

Abstract
Nuclear receptors (NR) are a superfamily of ligand-activated transcription factors that regulate development, reproduction, and metabolism of lipids, drugs and energy. The importance of this family of proteins in metabolic disease is exemplified by NR ligands used in the clinic or under exploratory development for the treatment of diabetes mellitus, dyslipidemia, hypercholesterolemia, or other metabolic abnormalities. Genetic studies in humans and rodents support the notion that NRs control a wide variety of metabolic processes by regulating the expression of genes encoding key enzymes, transporters and other proteins involved in metabolic homeostasis. Current knowledge of complex NR metabolic networks is summarized here.
Keywords: orphan nuclear receptors, metabolic syndrome, obesity, insulin resistance, dyslipidemia
Introduction
Nuclear receptors (NR) are a large family of ligand-activated transcription factors that act as transcriptional switches responding to lipophilic hormones, vitamins, dietary lipids, or other intracellular signals [1]. By virtue of this ligand-dependent activity, the nuclear hormone receptors serve as an interface between cellular or the whole body environment and our genome. The elucidation of the full length sequence of the first two founding members of the family, the human glucocorticoid receptor (GR) and estrogen receptor α (ERα) (cloned in 1985 and 1986 respectively) revealed a common domain structure as well as homology to the viral oncogene v-erbA, suggesting the existence of a receptor protein family [2]. Also in 1986, the cellular homologue of v-erb was discovered to be a receptor for thyroid hormone, indicating that ligands for the NRs are not limited to steroid hormones. Within 3 years, receptors for mineralocorticoid, progesterone, androgen and fat-soluble vitamins A and D were cloned, revealing extensive sequence similarity among these lipophilic hormone receptors [3]. Cloning efforts based on sequence homology have subsequently revealed the existence of numerous so-called “orphan NRs”, whose physiological ligands and functions were not initially known.
Availability of genomic sequence information reveals that humans encode 48 NR family members and mice encode 49 NR family members [4–7]. During the last two decades, remarkable progress has been made toward the understanding of both classical endocrine receptors as well as orphan receptors in animal physiology, due to technological advancement in molecular genetics, genome wide target gene identification, single nucleotide polymorphism identification, and high throughput ligand identification. NRs play important functions in virtually every aspect of mammalian physiology, including development, reproduction, immune response, vascular and cardiac function, tissue growth and tumor formation, toxin clearance, and carbohydrate and lipid metabolism. The notion that NRs integrate complex metabolic homeostasis is supported by systematic analyses of tissue-, time-, and stimuli-dependent NR gene expression [5–7]. As an obesity pandemic and associated illnesses such as type 2 diabetes and cardiovascular disease spread around the globe, particular attention has been focused on the role of NRs in the pathophysiology and/or treatment of metabolic diseases including the metabolic syndrome. Here we review the current understanding of NR biology in the context of metabolic regulation and disease.
NR structure and activation mechanisms
The NRs are defined by common structural motifs as depicted in Figure 1A [1]. A highly variable NH3-terminal region contains a ligand-independent activation domain called AF-1. The central DNA-binding domain (DBD), consisting of two highly conserved zinc-finger motifs unique to NRs, targets the receptor to specific DNA sequences called hormone response elements (HRE). A typical HRE consists of two hexa-nucleotide motifs AGGTCA or its variants, separated by a gap of several nucleotides. Binding specificity by various receptors is largely achieved by the spacing (the 3-4-5 rule) and the orientation of two half-sites (direct-, inverted- or everted-repeat) (Figure 1B). The hinge region confers structural flexibility in the receptor dimers allowing a single receptor dimer to interact with multiple HRE sequences. The C-terminal ligand-binding domain (LBD) is functionally very unique to NRs and responsible for: (1) receptor dimerization; (2) ligand recognition and (3) cofactor interaction.
Figure 1.
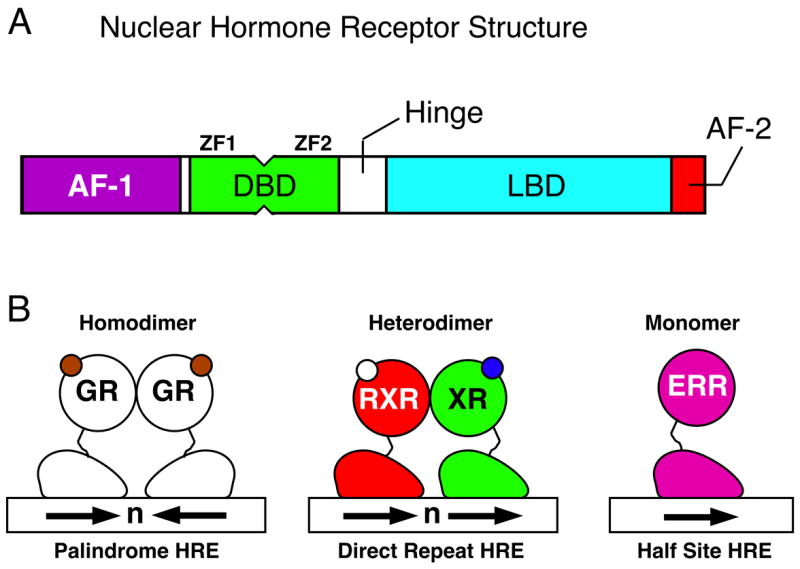
(A) Schematic diagrams for a common domain structure of NRs which include N-terminal activation function 1 (AF-1), DNA binding domain (DBD) consisting of two zinc fingers (ZF), nonconserved hinge-region (Hinge), ligand binding domain (LBD), and C-terminal AF-2 helix. (B) Schematic diagrams for NR dimerization and DNA binding sequences. From the left, homodimeric endocrine receptor (Palindrome HRE), RXR heterodimers (Direct Repeat HRE) and monomeric orphan receptor (Half Site HRE). Arrows: the consensus NR recognition sequence AGGTCA or a variant.
In the absence of ligand, NRs are either present in the cytoplasm forming a complex with heat shock proteins and immunophilin chaperones [8], or in the nucleus constitutively bound to a HRE, forming a repressive complex with a corepressor such as SMRT/NCOR and HDAC complexes [9]. The LBDs consist of approximately 12 helices; the last helix is called the AF-2 or Helix12 and is structurally dynamic. Binding of ligand in the ligand-binding pocket induces conformational changes in the AF-2, which mechanistically facilitates the release of corepressors and HDAC complexes and the recruitment of coactivators and HAT complexes (Figure 2) [1,10]. In some cases, the AF-2 peptide is fixed in an active conformation, resulting in constitutive receptor activation. In these cases, the activity of the NR is regulated by nuclear availability of the receptor itself or coactivators, or by signal-induced receptor modifications such as phosphorylation or acetylation.
Figure 2.
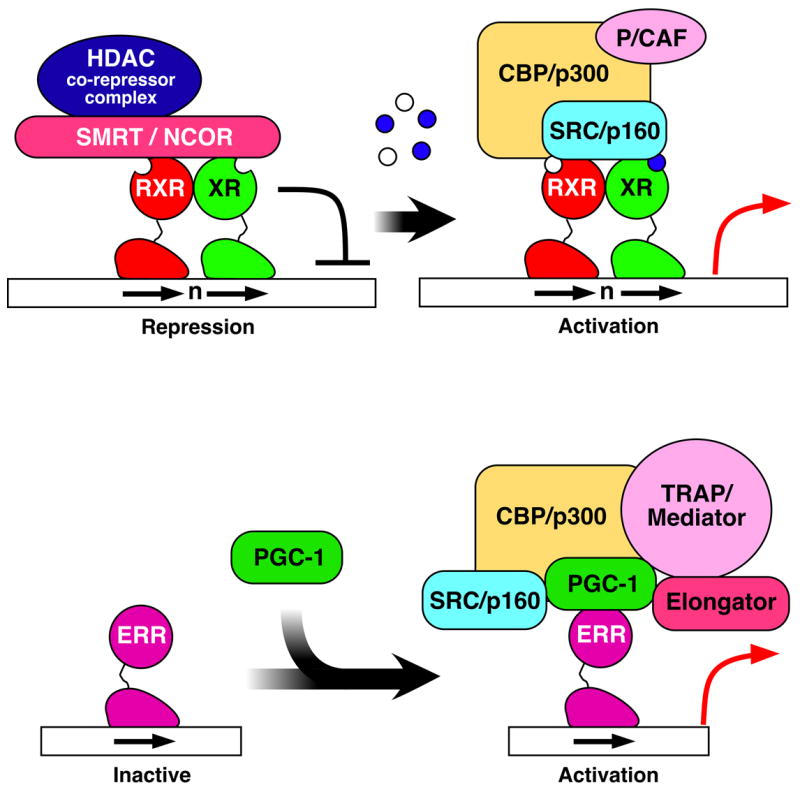
Schematic diagram for a typical NR activation mechanism. Top: In the absence of ligand, NRs form a repressive complex with HDACs (histone deacetylases) via corepressor SMRT or NCOR (left). Ligand binding induces a dissociation of corepressors and a recruitment of coactivators including HAT (histone acetyl-transferase) and chromatin remodeling complexes (right). Bottom: some nuclear receptors are activated by ligand-independendent binding of the PGC-1 coactivator and subsequent recruitment of additional coactivator complexes.
NR subtypes
The NRs can be broadly classified into three sub-groups based on their physiologic ligands and potential functions (Figure 3). The first class is endocrine receptors which all act as high affinity (Kd = nM range) receptors for fat-soluble hormones and vitamins. This class includes receptors for the steroid hormones, thyroid hormone (thyroid hormone receptor; TR), and vitamins A (retinoic acid receptors; RAR) and D (vitamin D receptor; VDR), which are all essential for homeostatic control of endocrine system. Mechanistically, the steroid receptors function as homodimers and TR, VDR, and RAR form heterodimers with the Retinoid X Receptor (RXR). The endocrine receptors are very successful drug targets, and ligands for each of these receptors are in use clinically. In addition, selective modulator ligands that target only a specific receptor isoform or that target a receptor only in a specific tissue are being investigated for an increased benefit, as full pan-agonists tend to have both beneficial and adverse effects [11,12].
Figure 3.
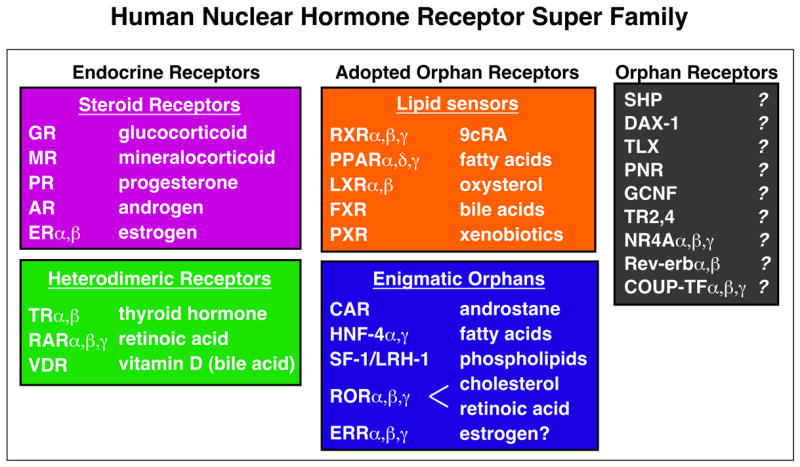
Human NR superfamily including 48 family members. Representative natural ligands are shown at right. Physiological importance of ligand-induced NR activation has been established for “Endocrine Receptors” (Pink: homodimerizing steroid receptors, and Green: RXR heterodimers) and “Adopted Orphan Receptors” (Orange), whereas it is not known for “Enigmatic Adopted Orphans” (Blue) and “Orphan receptors” (Grey). See text for details.
The second receptor class is the adopted orphans, which were identified originally based on sequence homology to endocrine receptors, thus called orphan receptors, but “de-orphanized” by the identification of a naturally occurring ligand. Identification of a vitamin A derivative, 9-cis retinoic acid, as an endogenous high-affinity ligand for RXR represented the first true adoption of an orphan NR and ushered in the age of “reverse endocrinology” in which a receptor is used to discover its natural ligand [13]. This class now includes low affinity (Kd = μM range) receptors for dietary lipids and xenobiotics, which all function as heterodimers with RXR. Some RXR heterodimers can be activated by RXR ligands (permissive) but others cannot (non-permissive) [14]. Receptors of this class are the most promising drug targets for metabolic disorders as they regulate lipid and/or glucose homeostasis by controlling uptake, synthesis, storage or clearance. PPARγ and PPARα are proven drug targets for amelioration of insulin resistance and dyslipidemia, respectively [15,16]. Ligands for other lipid receptors such as PPARδ, LXR, FXR and PXR are under extensive investigation for potential clinical use.
Included within adopted orphans are “enigmatic” adopted orphans, for which a ligand has been identified, at least for one of the subtypes, but the nature of ligand-dependent regulation in physiology has not been established. This group includes receptors whose activity and/or cofactor interaction can be modulated in non-physiological conditions by synthetic estrogens (estrogen related receptors; ERRβ and γ) and/or by endogenous molecules such as cholesterol (Retinoid-Related Orphan Receptor; RORα), retinoids (RORβ), androstane (Constitutive Androstane Receptor; CAR), or phospholipids (Steroidgenic Factor-1; SF-1 and human Liver Receptor Homologue-1; LRH-1) through binding to the ligand binding pocket [1,10,17–20]. Also included in this class is a receptor which is constitutively bound by a fatty acid molecule most likely as a structural component (Hepatocyte nuclear factor 4α; HNF4α). As receptors of this class could potentially be activated in animals by synthetic ligands, as demonstrated for CAR, they also constitute potential drug targets.
The third class is comprised of true orphans whose (natural or synthetic) ligand has not been identified. This class includes receptors which are most likely not regulated by ligands based on the size of the ligand binding pocket and the position of AF-2 helix in the apo-form in X-ray crystal structures (NR4A) [1,10]. These receptors are most likely regulated by coactivator availability, receptor expression itself, covalent modification or a combination. Although receptors of this class may be difficult to directly manipulate pharmacologically, genetic evidence indicates that some of them are also involved in metabolic regulation and thus are currently under extensive investigation.
Endocrine NRs - mediators of hormonal nutrient regulation
The importance of NR function in regulation of metabolic homeostasis and disease pre-dated their cloning. Excess glucocorticoid, sex steroids, thyroid hormone or their deficiency has complex and profound effects on human physiology and energy metabolism. Genetic studies and identification of selective ligands in the past 20 years have revealed some of the tissue- and subtype-specific activity of endocrine receptors.
The two primary adrenal steroids are glucocorticoids and mineralocorticoids. Glucocorticoids (hydrocortisone in humans) are produced during starvation or other stress conditions, and exert a variety of metabolic functions through GR, which was the first cloned NR [21]. During fasting, glucocorticoid promotes hepatic gluconeogenesis and glycogen synthesis. In peripheral tissues, it promotes amino acid catabolism, adipogenesis and insulin resistance. Thus, excess glucocorticoids in Cushing’s syndrome produces visceral obesity and hyperglycemia. In mice, adipose-specific glucocorticoid activation results in visceral obesity, hyperphagia, hyperlipidemia, hypertension, and insulin resistance, whereas its inactivation increases energy expenditure and protects the animal from diet-induced obesity and diabetes [22]. Liver specific inactivation of GR in mice leads to fasting hypoglycemia and ameliorates hyperglycemia in streptozotocin-induced diabetes mellitus [23]. Despite its central role in regulating glucose and energy metabolism and its known activity for cardioprotection [24], adverse effects potently elicited by glucocorticoids make its usage for the treatment of metabolic disease difficult. Alternatively, pharmacological inhibition of local glucocorticoid production (by a selective inhibitor for 11β-hydroxysteroid dehydrogenase type 1 under development) may be beneficial in metabolic syndrome and atherosclerosis [25].
The main action of mineralocorticoids is to modulate water and electrolyte balance through the activation of the mineralocorticoid receptor (MR). The principal endogenous mineralocorticoid is aldosterone. Glucocorticoids can also activate MR, although it is enzymatically inactivated in the epithelial tissues or heart where MR plays predominant roles. Excess aldosterone secretion causes hypertension and contributes to the progression of heart failure, at least in part by inducing sodium channels and sodium/potassium-ATPase in epithelial tissues [26]. Conversely, recently developed MR antagonists such as Eplerenone have clinically been proven to ameliorate hypertension and reduce mortality caused by heart failure [27]. In mice, global MR deficiency results in renal sodium wasting [28]. Heart specific reduction of MR expression results in severe heart failure without hypertension or chronic hyperaldosteronism, indicating the direct action of MR on heart function [29].
Two thyroid hormone receptors (TRα and TRβ) mediate the activity of thyroid hormone (T3: triiodothyronine) as a heterodimer with RXR [30]. Thyroid hormone signaling is remarkably complex in part due to a feedback mechanism for hormone production, the diverse nature of its signaling pathways involving multiple organs and multiple receptor isoforms, and complex modes of receptor action involving both repression and activation of gene transcription as well as non-genomic action. However, for amelioration of metabolic disease, a class of recently developed TRβ selective agonists, at least in model animals, have shown some promise since TRβ mediates most of the beneficial effects of T3, such as a reduction in serum LDL cholesterol, triglyceride and proatherogenic lipoprotein (a) [11]. In contrast, TRα mediates most of the adverse effects of T3, such as heart rate elevation or irregular heartbeat. Some T3 effects such as an increase in metabolic rate and resulting weight loss are mediated by both TRα and TRβ. Gene expression studies in euthyroid mice suggested that TRβ selective agonists reduce serum cholesterol by activating hepatic reverse cholesterol transport [31].
PPAR - FA receptors regulating lipid homeostasis and insulin action
Peroxisome proliferator-activated receptors (PPARs) include 3 members: α, β/δ, and γ, and each of them act as a heterodimer with RXR. As the name suggests, the founding member PPARα was identified as the target of the fibrate-class of anti-hyperlipidemic drug or peroxisome proliferators [16]. In fact, PPARα was the first orphan receptor whose (synthetic) ligand was identified as an existing prescription drug (fenofibrate). Shortly afterwards, PPARα was shown to bind to and function as an endogenous sensor for polyunsaturated fatty acids. PPARα is highly expressed in liver, heart, muscle and kidney where it regulates fatty acid oxidation and apolipoprotein synthesis. In particular, it induces hepatic peroxisomal fatty acid oxidation during fasting and PPARα deficient mice develop dramatic steatosis upon prolonged fasting or high-fat diet feeding. It is also abundantly present in the vascular wall and in human macrophage foam cells where it is thought to exert anti-inflammatory and anti-atherogenic effects [32]. Though the effects in rodents are still debatable, clinical studies in humans indicated that PPARα agonists are most likely effective not only in correcting dyslipidemia, but also cardiovascular mortality and morbidity [16]. In addition, fibrates suppress satiety and improve insulin resistance in mice, although they are not generally viewed as insulin sensitizers in humans.
PPARγ is the target of the thiazolidinedione (TZD)-class of insulin sensitizers, which commands the largest share of the current oral anti-diabetic drug market [15,33,34]. PPARγ is a master regulator of adipogenesis and is most abundantly expressed in adipose tissue. The mechanisms and location of insulin sensitization by TZDs are believed to involve PPARγ activation in adipocytes, an increase in adipocyte fat storage and secretion of insulin sensitizing adipokines such as adiponectin. In addition, studies of tissue-specific PPARγ knockout mice indicated that PPARγ in liver and skeletal muscle also contribute to the overall beneficial effects of TZDs, although these tissues express PPARγ only at very low levels. Consistent with the paradox that PPARγ promotes insulin sensitivity while promoting fat differentiation which could precipitate insulin resistance, a partial loss-of-function Pro12Ala mutation in humans or heterozygosity in mice results in improved insulin sensitivity, while the gain-of-function Pro115Gln mutation in humans results in obesity and insulin resistance [15]. These genetic studies suggest that a PPARγ modulator might exhibit a better insulin sensitizing profile than a full agonist. PPARγ is also abundantly expressed in foam cell macrophages in human aortic atherosclerotic lesions and is activated by pro-atherogenic lipoprotein, oxidized LDL [32]. PPARγ ligands decrease atherosclerosis in mice, whereas loss-of-PPARγ in bone marrow cells promotes it. The anti-atherogenic activity of PPARγ in macrophages involves anti-inflammatory effects as well as activation of reverse cholesterol transport. Cardiovascular effects of TZD in diabetic patients are still controversial [35].
PPARδ is the least studied subtype of PPARs. It is ubiquitously expressed and acts as a sensor for polyunsaturated fatty acids and VLDL lipoprotein particle [36]. Generally, PPARδ activation promotes mitochondrial fatty acid oxidation, energy expenditure and thermogenesis. PPARδ deficient mice are prone to obesity and insulin resistance. Transgenic overexpression of a constitutively active PPARδ in adipose tissue or skeletal muscle on the other hand protects mice from diet-induced obesity, insulin resistance, steatosis and dyslipidemia [33]. In addition, overexpression in skeletal muscle results in oxidative fiber type switch and improves running endurance. PPARδ agonists are under extensive exploration aimed at raising serum HDL in patients with atherogenic dyslipidemia as they reduce serum triglyceride and increase HDL-cholesterol in multiple animal models, as well as humans. Moreover, they suppress diet-induced obesity and insulin resistance in rodents. PPARδ agonists, like PPARγ and PPARα agonists, suppresses inflammatory gene expression in macrophages, and thereby supresses the development of atherosclerosis in animal models [32].
LXR and FXR -the Ying and Yang for cholesterol and triglyceride metabolism
LXR (liver X receptor) α and β serve as oxysterol (hydroxycholesterol) sensors and are critical for whole body cholesterol homeostasis [37,38]. Mice lacking LXRs accumulate cholesterol in multiple tissues, including liver, brain, adrenal tissues and arterial walls. In the liver, LXRs mediate cholesterol catabolism through the conversion of cholesterol to bile acids via induction of a key enzyme in cholesterol catabolism CYP7A1 (in rodents but not in human), as well as the promotion of cholesterol clearance by elevating fatty acid synthesis (through SREBP1c) and VLDL secretion. In the peripheral tissues and macrophages, LXR facilitates cholesterol removal by inducing reverse cholesterol transport. Synthetic LXR agonists inhibit the development of atherosclerosis in mice at least in part through this metabolic modulation as well as suppression of inflammation. LXR ligand has also been shown to ameliorate diet-induced insulin resistance, in part by suppressing hepatic gluconeogenesis and by stimulating insulin secretion by pancreatic β cells. The main adverse effect of LXR ligands is an increase in SREBP-1c mediated hepatic lipogenesis and resulting serum triglyceride levels, which are believed to be mediated by hepatic LXRα, whereas beneficial effects are mediated by both α and β subtypes. Thus LXRβ-selective agonists may have cardioprotective anti-diabetic effects without causing liver triglyceride accumulation. Very recently, LXR was reported to act as a sensor for glucose [39]. Though the physiological importance of this observation is not yet clear, it may provide insight to our understanding of glucose homeostasis.
FXR (farnesoid X receptor) serves as a sensor for bile acids, the end products of hepatic cholesterol catabolism, and counteracts LXR in both cholesterol and triglyceride metabolism [38,40]. On one hand, FXR blocks cholesterol catabolism in response to increased bile acid levels via suppression of the LXR target CYP7A. FXR also promotes enterohepatic clearance of bile acids by controlling the expression of bile acid transporters as well as fibroblast growth factors. On the other hand, FXR promotes lipid clearance through inducing genes involved in lipoprotein metabolism and inhibits hepatic lipogenesis via repression of SREBP-1c. Repression of CYP7A and SREBP-1c, two LXR regulated genes, occurs at least in part through the induction of an atypical NR SHP (Small Heterodimer Partner) which lacks DBD and inhibits other NRs by forming a non-functional heterodimer [41]. FXR deficient mice show increased serum bile acids, triglyceride and VLDL and LDL cholesterol levels, in addition to other metabolic abnormalities. Conversely, synthetic FXR agonists (or bile acids) dramatically decrease serum triglyceride levels, and have therefore been proposed for clinical use against dyslipidemia. Synthetic FXR agonists also prevent cholesterol gallstone disease and ameliorate cholestasis in rodent models by promoting hepatobiliary bile acid clearance. In addition, the cholesterol-lowering plant sterol guggulsterone has been suggested to act by antagonizing FXR.
ERR and PGC-1 - regulators of mitochondrial energy homeostasis
There are three subtypes of ERRs, α, β, and γ, that share a high degree of sequence similarity with estrogen receptors, and can bind to estrogen receptor binding sites in vitro as dimers [19]. However these receptors often act as monomers in physiological settings through consensus half sites found in many genes involved in mitochondrial function [42,43]. The activity of ERRβ and γ can be modulated by synthetic agonists and antagonists, but it is believed that all of the ERRs are regulated not by ligands, but by ligand-independent binding of inducible coactivators PGC-1α and PGC-1β [44,45]. PGC-1 expression is abundant in oxidative tissues and under complex regulation by physiological stimuli such as exercise, fasting, and cytokines. The activity of PGC-1 is further regulated by covalent modification such as phosphorylation and acetylation. Upon activation by direct binding of PGC-1, ERR induces mitochondrial fatty acid β-oxidation and oxidative coupled respiration. PGC-1 and ERR may be dysregulated in diabetic muscle, as reduction in mitochondrial oxidative metabolism and increased intramuscular lipid are proposed to play a causative role in the development of insulin resistance [46]. Decreases in PGC-1α activity and the resulting mitochondrial dysfunction are also implicated in the development of neurodegenerative disorders [47]. Although an ERR agonist that can promote mitochondrial metabolism has not been identified, the SIRT-1 activator resveratrol has been proposed to ameliorate high-fat diet induced metabolic syndrome by activating PGC-1α [48].
In addition to ERR, PGC-1α and PGC-1β target other NR and non-NR transcription factors [44,45]. In particular, PGC-1 may activate PPAR α and PPARδ in heart, muscle and white adipose tissue to induce mitochondrial fatty acid oxidation. In addition, PGC-1α activates FXR and HNF-4α in the liver to induce triglyceride metabolism and hepatic gluconeogenesis during fasting respectively. PGC-1β activates SREBP-1c and FOXA2 to promote lipogenesis and VLDL secretion. Paradoxically, mice deficient for PGC-1α or PGC-1β show increased expression of hepatic gluconeogenic genes and lipogenic genes respectively, although in both cases mitochondrial gene expression was coordinately decreased [49,50].
PXR/CAR - xenobiotic receptors protecting the body from harmful metabolites
PXR and CAR are highly expressed in the liver and intestine, where they act as mediators of drug-induced multi-drug clearance [18,51]. PXR has a large flexible ligand binding pocket and binds numerous structurally unrelated chemicals, making it a true drug sensor [1]. On the other hand, CAR is constitutively active in the nucleus, and indirectly regulated by nuclear localization elicited by activating drugs through a membrane-juxtaposed kinase signaling pathway. Nonetheless, PXR and CAR regulate both overlapping and distinct sets of target genes, most of which are involved in oxidative and conjugative drug metabolism and drug transport. In addition to exogenous chemicals, both PXR and CAR are believed to be involved in the clearance of excess steroid hormones and detoxification of endogenously generated toxic compounds, such as bilirubin and toxic cholesterol metabolites. For example, the PXR agonist rifampin is reported to be effective in relieving cholestasis associated-pruritus. Likewise, the Chinese herb Yin Zhi Huang activates CAR and is effective to treat neonatal jaundice. Thus PXR and CAR activators could be used as potential therapeutics for hepatobiliary diseases such as cholestasis.
Orphan NRs implicated in metabolic diseases
Mutations in HNF4α protein cause Maturity Onset Diabetes of the Young (MODY1) in humans [52]. HNF4α is highly expressed in liver, kidney, and intestine as well as in pancreatic islets. Single nucleotide polymorphisms in both liver-specific and pancreatic-specific promoter regions are also associated with adult onset type 2 diabetes. HNF4α is a key regulator of hepatic gene expression and a major activator of HNF1α (MODY3), which in turn activates the expression of a large number of liver-specific genes, including those involved in glucose, cholesterol, and fatty acid metabolism. Indeed, mutations in the HNF4α binding site in HNF1α also cause MODY. The HNF4α knockout mouse is embryonic lethal. β-cell specific inactivation of HNF4α results in insulin resistance in mice, while mice lacking hepatic HNF4α expression accumulate lipids in the liver and exhibit greatly reduced serum cholesterol and triglyceride levels and increased serum bile acid concentrations [52–54]. Interestingly, endogenous fatty acids are able to bind to the ligand-binding pocket of HNF4α and result in a constitutively active conformation, although this is not believed to have a regulatory function [1,10].
The three members of the NR4A subfamily have recently been shown to act as important regulators of gluconeogenesis in fasting and diabetes [55]. NR4As are transcriptionally induced by the glucagon/cAMP axis in the liver, where they control the expression of many gluconeogenic genes. Elevated expression of NR4A is observed in diabetic animal models and also associated with insulin resistance and diabetes in some human studies. NR4A proteins have also been shown to modulate macrophage inflammatory responses and smooth muscle proliferation, highlighting a potential role for NR4As in cardiovascular disease [56,57]. Initial evidence suggests that NR4A activation protects mice against neointima formation, but their roles in atherosclerosis need to be further addressed in relevant animal models. Crystallography studies suggest that the ligand-binding pocket of NR4As is too small to bind any reasonably sized ligand, and indeed no ligand has been identified for NR4As [1,10].
As described above, SHP (along with another NR, DAX-1) is an atypical NR that does not have a DBD and acts by inhibiting other NR pathways [41]. In addition to mediating FXR-dependent repression of cholesterol catabolism and lipogenic programs by binding to LXR, LRH-1 and HNF4α on the CYP7A and SREBP-1c promoters, SHP can interact with many other NR and control broad aspects of nutrient metabolism. In humans, mutations in the SHP gene are associated with mild obesity [41], whereas SHP deficiency in mice causes increased energy expenditure, improved pancreatic β cell function and improved glucose homeostasis [58,59]. Regulation of energy expenditure by SHP is achieved by binding to ERRγ and the resulting suppression of PGC-1α expression in brown adipocytes [59].
In addition to HNF4α, NR4A and SHP, some less characterized orphan receptors are also likely to be involved in metabolic regulation. Rev-erbβ regulates the expression of genes involved in lipid absorption in skeletal muscle cells [60]. RORα binds to cholesterol and regulates the expression of apolipoproteins and RORα deficient staggerer mice show premature atherosclerois lesion development [61]. Although it is not clear whether activity of orphan NRs described here could be pharmacologically modulated, current evidence clearly indicates the importance of these proteins in metabolic regulation. Further study on the regulation of orphan NRs may uncover possible therapeutic strategies through these receptor functions.
Conclusion and perspectives
The transcriptional activity of many NRs can be modulated by lipophilic ligands, and in turn NRs control fundamental processes important for metabolic and energy homeostasis. Thus, NRs provide a powerful platform for drug discovery to treat metabolic disease. Indeed, a number of synthetic ligands for endocrine receptors as well as adopted orphan receptors are currently in clinical use or under clinical trial (Table 1). In addition to the receptors that we described here, there are a number of orphan receptors about which little is known. Based on the therapeutic promise of the adopted orphan receptors, we predict an auspicious future for orphan receptor research. In addition, the activities of NRs are further modulated by cofactors and membrane juxtaposed signaling pathways, which can also be targeted for pharmacological intervention. This was perhaps best illustrated by recent demonstrations that the SIRT-1 activator resveratrol promotes mitochondrial oxidative metabolism and ameliorates diet-induced insulin resistance by activating NR coactivator PGC-1α via deacetylation [48]. In addition to PGC-1, a number of NR cofactors such as p160/SRC proteins, p300/CBP, Mediator/TRAP220, SMRT/NCOR, RIP140 modulate multiple metabolic programs including adipogenesis, oxidative lipid metabolism, or glucose homeostasis, suggesting a possibility for therapeutic intervention via coactivator modulation [62]. In addition to typical transcriptional mechanisms via NR binding to elements in promoter regions, NRs can elicit their activity through binding to other transcription factors, signaling molecules, or metabolic enzymes. Indeed, the anti-inflammatory effects of GR, for which synthetic glucocorticoids are most often prescribed, are exerted through interactions with non-NR transcription factors such as NF-κB [63]. Anti-inflammatory effects of PPARs and LXRs are also likely mediated by non-DBD dependent protein-protein interaction.
Table 1.
NRs associated with metabolic disease, and their synthetic ligands. Refer to text for details.
| Receptor (or coactivator) | Associated Metabolic Disease or Pathologies | Synthetic ligand or modulator being used to ameliorate metabolic disease | Other representative synthetic ligands |
|---|---|---|---|
| GR | Viceral obesity, hyperglycemia | N/A | Dexamethasone, RU38486 |
| MR | Hypertension
Cardiac failure |
Aldosterone antagonist (Spironolactone, Eplerenone) | fludrocortisone |
| TRβ | Hypercholesterolemia | N/A | GC-1, KB-141 |
| PPARα | Dyslipidemia
Atherosclerosis |
fibrate (Gemfibrozil, fenofibrate, clofibrate) | WY14643 |
| PPARγ | Diabetes Mellitus, insulin-resistant
Atherosclerosis |
thiazolidinedione (pioglitazone, rosiglitazone) | FMOC-L-Leucine |
| PPARδ | Dyslipidemia
Atherosclerosis |
N/A | GW501516, KD3010 |
| LXRα/β | Dyslipidemia
Atherosclerosis |
N/A | T0901317, GW3965 |
| FXR | Hypercholesterolemia
Bilary cholestasis Dyslipidemia |
guggulsterone (antagonist) chenodeoxycholic acid (agonist) | Fexaramine, GW4046 |
| PXR | Bilary cholestasis, jaundice | St. John’s wort (hyperforin), rifampin | SR12813 |
| CAR | Bilary cholestasis, jaundice | Yin Zhi Huang (6,7-dimethylesculetin) | CITCO |
| ERRα/γ | Insulin resistance | N/A | XCT790, 4-hydroxytamoxifen |
| HNF4α | MODY, type I
Non-Insulin Dependent Diabetes Mellitus |
N/A | N/A |
| SHP | Mild obesity | N/A | N/A |
| RORα | Atherosclerosis | N/A | N/A |
In summary, although NR ligands already serve as a powerful tool to treat a number of human diseases, a deepening of our understanding of NR signaling pathways in mammalian physiology will help us better combat metabolic disease in this emerging era of pandemic obesity.
Acknowledgments
Owing to space constraints, numerous primary references could not be cited directly, but instead were included in other review articles cited in the text. We thank Grant Barish and Ruth Yu for critical reading of the manuscript. J.S. is supported by the American Heart Association. R.M.E. is an Investigator of the Howard Hughes Medical Institute and March of Dimes Chair in Molecular and Developmental Biology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nagy L, Schwabe JW. Mechanism of the nuclear receptor molecular switch. Trends Biochem Sci. 2004;29:317–24. doi: 10.1016/j.tibs.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 2.Evans RM. A transcriptional basis for physiology. Nat Med. 2004;10:1022–1026. doi: 10.1038/nm1004-1022. [DOI] [PubMed] [Google Scholar]
- 3.Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–95. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maglich JM, et al. Comparison of complete nuclear receptor sets from the human, Caenorhabditis elegans and Drosophila genomes. Genome Biol. 2001;2:RESEARCH0029. doi: 10.1186/gb-2001-2-8-research0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126:789–99. doi: 10.1016/j.cell.2006.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang X, Downes M, Yu RT, Bookout AL, He W, Straume M, Mangelsdorf DJ, Evans RM. Nuclear receptor expression links the circadian clock to metabolism. Cell. 2006;126:801–10. doi: 10.1016/j.cell.2006.06.050. [DOI] [PubMed] [Google Scholar]
- 7.Barish GD, Downes M, Alaynick WA, Yu RT, Ocampo CB, Bookout AL, Mangelsdorf DJ, Evans RM. A Nuclear Receptor Atlas: macrophage activation. Mol Endocrinol. 2005;19:2466–77. doi: 10.1210/me.2004-0529. [DOI] [PubMed] [Google Scholar]
- 8.Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev. 1997;18:306–60. doi: 10.1210/edrv.18.3.0303. [DOI] [PubMed] [Google Scholar]
- 9.Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006;20:1405–28. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Lambert MH, Xu HE. Activation of nuclear receptors: a perspective from structural genomics. Structure. 2003;11:741–6. doi: 10.1016/s0969-2126(03)00133-3. [DOI] [PubMed] [Google Scholar]
- 11.Baxter JD, Webb P, Grover G, Scanlan TS. Selective activation of thyroid hormone signaling pathways by GC-1: a new approach to controlling cholesterol and body weight. Trends Endocrinol Metab. 2004;15:154–7. doi: 10.1016/j.tem.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 12.Ma Y, et al. Identification and characterization of noncalcemic, tissue-selective, nonsecosteroidal vitamin D receptor modulators. J Clin Invest. 2006;116:892–904. doi: 10.1172/JCI25901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szanto A, Narkar V, Shen Q, Uray IP, Davies PJ, Nagy L. Retinoid X receptors: X-ploring their (patho)physiological functions. Cell Death Differ. 2004;11(Suppl 2):S126–43. doi: 10.1038/sj.cdd.4401533. [DOI] [PubMed] [Google Scholar]
- 14.Shulman AI, Mangelsdorf DJ. Retinoid × receptor heterodimers in the metabolic syndrome. N Engl J Med. 2005;353:604–15. doi: 10.1056/NEJMra043590. [DOI] [PubMed] [Google Scholar]
- 15.Semple RK, Chatterjee VK, O’Rahilly S. PPAR gamma and human metabolic disease. J Clin Invest. 2006;116:581–9. doi: 10.1172/JCI28003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lefebvre P, Chinetti G, Fruchart JC, Staels B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J Clin Invest. 2006;116:571–80. doi: 10.1172/JCI27989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kallen J, Schlaeppi JM, Bitsch F, Delhon I, Fournier B. Crystal structure of the human RORalpha Ligand binding domain in complex with cholesterol sulfate at 2.2 A. J Biol Chem. 2004;279:14033–8. doi: 10.1074/jbc.M400302200. [DOI] [PubMed] [Google Scholar]
- 18.Timsit YE, Negishi M. CAR and PXR: The xenobiotic-sensing receptors. Steroids. 2007;72:231–46. doi: 10.1016/j.steroids.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giguere V. To ERR in the estrogen pathway. Trends Endocrinol Metab. 2002;13:220–5. doi: 10.1016/s1043-2760(02)00592-1. [DOI] [PubMed] [Google Scholar]
- 20.Forman BM. Are those phospholipids in your pocket? Cell Metab. 2005;1:153–5. doi: 10.1016/j.cmet.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 21.Chrousos GP, Kino T. Intracellular glucocorticoid signaling: a formerly simple system turns stochastic. Sci STKE. 2005;2005:pe48. doi: 10.1126/stke.3042005pe48. [DOI] [PubMed] [Google Scholar]
- 22.Seckl JR. 11beta-hydroxysteroid dehydrogenases: changing glucocorticoid action. Curr Opin Pharmacol. 2004;4:597–602. doi: 10.1016/j.coph.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 23.Opherk C, Tronche F, Kellendonk C, Kohlmuller D, Schulze A, Schmid W, Schutz G. Inactivation of the glucocorticoid receptor in hepatocytes leads to fasting hypoglycemia and ameliorates hyperglycemia in streptozotocin-induced diabetes mellitus. Mol Endocrinol. 2004;18:1346–53. doi: 10.1210/me.2003-0283. [DOI] [PubMed] [Google Scholar]
- 24.Thiemermann C. Corticosteroids and cardioprotection. Nat Med. 2002;8:453–5. doi: 10.1038/nm0502-453. [DOI] [PubMed] [Google Scholar]
- 25.Walker BR, Andrew R. Tissue production of cortisol by 11beta-hydroxysteroid dehydrogenase type 1 and metabolic disease. Ann N Y Acad Sci. 2006;1083:165–84. doi: 10.1196/annals.1367.012. [DOI] [PubMed] [Google Scholar]
- 26.Connell JM, Davies E. The new biology of aldosterone. J Endocrinol. 2005;186:1–20. doi: 10.1677/joe.1.06017. [DOI] [PubMed] [Google Scholar]
- 27.Funder JW. Mineralocorticoid-receptor blockade, hypertension and heart failure. Nat Clin Pract Endocrinol Metab. 2005;1:4–5. doi: 10.1038/ncpendmet0016. [DOI] [PubMed] [Google Scholar]
- 28.Berger S, et al. Mineralocorticoid receptor knockout mice: pathophysiology of Na+ metabolism. Proc Natl Acad Sci U S A. 1998;95:9424–9. doi: 10.1073/pnas.95.16.9424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beggah AT, et al. Reversible cardiac fibrosis and heart failure induced by conditional expression of an antisense mRNA of the mineralocorticoid receptor in cardiomyocytes. Proc Natl Acad Sci U S A. 2002;99:7160–5. doi: 10.1073/pnas.102673599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng SY. Thyroid hormone receptor mutations and disease: beyond thyroid hormone resistance. Trends Endocrinol Metab. 2005;16:176–82. doi: 10.1016/j.tem.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 31.Johansson L, Rudling M, Scanlan TS, Lundasen T, Webb P, Baxter J, Angelin B, Parini P. Selective thyroid receptor modulation by GC-1 reduces serum lipids and stimulates steps of reverse cholesterol transport in euthyroid mice. Proc Natl Acad Sci U S A. 2005;102:10297–302. doi: 10.1073/pnas.0504379102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li AC, Palinski W. Peroxisome proliferator-activated receptors: how their effects on macrophages can lead to the development of a new drug therapy against atherosclerosis. Annu Rev Pharmacol Toxicol. 2006;46:1–39. doi: 10.1146/annurev.pharmtox.46.120604.141247. [DOI] [PubMed] [Google Scholar]
- 33.Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004;10:355–61. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 34.Smyth S, Heron A. Diabetes and obesity: the twin epidemics. Nat Med. 2006;12:75–80. doi: 10.1038/nm0106-75. [DOI] [PubMed] [Google Scholar]
- 35.Krall RL. Cardiovascular safety of rosiglitazone. Lancet. 2007;369:1995–6. doi: 10.1016/S0140-6736(07)60824-1. [DOI] [PubMed] [Google Scholar]
- 36.Barish GD, Narkar VA, Evans RM. PPAR delta: a dagger in the heart of the metabolic syndrome. J Clin Invest. 2006;116:590–7. doi: 10.1172/JCI27955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tontonoz P, Mangelsdorf DJ. Liver X receptor signaling pathways in cardiovascular disease. Mol Endocrinol. 2003;17:985–93. doi: 10.1210/me.2003-0061. [DOI] [PubMed] [Google Scholar]
- 38.Kalaany NY, Mangelsdorf DJ. LXRS and FXR: the yin and yang of cholesterol and fat metabolism. Annu Rev Physiol. 2006;68:159–91. doi: 10.1146/annurev.physiol.68.033104.152158. [DOI] [PubMed] [Google Scholar]
- 39.Mitro N, Mak PA, Vargas L, Godio C, Hampton E, Molteni V, Kreusch A, Saez E. The nuclear receptor LXR is a glucose sensor. Nature. 2007;445:219–23. doi: 10.1038/nature05449. [DOI] [PubMed] [Google Scholar]
- 40.Lee FY, Lee H, Hubbert ML, Edwards PA, Zhang Y. FXR, a multipurpose nuclear receptor. Trends Biochem Sci. 2006;31:572–80. doi: 10.1016/j.tibs.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 41.Bavner A, Sanyal S, Gustafsson JA, Treuter E. Transcriptional corepression by SHP: molecular mechanisms and physiological consequences. Trends Endocrinol Metab. 2005;16:478–88. doi: 10.1016/j.tem.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 42.Schreiber SN, Emter R, Hock MB, Knutti D, Cardenas J, Podvinec M, Oakeley EJ, Kralli A. The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proc Natl Acad Sci U S A. 2004;101:6472–7. doi: 10.1073/pnas.0308686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mootha VK, et al. Erralpha and Gabpa/b specify PGC-1alpha-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc Natl Acad Sci U S A. 2004;101:6570–5. doi: 10.1073/pnas.0401401101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–70. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 45.Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116:615–22. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Petersen KF, Shulman GI. Etiology of insulin resistance. Am J Med. 2006;119:S10–6. doi: 10.1016/j.amjmed.2006.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McGill JK, Beal MF. PGC-1alpha, a new therapeutic target in Huntington’s disease? Cell. 2006;127:465–8. doi: 10.1016/j.cell.2006.10.023. [DOI] [PubMed] [Google Scholar]
- 48.Koo SH, Montminy M. In vino veritas: a tale of two sirt1s? Cell. 2006;127:1091–3. doi: 10.1016/j.cell.2006.11.034. [DOI] [PubMed] [Google Scholar]
- 49.Lin J, et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–35. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 50.Sonoda J, Mehl IR, Chong LW, Nofsinger RR, Evans RM. PGC-1beta controls mitochondrial metabolism to modulate circadian activity, adaptive thermogenesis, and hepatic steatosis. Proc Natl Acad Sci U S A. 2007;104:5223–8. doi: 10.1073/pnas.0611623104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sonoda J, Evans RM. Biological function and mode of action of nuclear xenobiotic receptors. Pure Appl Chem. 2003:11–12. 1733–1742. [Google Scholar]
- 52.Gupta RK, Kaestner KH. HNF-4alpha: from MODY to late-onset type 2 diabetes. Trends Mol Med. 2004;10:521–4. doi: 10.1016/j.molmed.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 53.Miura A, et al. Hepatocyte nuclear factor-4alpha is essential for glucose-stimulated insulin secretion by pancreatic beta-cells. J Biol Chem. 2006;281:5246–57. doi: 10.1074/jbc.M507496200. [DOI] [PubMed] [Google Scholar]
- 54.Gupta RK, Vatamaniuk MZ, Lee CS, Flaschen RC, Fulmer JT, Matschinsky FM, Duncan SA, Kaestner KH. The MODY1 gene HNF-4alpha regulates selected genes involved in insulin secretion. J Clin Invest. 2005;115:1006–15. doi: 10.1172/JCI200522365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pei L, Waki H, Vaitheesvaran B, Wilpitz DC, Kurland IJ, Tontonoz P. NR4A orphan nuclear receptors are transcriptional regulators of hepatic glucose metabolism. Nat Med. 2006;12:1048–55. doi: 10.1038/nm1471. [DOI] [PubMed] [Google Scholar]
- 56.Pei L, Castrillo A, Tontonoz P. Regulation of macrophage inflammatory gene expression by the orphan nuclear receptor Nur77. Mol Endocrinol. 2006;20:786–94. doi: 10.1210/me.2005-0331. [DOI] [PubMed] [Google Scholar]
- 57.Bonta PI, et al. Nuclear receptors Nur77, Nurr1, and NOR-1 expressed in atherosclerotic lesion macrophages reduce lipid loading and inflammatory responses. Arterioscler Thromb Vasc Biol. 2006;26:2288–94. doi: 10.1161/01.ATV.0000238346.84458.5d. [DOI] [PubMed] [Google Scholar]
- 58.Wang L, et al. Orphan receptor small heterodimer partner is an important mediator of glucose homeostasis. Mol Endocrinol. 2006;20:2671–81. doi: 10.1210/me.2006-0224. [DOI] [PubMed] [Google Scholar]
- 59.Wang L, Liu J, Saha P, Huang J, Chan L, Spiegelman B, Moore DD. The orphan nuclear receptor SHP regulates PGC-1alpha expression and energy production in brown adipocytes. Cell Metab. 2005;2:227–38. doi: 10.1016/j.cmet.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 60.Ramakrishnan SN, Lau P, Burke LJ, Muscat GE. Reverbbeta regulates the expression of genes involved in lipid absorption in skeletal muscle cells: evidence for cross-talk between orphan nuclear receptors and myokines. J Biol Chem. 2005;280:8651–9. doi: 10.1074/jbc.M413949200. [DOI] [PubMed] [Google Scholar]
- 61.Boukhtouche F, Mariani J, Tedgui A. The “CholesteROR” protective pathway in the vascular system. Arterioscler Thromb Vasc Biol. 2004;24:637–43. doi: 10.1161/01.ATV.0000119355.56036.de. [DOI] [PubMed] [Google Scholar]
- 62.Yu S, Reddy JK. Transcription coactivators for peroxisome proliferator-activated receptors. Biochim Biophys Acta. 2007 doi: 10.1016/j.bbalip.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 63.Glass CK, Ogawa S. Combinatorial roles of nuclear receptors in inflammation and immunity. Nat Rev Immunol. 2006;6:44–55. doi: 10.1038/nri1748. [DOI] [PubMed] [Google Scholar]