

Abstract
Microglial cell activation and migration play an important role in neuroinflammation propagation. While it is known that the lipid transmitter palmitoylethanolamide (PEA) regulates microglial migration by interacting with a cannabinoid-like receptor, the production and inactivation of this lipid by microglia has never been addressed directly. Here we show that the mouse microglial cell line BV-2 produces and hydrolyzes PEA. The carbamate compound URB602 inhibits PEA hydrolysis in BV-2 cell homogenates and increases PEA levels in intact cells, whereas the FAAH inhibitor URB597 and serine-hydrolase inhibitor MAFP do not affect PEA levels in intact cells. This unique pharmacological profile of inhibitors on PEA hydrolysis suggests the involvement of a previously undescribed enzyme that degrades PEA. This enzyme expressed by microglia constitutes a promising target for controlling the propagation of neuroinflammation.
Keywords: Cannabinoids, Lipids, Lipase, Microglial cells, Hydrolysis, NAAA
INTRODUCTION
Anandamide (arachidonoylethanolamide, AEA) and its analogue palmitoylethanolamide (PEA) regulate many of the same pathophysiological processes, including pain perception, convulsions, neurotoxicity and inflammation (Calignano et al. 1998; Lo Verme et al. 2005; Jaggar et al. 1998; Lambert et al. 2001; Skaper et al. 1996). They both fulfill the three criteria required to be considered bona fide lipid transmitters: stimulus-dependent production, interaction with specific receptors and enzymatic inactivation. Yet recent reports suggest that AEA and PEA likely belong to independent signaling pathways, with distinct synthesis, receptors and inactivation (reviewed in Mackie and Stella 2006). Thus increasing our understanding of the molecular steps involved in either AEA or PEA biosynthesis or inactivation may lead to the identification of unique targets that will independently control AEA and PEA signaling.
Few studies exist on AEA and PEA biosynthesis. They both are present in the CNS and peripheral tissues, with PEA often being ten times more abundant than AEA (Cadas et al. 1997; Calignano et al. 1998; Franklin et al. 2003). Specific stimuli may lead to their independent accumulation. For example, in neurons in primary culture, activation of α7 nicotinic receptors increases AEA without changing PEA levels, whereas activation of muscarinic receptors increases PEA without affecting AEA levels (Stella and Piomelli 2001). In astrocytes in primary culture, ionomycin increases AEA without affecting PEA levels (Walter et al. 2002). In mouse brain, experimental autoimmune encephalomylitis leads to a 30-fold increase in PEA without changing AEA levels (Witting et al. 2006) (A. Witting and N. Stella, personal communication). Independent increases in AEA and PEA likely reflect independent biosynthetic pathways. Accordingly, PEA levels are reduced in NAPE-PLD knockout mouse brain, while AEA levels are unchanged (Leung et al. 2006). Whether NAPE-PLD is involved in stimuli-induced increases in either PEA or AEA is not known. Other biosynthetic pathways, including PLC, certain phosphatases (e.g. PTPN22), α/β-hydrolase 4 (Abh4) and metal-dependent phosphatases, may play a role in AEA synthesis (Liu et al. 2006; Simon and Cravatt 2006). Thus, both in vitro and in vivo evidence suggest independent pathways for the biosynthesis of PEA and AEA; but the precise molecular steps of their biosynthesis are unclear.
The receptors mediating the biological effects of AEA and PEA are also distinct, even though these lipids differ only by their fatty acid moiety (20:4 versus 16:0, respectively). AEA binds CB1 and CB2 cannabinoid receptors with high affinity, while PEA does not (Lambert et al. 1999; Sheskin et al. 1997). Several unique biological responses have been attributed to PEA. In BV-2 cells, PEA inhibits cAMP accumulation with an IC50 of 7 nM, and this response is insensitive to CB1 receptor antagonist SR141716A and the CB2 receptor antagonist SR144528 (Franklin et al. 2003). PEA binds PPAR-α and blocks inflammation in wild-type but not PPAR-α knockout mice, suggesting that it specifically interacts with this receptor (Lo Verme et al. 2005; Lo Verme et al. 2006). Thus, AEA mediates most of its biological effects by activating CB1 and CB2 receptors, while PEA activates either PPAR-α and/or an unknown Gi/o protein-coupled receptor.
At least two enzymes capable of hydrolyzing AEA and PEA have been reported. Fatty acid amide hydrolase (FAAH) preferentially hydrolyzes AEA over PEA (Ueda et al. 1995; Desarnaud et al. 1995; Cravatt et al. 1996), while the newly identified N-acylethanolamine-hydrolyzing acid amidase (NAAA) preferentially hydrolyzes PEA over AEA (Tsuboi et al. 2005). While FAAH and NAAA are both expressed in brain and have different pH sensitivities and pharmacological profiles (Sun et al. 2005), depending on the cell type and pathophysiological condition, PEA hydrolysis may occur through FAAH and/or NAAA. Thus, identification of selective inhibitors of either enzyme is necessary to selectively boost PEA or AEA signaling.
We have previously shown that BV-2 cells express a fully functional AEA signaling system, as these cells produce and inactivate AEA, and AEA modulates their migration (Walter et al. 2003). We have also shown that PEA regulates BV-2 cell migration through an unknown receptor (Franklin et al. 2003), but had not determined if these cells produce and inactivate PEA. Here we sought to address these questions and test the hypothesis that PEA signaling is independent of AEA signaling in microglial cells.
METHODS
Materials
URB597 (3’carbamoyl-biphenyl-3-yl-cyclohexylcarbamate), URB602 ((1,1-biphenyl)-3-yl-carbamic acid cyclohexyl ester), and MAFP (methylarachidonyl fluorophosphate) were from Cayman Chemical (Ann Arbor, MI). [3H]-PEA (radiolabeled on the ethanolamine) was from American Radiolabeled Chemicals (St. Louis, MO) and the National Institute on Drug Abuse drug supply system. Anandamide, PEA, and d4-PEA were synthesized in the lab (Walter et al. 2002).
Homogenate preparation and measurement of [3H]-PEA hydrolysis
Eight × 106 BV-2 cells (in 100 mm dishes) were rinsed once with PBS, lysed in 1 ml of ice-cold Hepes (250 mM) – Sucrose (10 mM) buffer (pH 7.4) and homogenized on ice with a Dounce tissue homogenizer. Homogenates (20 μg of proteins in 400 μl of Tris.HCl (100 mM, pH7.4) were added to silanized glass tubes placed on ice and containing either 0.5 μl of drug in DMSO or DMSO alone (0.1%, control). Hydrolysis was initiated by adding 100 μl of [3H]-PEA (3 nM, ≈ 70,000 dpm) in Tris HCl (0.1% fatty acid-free BSA). Tubes were incubated in a shaking water bath at 37°C. Reactions were stopped by adding 2 ml of ice-cold MeOH-CHCl3 (1:1) and the products of hydrolysis extracted by vigorous mixing and subsequent centrifugation at 800 × g (10 min). One milliliter of the upper phase was recovered, mixed with Ecoscint (4 ml), and radioactivity therein determined by liquid scintillation. Tubes containing only buffer were used as control for chemical hydrolysis (blank) and this value was systematically subtracted.
Quantification of PEA by chemical ionization gas chromatography/mass spectrometry (CI-GC/MS)
PEA amounts in BV-2 cells (3 × 106 cells/100 mm dish) were measured as described with some modifications (Walter and Stella 2003; Walter et al. 2002). Briefly, cell media was replaced by MEM + CellGro® (10 mL). After 12 hrs, MAFP, URB597, URB602 or 0.1% DMSO were added (in 1 mL MEM + CellGro®) to the cells for 20 min under gentle agitation in a shaking water bath kept at 37°C. Media was then removed and cells fixed with ice-cold MeOH (5 mL). The homogenates were recovered in glass vials containing 200 pmol of d4-PEA in CHCl3 (10 mL). PBS (2.5 mL) was then added to obtain a 4:2:1 ratio of CHCl3, MeOH and water. Following vigorous mixing and centrifugation at 1000 × g (5 min), the organic phase was recovered and dried under a nitrogen stream. The residue was recovered in CHCl3 and purified by silica open-bed chromatography using EtOAc-Acetone (1:1) as elution solvent. PEA was then converted to its trimethylsilyl derivative by BSTFA and quantified by isotope dilution using CI-GC/MS.
Data analysis
GraphPad PRISM® (version 4) was used to analyze the data and generate dose-response curves.
RESULTS
We used CI-GC/MS and isotope dilution to determine whether BV-2 cells produce PEA. Our first set of experiments was designed to determine the number of BV-2 cells required to reliably detect endogenously produced PEA. Indeed, endogenous PEA is quantified by spiking lipids extracted from BV-2 cells with deuterated PEA (200 pmol). Because deuterated PEA always contains a small amount of non-deuterated PEA, we wanted to determine the number of BV-2 cells required to generate a PEA/d4-PEA ratio that is statistically greater than the PEA/d4-PEA ratio generated by deuterated PEA alone. We found that our batch of d4-PEA contained 14 ± 0.67 % of non-deuterated PEA (Fig 1, white bar) and that 3 × 106 BV-2 cells generated a ratio (17.5 ± 0.68 %) that was statistically greater than the ratio generated by deuterated PEA alone (Fig 1). According to our isotope dilution calibration curve, this ratio corresponds to 1.83 pmol of PEA per 106 cells.
Figure 1. BV-2 cells produce PEA.
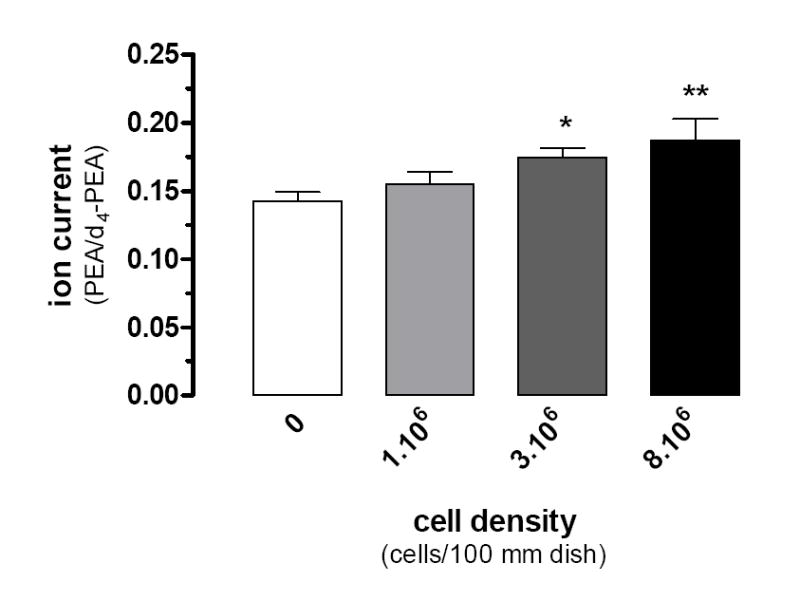
Increasing numbers of BV-2 cells (1, 3, and 8×106/100 mm dish) in MEM+Cellgro® were incubated for 20 min (37°, 0.1% DMSO) before lipid extraction and PEA quantification by CI-GC/MS. The PEA/d4-PEA ratios were determined and compared to the ratio found with the d4-PEA standard alone. Data are the mean of 3 experiments performed in duplicate (i.e. 6 dishes/condition) ± SEM. ANOVA followed by Dunnett’s post-test was used (* = P < 0.05; ** = P < 0.01).
Our second set of experiments was designed to verify the chromatographic properties and fragmentation pattern of the PEA peak generated when analyzing 3 × 106 BV-2 cells. Under the GC conditions used in our study, deuterated PEA eluted at 9.35 minutes (Fig 2). Its mass spectra included two predominant fragments that we had previously reported and have high diagnostic value (Walter et al. 2002): ions at m/z 376, corresponding to the protonated TMS molecules ([M+H]+) and ions at m/z of 448, corresponding to the protonated di-TMS molecules (the second TMS being added to the nitrogen of the ethanolamine moiety) (Fig 2). When analyzing BV-2 cell lipid extracts and following fragments at m/z 372 and 444 (i.e. resulting from endogenous PEA), we found a predominant peak at 9.35 min (Fig 2). Together, these results show that BV-2 cells do indeed produce PEA.
Figure 2. CI-GC/MS characterization of endogenous PEA in BV-2 cells.
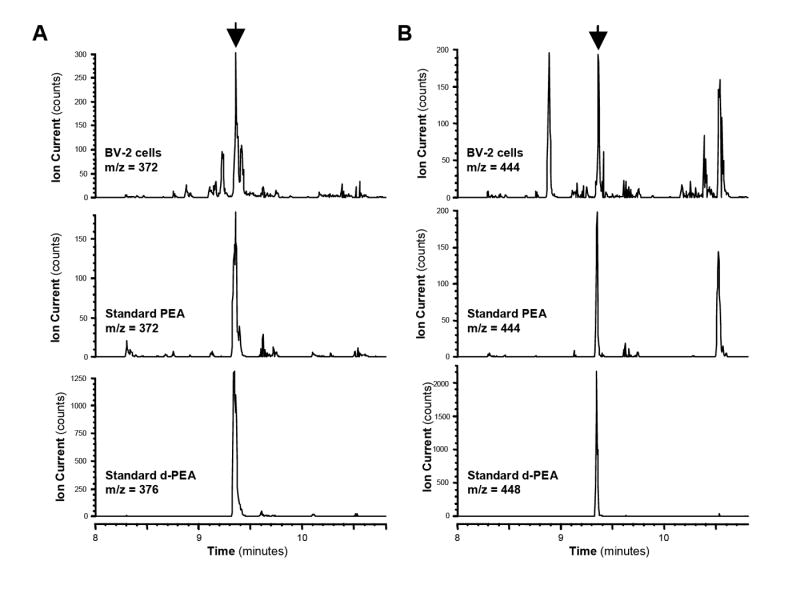
GC/MS chromatogram peak (arrow) from BV-2 cells lipid extracts (upper panels), PEA standard (middle panels), and d4-PEA standard (lower panels) corresponding to the mono-TMS adduct (A, m/z of 372 and 376 for PEA and d4-PEA, respectively) and di-TMS adduct (B, m/z of 444 and 448 for PEA and d4-PEA, respectively).
We then sought to determine if BV-2 cells hydrolyze PEA. BV-2 cell homogenates hydrolyzed [3H]-PEA in a protein-and time-dependent manner, yielding a specific activity of 0.2 pmol/min/mg of protein (Fig. 3a,b). This activity started to saturate at 35 μg of protein when using 20 min of incubation at pH 7 (Fig 3a). Note that [3H]-PEA hydrolysis was optimal at pH 8 (Fig 4a), which is reminiscent of the pH dependence described for FAAH, and minimal at pH 5 (Fig 3a and 4a), the optimal pH for NAAA activity (Ueda et al. 1999; Ueda et al. 1995). Both PEA and AEA competed for [3H]-PEA hydrolysis with IC50 values of 1.1 μM and 0.4 μM, respectively, which again is reminiscent of FAAH (Jonsson et al. 2001) (Fig. 4b). To further characterize this PEA hydrolyzing activity, we tested the effect of three commonly used inhibitors of endocannabinoid hydrolysis: MAFP, URB597 and URB602. We found that MAFP, a non-selective fluorophosphonate inhibitor of numerous serine hydrolases, inhibited [3H]-PEA hydrolysis with an IC50 of 0.07 nM (Fig. 4c). URB597, the rather selective FAAH inhibitor (Lichtman et al. 2004; Zhang et al. 2007) inhibited [3H]-PEA hydrolysis with an IC50 of 1.3 nM. URB602, a carbamate compound that inhibits both 2-AG and AEA hydrolysis (Hohmann et al. 2005; Vandevoorde et al. 2007; Muccioli et al. 2007), inhibited [3H]-PEA hydrolysis with an IC50 of 4.3 μM. These results show that PEA hydrolysis in BV-2 cell homogenates is mediated by a serine hydrolase activity that is sensitive to both URB597 and URB602.
Figure 3. BV-2 cell homogenates hydrolyze PEA.
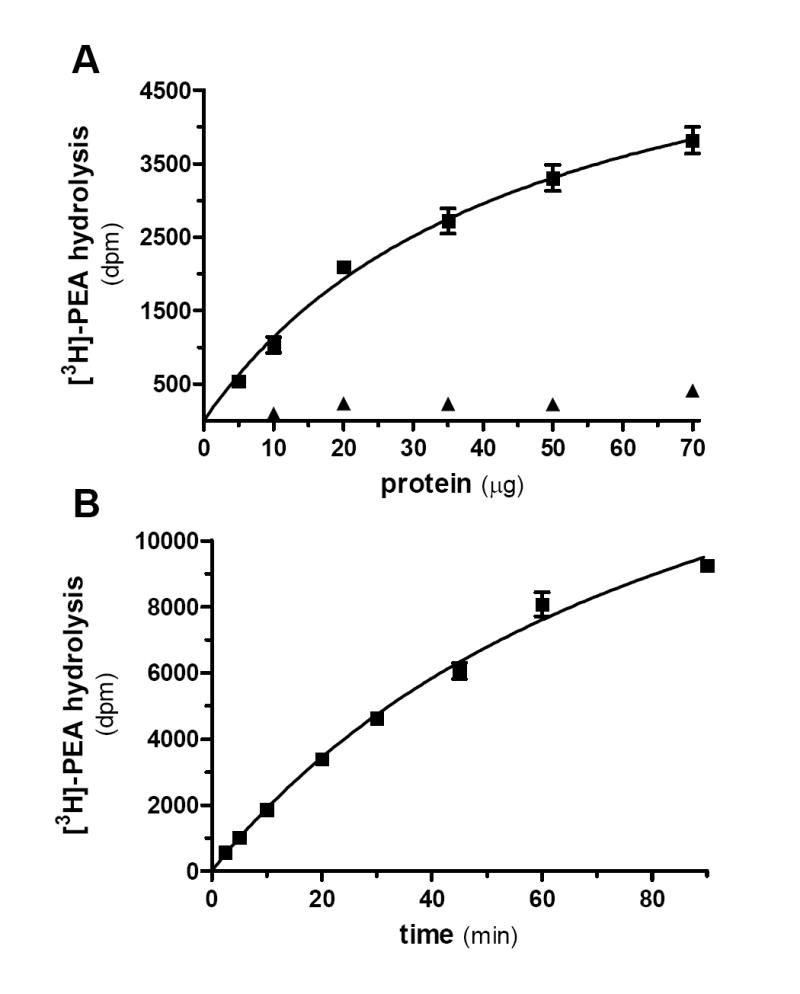
[3H]-PEA hydrolysis by BV-2 whole cell homogenate was measured at pH 7.4 using increasing amounts of homogenate (A, incubating 20 minutes) or increasing duration of incubation (B, using 20 μg of protein). When incubated at pH 5, the activity is almost completely absent (A, triangles). Values are mean ± SEM (3 experiments performed in duplicate).
Figure 4. Characterization of PEA hydrolysis by BV-2 cell homogenates.
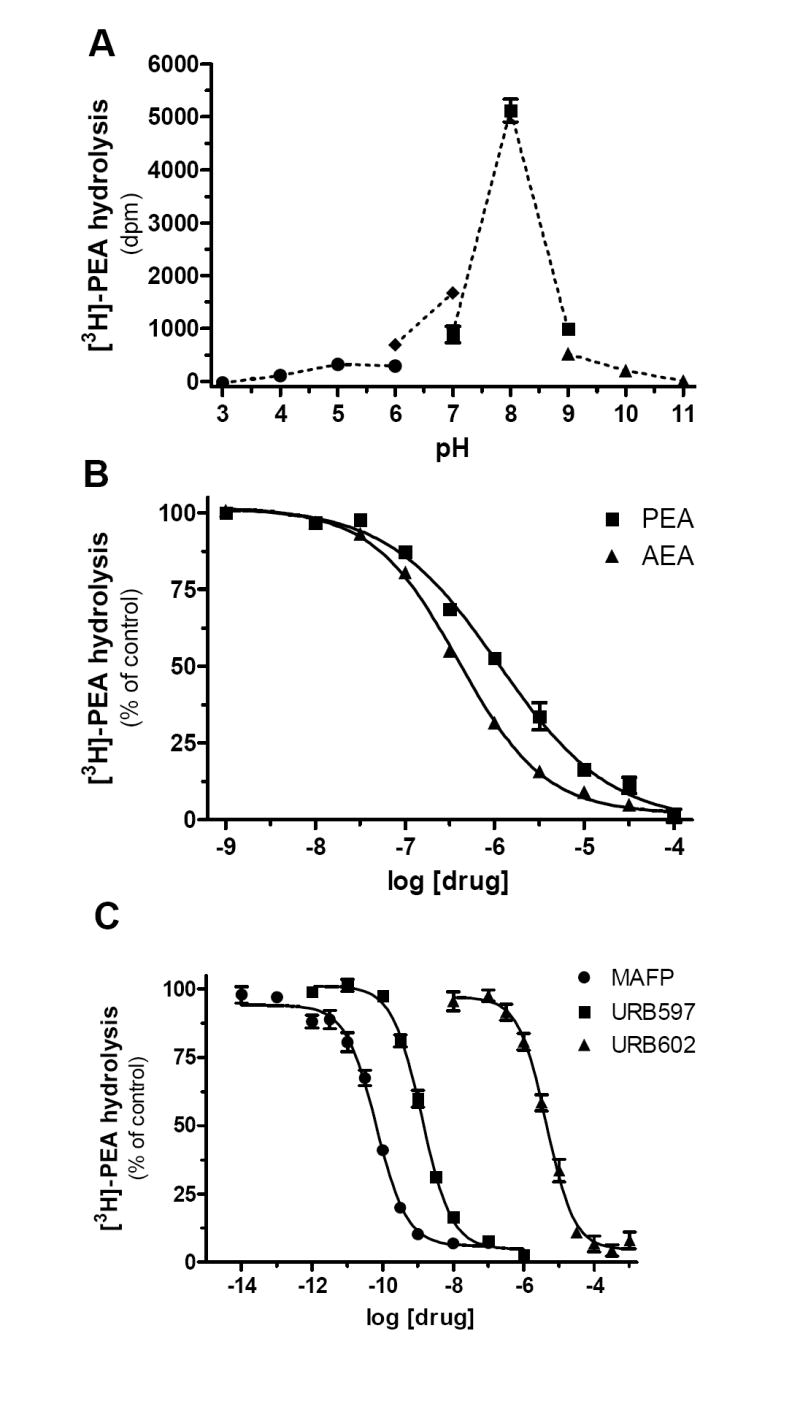
(A) Influence of the pH on [3H]-PEA hydrolysis. pH was adjusted with the following buffers (100 mM): NaAcetate (from pH 3 to pH 6), HEPES (from pH 6 to pH 7), Tris (from pH 7 to pH 9) and Na2B4O7 (from pH 9 to pH 11). (B) Effect of increasing concentrations of PEA (squares) or AEA (triangles) on [3H]-PEA hydrolysis by BV-2 whole cell homogenates. (C) [3H]-PEA hydrolysis is completely, and dose-dependently, inhibited by MAFP (circles), URB597 (squares), and URB602 (triangles). Values are mean ± SEM (3 experiments performed in duplicate) and are expressed as percentage of control.
Because of the pharmacological profile of PEA hydrolysis in BV-2 cell homogenates, we sought to test the effect of these inhibitors on PEA levels in intact BV-2 cells. Thus we treated BV-2 cells in culture with MAFP, URB597 or URB602, and quantified PEA levels by CI-GC/MS. Figure 5 shows that, in contrast to our results obtained in cell homogenates, MAFP (1 μM) and URB597 (0.1 μM) did not significantly affect PEA levels in intact BV-2 cells (although MAFP had a trend to increase PEA levels, but this effect did not reach statistical significance). Remarkably, URB602 at 100 μM induced a 5-fold increase in PEA levels in BV-2 cells. This result shows that the pharmacological profile of PEA hydrolysis in homogenates does not correspond to the pharmacological profile of inhibiting PEA hydrolysis in intact cells and that URB602 efficiently increases PEA levels in intact cells.
Figure 5. PEA levels in intact BV-2 cells are increased by URB602 but not by URB597 or MAFP.
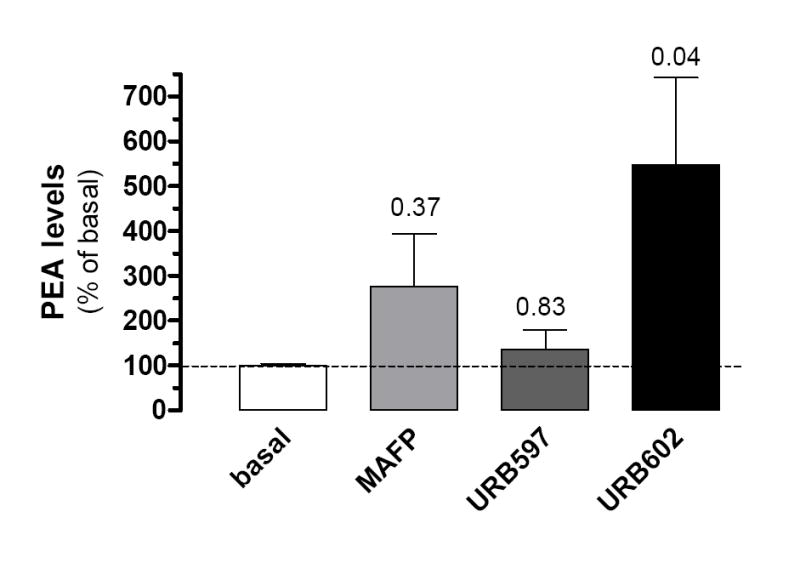
BV-2 cells (3×106/dish) were incubated (37°, 20 min) with MAFP (1 μM), URB597 (100 nM), URB602 (100 μM) or 0.1% DMSO (basal). Lipids were then extracted and PEA quantified by CI-GC/MS. PEA levels in the presence of inhibitors are compared to PEA basal level and are the mean of 4-5 experiments performed in duplicate (i.e. 8 to 10 dishes/condition) ± SEM. ANOVA followed by Dunnett’s post-test was used and P values are indicated.
DISCUSSION
We show that BV-2 cells, a cell line commonly used to study microglial cell function, produce 1.5 fold more PEA than 2-AG and 1.3 fold less than AEA (Muccioli et al. 2007), and degrade PEA through a URB602-sensitive enzymatic pathway. Since BV-2 cells also respond to PEA (Franklin et al. 2003), our results suggest that these cells express a fully functional PEA signaling system. Because URB602 increases PEA levels without affecting AEA levels (Muccioli et al. 2007), our results confirm the existence of independent PEA and AEA signaling pathways in microglia.
A recent study reported the identification of NAAA, an enzyme capable of hydrolyzing PEA more efficiently than AEA (Tsuboi et al. 2005). While NAAA is expressed by peripheral macrophages and RAW264.7 cells and is thought to degrade PEA in these cells (Sun et al. 2005), we provide evidence that NAAA does not participate in PEA hydrolysis by BV-2 cells, which is also a macrophage-like cell line. Specifically, while NAAA activity is optimal at pH 5 and insensitive to URB597 and MAFP (up to 10 and 1 μM, respectively) (Sun et al. 2005; Tsuboi et al. 2005), we found that [3H]-PEA hydrolysis by BV-2 cell homogenates is minimal at pH 5, and entirely inhibited by URB597 and MAFP at pH 7. Furthermore, we found that BV-2 cell homogenates hydrolyze PEA with a lower specific activity (0.2 pmol/min/mg of protein) than AEA and 2-AG, 0.4 and 1.2 pmol/min/mg of protein, respectively (Muccioli et al. 2007). Thus, both the pH dependence and pharmacological profile of [3H]-PEA hydrolysis by BV-2 homogenates allowed us to rule out the contribution of NAAA in this activity.
When focusing on PEA hydrolysis by BV-2 cell homogenates, three sets of evidence suggest the involvement of FAAH. First, the pH profile of [3H]-PEA hydrolysis parallels the pH profile described for [3H]-AEA hydrolysis by FAAH, both of which reach optimal activity at pH 8-9 (Omeir et al. 1995; Ueda et al. 1995). Second, MAFP and URB597 inhibit [3H]-PEA hydrolysis with IC50 values corresponding to those reported for recombinant FAAH (Lichtman et al. 2004). Third, both PEA and AEA compete for [3H]-PEA hydrolysis with similar micromolar IC50 values. However, our results obtained with intact cells clearly refute the involvement of FAAH in controlling PEA levels. Indeed, treating intact BV-2 cells with URB597 and MAFP at concentrations known to increase AEA by 2-3 fold (Muccioli et al. 2007) does not significantly affect PEA levels. Because AEA levels measured in intact cells are increased by URB597 and MAFP when using the same protocol (Muccioli et al. 2007), we are confident that these inhibitors reached FAAH. Thus we conclude that FAAH can hydrolyze PEA when experiments are performed with homogenates, but FAAH does not control PEA levels in intact BV-2 cells. A similar scenario is also true for 2-AG. Several reports have shown that FAAH hydrolyzes 2-AG when experiments are performed with homogenates, but pharmacological inhibition and genetic deletion of FAAH does not affect 2-AG levels in intact cells and tissue (Kathuria et al. 2003; Patel et al. 2005).
We found that URB602 inhibits [3H]-PEA hydrolysis by BV-2 homogenates as well as increases PEA levels in intact BV-2 cells. Yet in our previous study, we had found that URB602 also inhibits [3H]-AEA hydrolysis by BV-2 homogenates but does not lead to an increase in AEA levels in intact BV-2 cells (Muccioli et al. 2007). How can the discrepancy between results obtained with homogenates and intact cells be reconciled? We would like to propose the following two interpretations, which are not mutually exclusive. First, it is possible that AEA and PEA synthesis have distinct sub-cellular locations in intact cells and that FAAH is only expressed where AEA is synthesized. Accordingly, treating intact cells with URB597 and MAFP increases AEA levels, without affecting PEA levels (Muccioli et al. 2007). However, when cell homogenates are prepared and incubated with exogenously added [3H]-AEA and [3H]-PEA, both lipids may now reach FAAH and be hydrolyzed. Accordingly, recombinant FAAH is capable of using both AEA and PEA as substrates (Boger et al. 2000; Wei et al. 2006). Note that one study showed that recombinant FAAH activity measured in homogenates is inhibited by micromolar concentrations of URB602 ((Vandevoorde et al. 2007). Second, it is possible that an unknown, URB602-sensitive, enzymatic activity is expressed by BV-2 cells and is located where PEA is synthesized in intact cells. Accordingly, treating intact cells with URB602 increases PEA levels without affecting AEA levels. However, when cell homogenates are prepared and incubated with exogenously added [3H]-AEA and [3H]-PEA, both lipids may now reach this unknown enzyme and be hydrolyzed. Thus our results suggest that BV-2 cells express a novel PEA-hydrolyzing enzyme that is sensitive to URB602, which further emphasizes that this compound is not a specific MGL inhibitor. The definite proof for the existence of such enzyme will have to wait for its molecular cloning. Note that an enzyme capable of hydrolyzing PEA and insensitive to URB597 has been identified in rat duodenum (Fegley et al. 2005). Whether this enzyme is NAAA or the same enzyme as the one described here is not known.
We have previously shown that BV-2 cells also express a novel 2-AG-hydrolyzing activity that is also sensitive to URB602 (Muccioli et al. 2007). Two lines of evidence suggest that the novel 2-AG hydrolyzing enzyme is yet also distinct from the novel PEA hydrolyzing enzyme described here. First, MAFP inhibits the novel 2-AG hydrolyzing enzyme and increases 2-AG levels in intact cells without changing PEA levels (Muccioli et al. 2007). Second, URB602 inhibits the novel 2-AG hydrolyzing enzyme with an IC50 value of 7.6 mM, while inhibiting the PEA hydrolyzing enzyme described here with an IC50 value of 4.3 μM. Thus, our results suggest that BV-2 cells express three distinct endocannabinoid-hydrolyzing enzymes: FAAH that controls AEA levels, a novel 2-AG hydrolyzing enzyme that controls 2-AG levels and a novel PEA hydrolyzing enzyme that controls PEA levels.
The physiological role and pharmacological properties of PEA in the CNS are poorly understood compared to those of AEA and 2-AG. While PEA is found in significant levels in whole mouse or rat brains (100-550 pmol/g) (Cravatt et al. 2004; Fegley et al. 2004; Franklin et al. 2003; Patel et al. 2005), pathophysiological stimuli may selectively increase its levels. Neurons in culture produce PEA (Stella and Piomelli 2001) and astrocytes produce 2 to 3 times more PEA than AEA (Walter et al. 2002). Activated microglial cells migrate toward damaged cells to either repair or further damage these cells depending on their activation profile. PEA potentiates microglial cell motility through a Gi/o-protein mechanism distinct from CB1 and CB2 cannabinoid receptors. Thus compounds that selectively target the PEA signaling system might constitute promising leads for the identification of novel therapies aimed at controlling the propagation of chronic neuroinflammation.
In summary, the BV-2 microglial cell line produces, responds to and hydrolyzes PEA, and URB602 increases PEA levels in these cells. Although the precise target of URB602 is unknown, the relevance of this result is highlighted by the fact that PEA enhances AEA-induced migration of BV-2 cells through a non-CB1, non-CB2, Gi/o protein-coupled receptor.Thus, our study suggests that BV-2 cells constitute a viable cellular model for developing PEA-based therapeutics for chronic neuroinflammation.
Acknowledgments
This work was supported by the National Institute on Drug Abuse DA014486 and DA022469 (to N.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Boger DL, Fecik RA, Patterson JE, Miyauchi H, Patricelli MP, Cravatt BF. Fatty acid amide hydrolase substrate specificity. Bioorganic & Medicinal Chemistry Letters. 2000;10:2613–2616. doi: 10.1016/s0960-894x(00)00528-x. [DOI] [PubMed] [Google Scholar]
- Cadas H, di Tomaso E, Piomelli D. Occurrence and biosynthesis of endogenous cannabinoid precursor, N-arachidonoyl phosphatidylethanolamine, in rat brain. Journal of Neuroscience. 1997;17:1226–1242. doi: 10.1523/JNEUROSCI.17-04-01226.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Saghatelian A, Hawkins EG, Clement AB, Bracey MH, Lichtman AH. Functional disassociation of the central and peripheral fatty acid amide signaling systems. Proceedings of the National Academy of Sciences USA. 2004;101:10821–10826. doi: 10.1073/pnas.0401292101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desarnaud F, Cadas H, Piomelli D. Anandamide amidohydrolase activity in rat brain microsomes. Identification and partial characterization. Journal of Biological Chemistry. 1995;270:6030–6035. doi: 10.1074/jbc.270.11.6030. [DOI] [PubMed] [Google Scholar]
- Fegley D, Kathuria S, Mercier R, Li C, Goutopoulos A, Makriyannis A, Piomelli D. Anandamide transport is independent of fatty-acid amide hydrolase activity and is blocked by the hydrolysis-resistant inhibitor AM1172. Proceedings of the National Academy of Sciences USA. 2004;101:8756–8761. doi: 10.1073/pnas.0400997101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fegley D, Gaetani S, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. Characterization of the Fatty Acid Amide Hydrolase Inhibitor Cyclohexyl Carbamic Acid 3’-Carbamoyl-biphenyl-3-yl Ester (URB597): Effects on Anandamide and Oleoylethanolamide Deactivation. Journal of Pharmacology And Experimental Therapeutics. 2005;313:352–358. doi: 10.1124/jpet.104.078980. [DOI] [PubMed] [Google Scholar]
- Franklin A, Parmentier-Batteur S, Walter L, Greenberg DA, Stella N. Palmitoylethanolamide increases after focal cerebral ischemia and potentiates microglial cell motility. Journal of Neuroscience. 2003;23:7767–7775. doi: 10.1523/JNEUROSCI.23-21-07767.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- Jaggar SI, Hasnie FS, Sellaturay S, Rice ASC. The anti-hyperalgesic actions of the cannabinoid anandamide and the putative CB2 receptor agonist palmitoylethanolamide in visceral and somatic inflammatory pain. Pain. 1998;76:189–199. doi: 10.1016/s0304-3959(98)00041-4. [DOI] [PubMed] [Google Scholar]
- Jonsson KO, Vandevoorde S, Lambert DM, Tiger G, Fowler CJ. Effects of homologues and analogues of palmitoylethanolamide upon the inactivation of the endocannabinoid anandamide. British Journal of Pharmacology. 2001;133:1263–1275. doi: 10.1038/sj.bjp.0704199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nature Medicine. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- Lambert DM, DiPaolo FG, Sonveaux P, Kanyonyo M, Govaerts SJ, Hermans E, Bueb JL, Delzenne NM, Tschirhart EJ. Analogues and homologues of N-palmitoylethanolamide, a putative endogenous CB2 cannabinoid, as potential ligands for the cannabinoid receptors. Biochimica et Biophysica Acta, Molecular and Cell Biology of Lipids. 1999;1440:266–274. doi: 10.1016/s1388-1981(99)00132-8. [DOI] [PubMed] [Google Scholar]
- Lambert DM, Vandevoorde S, Diependaele G, Govaerts SJ, Robert AR. Anticonvulsant activity of N-palmitoylethanolamide, a putative endocannabinoid, in mice. Epilepsia. 2001;42:321–327. doi: 10.1046/j.1528-1157.2001.41499.x. [DOI] [PubMed] [Google Scholar]
- Leung D, Saghatelian A, Simon GM, Cravatt BF. Inactivation of N-Acyl Phosphatidylethanolamine Phospholipase D Reveals Multiple Mechanisms for the Biosynthesis of Endocannabinoids. Biochemistry. 2006;45:4720–4726. doi: 10.1021/bi060163l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman AH, Leung D, Shelton CC, Saghatelian A, Hardouin C, Boger DL, Cravatt BF. Reversible Inhibitors of Fatty Acid Amide Hydrolase That Promote Analgesia: Evidence for an Unprecedented Combination of Potency and Selectivity. Journal of Pharmacology And Experimental Therapeutics. 2004;311:441–448. doi: 10.1124/jpet.104.069401. [DOI] [PubMed] [Google Scholar]
- Liu J, Wang L, Harvey-White J, Osei-Hyiaman D, Razdan R, Gong Q, Chan AC, Zhou Z, Huang BX, Kim HY, Kunos G. A biosynthetic pathway for anandamide. Proceedings of the National Academy of Sciences USA. 2006;103:13345–13350. doi: 10.1073/pnas.0601832103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Verme J, Fu J, Astarita G, La Rana G, Russo R, Calignano A, Piomelli D. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Molecular Pharmacology. 2005;67:15–19. doi: 10.1124/mol.104.006353. [DOI] [PubMed] [Google Scholar]
- Lo Verme J, Russo R, La Rana G, Fu J, Farthing J, Mattace-Raso G, Meli R, Hohmann A, Calignano A, Piomelli D. Rapid broad-spectrum analgesia through activation of peroxisome proliferator-activated receptor-alpha. Journal of Pharmacology And Experimental Therapeutics. 2006;319:1051–1061. doi: 10.1124/jpet.106.111385. [DOI] [PubMed] [Google Scholar]
- Mackie K, Stella N. Cannabinoid receptors and endocannabinoids: evidence for new players. AAPS Journal. 2006;8:E298–E306. doi: 10.1007/BF02854900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muccioli GG, Xu C, Cudaback E, Cisneros J-A, Lambert DM, Lopez-Rodriguez ML, Bajjalieh S, Stella N. Identification of a novel endocannabinoid-hydrolyzing enzyme expressed by microglial cells. Journal of Neuroscience. 2007;27:2883–2889. doi: 10.1523/JNEUROSCI.4830-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omeir RL, Chin S, Hong Y, Ahern DG, Deutsch DG. Arachidonoylethanolamide-[1,2-14C] as a substrate for anandamide amidase. Life Sciences. 1995;56:1999–2005. doi: 10.1016/0024-3205(95)00181-5. [DOI] [PubMed] [Google Scholar]
- Patel S, Carrier EJ, Ho W-SV, Rademacher DJ, Cunningham S, Reddy DS, Falck JR, Cravatt BF, Hillard CJ. The postmortal accumulation of brain N-arachidonylethanolamine (anandamide) is dependent upon fatty acid amide hydrolase activity. Journal of Lipid Research. 2005;46:342–349. doi: 10.1194/jlr.M400377-JLR200. [DOI] [PubMed] [Google Scholar]
- Sheskin T, Hanus L, Slager J, Vogel Z, Mechoulam R. Structural requirements for binding of anandamide-type compounds to the brain cannabinoid receptor. Journal of Medicinal Chemistry. 1997;40:659–667. doi: 10.1021/jm960752x. [DOI] [PubMed] [Google Scholar]
- Simon GM, Cravatt BF. Endocannabinoid biosynthesis proceeding through glycerophospho-N-acyl ethanolamine and a role for a/b-hydrolase 4 in this pathway. Journal of Biological Chemistry. 2006;281:26465–26472. doi: 10.1074/jbc.M604660200. [DOI] [PubMed] [Google Scholar]
- Skaper SD, Buriani A, Dal Toso R, Petrelli L, Romanello S, Facci L, Leon A. The ALIAmide palmitoylethanolamide and cannabinoids, but not anandamide, are protective in a delayed postglutamate paradigm of excitotoxic death in cerebellar granule neurons. Proceedings of the National Academy of Sciences USA. 1996;93:3984–3989. doi: 10.1073/pnas.93.9.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella N, Piomelli D. Receptor-dependent formation of endogenous cannabinoids in cortical neurons. European Journal of Pharmacology. 2001;425:189–196. doi: 10.1016/s0014-2999(01)01182-7. [DOI] [PubMed] [Google Scholar]
- Sun YX, Tsuboi K, Zhao LY, Okamoto Y, Lambert DM, Ueda N. Involvement of N-acylethanolamine-hydrolyzing acid amidase in the degradation of anandamide and other N-acylethanolamines in macrophages. Biochimica and Biophysica Acta. 2005;1736:211–220. doi: 10.1016/j.bbalip.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Tsuboi K, Sun YX, Okamoto Y, Araki N, Tonai T, Ueda N. Molecular characterization of N-acylethanolamine-hydrolyzing acid amidase, a novel member of the choloylglycine hydrolase family with structural and functional similarity to acid ceramidase. Journal of Biological Chemistry. 2005;280:11082–11092. doi: 10.1074/jbc.M413473200. [DOI] [PubMed] [Google Scholar]
- Ueda N, Yamanaka K, Terasawa Y, Yamamoto S. An acid amidase hydrolyzing anandamide as an endogenous ligand for cannabinoid receptors. FEBS Letters. 1999;454:267–270. doi: 10.1016/s0014-5793(99)00820-0. [DOI] [PubMed] [Google Scholar]
- Ueda N, Kurahashi Y, Yamamoto S, Tokunaga T. Partial Purification and Characterization of the Porcine Brain Enzyme Hydrolyzing and Synthesizing Anandamide. Journal of Biological Chemistry. 1995;270:23823–23827. doi: 10.1074/jbc.270.40.23823. [DOI] [PubMed] [Google Scholar]
- Vandevoorde S, Jonsson KO, Labar G, Persson E, Lambert DM, Fowler CJ. Lack of selectivity of URB602 for 2-oleoylglycerol compared to anandamide hydrolysis in vitro. British Journal of Pharmacology. 2007;150:186–191. doi: 10.1038/sj.bjp.0706971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter L, Franklin A, Witting A, Wade C, Xie Y, Kunos G, Mackie K, Stella N. Nonpsychotropic cannabinoid receptors regulate microglial cell migration. Journal of Neuroscience. 2003;23:1398–1405. doi: 10.1523/JNEUROSCI.23-04-01398.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter L, Stella N. Endothelin-1 increases 2-arachidonoyl glycerol (2-AG) production in astrocytes. Glia. 2003;44:85–90. doi: 10.1002/glia.10270. [DOI] [PubMed] [Google Scholar]
- Walter L, Franklin A, Witting A, Moller T, Stella N. Astrocytes in Culture Produce Anandamide and Other Acylethanolamides. Journal of Biological Chemistry. 2002;277:20869–20876. doi: 10.1074/jbc.M110813200. [DOI] [PubMed] [Google Scholar]
- Wei BQ, Mikkelsen TS, McKinney MK, Lander ES, Cravatt BF. A Second Fatty Acid Amide Hydrolase with Variable Distribution among Placental Mammals. Journal of Biological Chemistry. 2006;281:36569–36578. doi: 10.1074/jbc.M606646200. [DOI] [PubMed] [Google Scholar]
- Witting A, Chen L, Cudaback E, Straiker A, Walter L, Rickman B, Moller T, Brosnan C, Stella N. Experimental autoimmune encephalomyelitis disrupts endocannabinoid-mediated neuroprotection. Proceedings of the National Academy of Sciences USA. 2006;103:6362–6367. doi: 10.1073/pnas.0510418103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Saraf A, Kolasa T, Bhatia P, Zheng GZ, Patel M, Lannoye GS, Richardson P, Stewart A, Rogers JC, Brioni JD, Surowy CS. Fatty acid amide hydrolase inhibitors display broad selectivity and inhibit multiple carboxylesterases as off-targets. Neuropharmacology. 2007;52:1095–1105. doi: 10.1016/j.neuropharm.2006.11.009. [DOI] [PubMed] [Google Scholar]