An immune response that is secondary to some other initial defect is consistent with what has been observed with some other autoimmune diseases, such as systemic lupus erythematosis [14]. It remains to be determined why a mitochondrial defect in renal tubular epithelium leads to an immune response, whereas a similar defect in liver or heart mitochondria apparently does not. We speculate that the differences in energy requirements between tubular cells vs. hepatocytes or heart cells may differentially affect cell stability and immunogenicity. Because of the high rate of tubular transport in renal epithelia, they are very dependent on oxygen consumption for energy metabolism [15]. Liver cells, on the other hand, have alternative energy sources at low levels of oxygen supply [16]. These differences may hypothetically explain the relative tissue sensitivity of nephrons with PLMP mutations compared to other tissues. In this regard, there is evidence that apoptosis occurs in renal tubular epithelial cells of kd/kd homozygotes before leukocyte infiltration can be observed [10]. Apoptosis leads to externalization of signal molecules such as phosphatidylserine, which can be recognized by macrophages [17] and NK cells [18]. The immune response that is generated in the kd/kd mouse after apoptosis of renal tubular epithelium can involve either effector T cells or NK cells [10].
We suggest that the human disease with the greatest similarity to this mouse model is represented by several patients with tubulointerstitial nephritis who proved to have deletions or mutations in their mitochondrial DNA [19, 20]. In those cases the defects involved mitochondrial genes rather than a nuclear gene such as kd, and the symptoms were not identical to those of kd/kd mice. However, the similarities are quite interesting. All of these patients started life in an apparent state of health, but at some point developed a chronic tubulointerstitial nephritis. Ultrastructural analysis in all cases revealed abnormalities in the mitochondria of renal tubular epithelial cells. The kd/kd mouse may be a useful model, not only for studying the physiologic role of PLMP, but for recognizing the molecular defect in a number of human conditions that have not been fully characterized.
CONCLUSION
These results demonstrate that the kd/kd mouse has a mutation in a novel PLMP. Mitochondria in mutant mice have structural abnormalities in renal tubular epithelium, and to a lesser extent, in liver and heart. Evidence suggests that apoptosis occurs in the renal tubular epithelium, and this leads to an autoimmune response.
ACKNOWLEDGMENTS
This work was supported by grants DK 55852 and P30-DK 50306 from the National Institutes of Health. We thank Tsai-Lung Tsai for excellent technical assistance; Dr. Jean Richa and his colleagues in the Transgenic and Chimeric Mouse Facility for generating the BAC transgenic lines; and the members of the Morphology Core for Molecular Studies in Digestive and Liver Diseases for histologic preparations.
REFERENCES
- 1.Lyon MF, Hulse EV. An inherited kidney disease of mice resembling human nephronophthisis. J Med Genet. 1971;8:41–48. doi: 10.1136/jmg.8.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neilson EG, McCafferty E, Feldman A, et al. Spontaneous interstitial nephritis in kdkd mice. I. An experimental model of autoimmune renal disease. J Immunol. 1984;133:2560–2565. [PubMed] [Google Scholar]
- 3.Sibalic V, Fan X, Wuthrich RP. Characterization of cellular infiltration and adhesion molecule expression in CBA/CaH-kdkd mice with tubulointerstitial renal disease. Histochem Cell Biol. 1997;108:235–242. doi: 10.1007/s004180050163. [DOI] [PubMed] [Google Scholar]
- 4.Kelly CJ, Korngold R, Mann R, et al. Spontaneous interstitial nephritis in kdkd mice. II. Characterization of a tubular antigen-specific, H-2K-restricted Lyt-2+ effector T cell that mediates destructive tubulointerstitial injury. J Immunol. 1986;136:526–531. [PubMed] [Google Scholar]
- 5.Dell KM, Li Y-X, Peng M, et al. Localization of the mouse kidney disease (kd) gene to a YAC/BAC contig on Chromosome 10. Mammalian Genome. 2000;11:967–971. doi: 10.1007/s003350010188. [DOI] [PubMed] [Google Scholar]
- 6.Hildebrandt F, Otto E, Rensing C, et al. A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat Genet. 1997;17:149–153. doi: 10.1038/ng1097-149. [DOI] [PubMed] [Google Scholar]
- 7.Haider NB, Carmi R, Shalev H, et al. A Bedouin kindred with infantile nephronophthisis demonstrates linkage to chromosome 9 by homozygosity mapping. Am J Hum Genet. 1998;63:1404–1410. doi: 10.1086/302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Omran H, Fernandez C, Jung M, et al. Identification of a new gene locus for adolescent nephronophthisis, on chromosome 3q22 in a large Venezuelan pedigree. Am J Hum Genet. 2000;66:118–127. doi: 10.1086/302705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Otto E, Hoefele J, Ruf R, et al. A gene mutated in nephronophthisis and retinitis pigmentosa encodes a novel protein, nephroretinin, conserved in evolution. Am J Hum Genet. 2002;71:1161–1167. doi: 10.1086/344395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hancock WW, Tsai T-L, Madaio M, Gasser DL. Cutting edge: Multiple autoimmune pathways in kdkd mice. J Immunol. 2003;171:2778–2781. doi: 10.4049/jimmunol.171.6.2778. [DOI] [PubMed] [Google Scholar]
- 11.Smith RM, Jarett L, de Pablo F. Electron microscopic immunocytochemical approaches to the localization of ligands, receptors, transducers, and transporters. In: Scanes CG, Weintraub BD, editors. Handbook of Endocrine Research Techniques. Academic Press; London: 1993. pp. 227–264. [Google Scholar]
- 12.Ashby MN, Edwards PA. Elucidation of the deficiency in two yeast coenzyme Q mutants. J Biol Chem. 1990;265:13157–13164. [PubMed] [Google Scholar]
- 13.Grunler J, Ericsson J, Dallner G. Branch-point reactions in the biosynthesis of cholesterol, dolichol, ubiquinone and prenylated proteins. Biochim Biophys Acta. 1994;1212:259–277. doi: 10.1016/0005-2760(94)90200-3. [DOI] [PubMed] [Google Scholar]
- 14.Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179:1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mandel LJ, Balaban RS. Stoichiometry and coupling of active transport to oxidative metabolism in epithelial tissues. Am J Physiol. 1981;240:F357–F371. doi: 10.1152/ajprenal.1981.240.5.F357. [DOI] [PubMed] [Google Scholar]
- 16.Kekonen EM, Jauhonen VP, Hassinen IE. Oxygen and substrate dependence of hepatic cellular respiration: Sinusoidal oxygen gradient and effects of ethanol in isolated perfused liver and hepatocytes. J Cell Physiol. 1987;133:119–126. doi: 10.1002/jcp.1041330115. [DOI] [PubMed] [Google Scholar]
- 17.Fadok VA, Bratton DL, Frasch SC, et al. The role of phosphatidylserine in recognition of apoptotic cells by phagocytosis. Cell Death Differ. 1998;5:551–562. doi: 10.1038/sj.cdd.4400404. [DOI] [PubMed] [Google Scholar]
- 18.Blom WM, De Bont HJGM, Nagelkerke JF. Regional loss of the mitochondrial membrane potential in the hepatocyte is rapidly followed by externalization of phosphatidylserines at that specific site during apoptosis. J Biol Chem. 2003;278:12467–12474. doi: 10.1074/jbc.M201264200. [DOI] [PubMed] [Google Scholar]
- 19.Szabolcs MJ, Seigle R, Shanske S, et al. Mitochondrial DNA deletion: A cause of chronic tubulointerstitial nephropathy. Kidney Int. 1994;45:1388–1396. doi: 10.1038/ki.1994.181. [DOI] [PubMed] [Google Scholar]
- 20.Zsurka G, Ormos J, Ivanyi B, et al. Mitochondri al mutation as a probable causative factor in familial progressive tubulointerstitial nephritis. Hum Genet. 1997;99:484–487. doi: 10.1007/s004390050393. [DOI] [PubMed] [Google Scholar]