

Abstract
With many genomes sequenced, a pressing challenge in biology is predicting the function of the proteins that the genes encode. When proteins are unrelated to others of known activity, bioinformatics inference for function becomes problematic. It would thus be useful to interrogate protein structures for function directly. Here, we predict the function of an enzyme of unknown activity, Tm0936 from Thermotoga maritima, by docking high-energy intermediate forms of thousands of candidate metabolites. The docking hit list was dominated by adenine analogues, which appeared to undergo C6-deamination. Four of these, including 5-methylthioadenosine and S-adenosylhomocysteine (SAH), were tested as substrates, and three had substantial catalytic rate constants (105 M−1s−1). The X-ray crystal structure of the complex between Tm0936 and the product resulting from the deamination of SAH, S-inosylhomocysteine, was determined, and it corresponded closely to the predicted structure. The deaminated products can be further metabolized by T. maritima in a previously uncharacterized SAH degradation pathway. Structure-based docking with high-energy forms of potential substrates may be a useful tool to annotate enzymes for function.
For enzymes of unknown function, substrate prediction based on structural complementarity is, in principle, an alternative to bio-informatics inference of function1,2. Structure-based prediction becomes attractive when the target enzyme has little relationship to orthologues of known activity, making inference unreliable3,4. Whereas structure-based prediction has been used with some successes for inhibitor design, substrate prediction has proven difficult5–8. In addition to the well-known problems of sampling and scoring in docking, substrate prediction confronts several additional challenges. These include the many possible substrates to consider and the many reactions that an enzyme might catalyse9–11. Furthermore, enzymes preferably recognize transition states over the ground state structures that are usually represented in docking databases12–14.
Docking metabolites as high-energy intermediates
If, in its most general form, structure-based substrate prediction seems daunting, it may be simplified by several pragmatic choices. If we focus on a single class of reactions, here those catalysed by the amidohydrolase superfamily (AHS), of which Tm0936 is a member, we reduce the number of possible reactions from practically unbounded to a limited set of mechanistically related transformations. Thus, the 6,000 catalogued members of the AHS catalyse ∼30 reactions in biosynthetic and catabolic pathways15–17. All adopt a common (β/α)8-barrel fold and almost all are metallo-enzymes that cleave carbon-hetero-atom bonds. The problem of activity prediction may be further simplified by focusing on a single source of likely substrates, here the KEGG metabolite database18. Although substrate identification remains challenging—there are probably hundreds of molecules that are specifically recognized, not all of which are metabolites—it is at least a finite problem.
To address the challenge of transition state recognition, ground state structures were transformed into structures mimicking the high-energy intermediates that occur along the enzyme reaction coordinate. We will refer to these transition-state-like geometries as high-energy intermediates; this form of the substrate is among those that should best complement steric and electronic features of the enzyme active site14,19. All functional groups potentially recognized by AHS enzymes, for each of the 4,207 metabolites that bore them, were converted into high-energy intermediate geometries, with their appropriate charge distributions (Fig. 1). For instance, aromatic amines, which in the ground state are planar, are converted computationally into tetrahedral centres, representing the high-energy intermediate for deamination. Similarly, tetrahedral phosphates are converted into trigonal, bipyramidal forms. Overall, 28 amidohydrolase reactions operating on 19 functional groups were modelled by these high-energy structures, leading to the calculation of about 22,500 different forms of the metabolites. In retrospective calculations, docking these high-energy intermediate structures into seven well-studied amidohydrolases consistently identified the correct substrate from among the thousands of decoy molecules, typically outperforming docking of the ground state forms of the same molecules20,21.
Figure 1. Sample transformations of metabolites from their ground state structure into the high-energy intermediate forms that were used for docking.

Transformations were computed according to the conserved reaction mechanism of amidohydrolases, a nucleophilic attack of a hydroxide at an electrophilic centre atom. Every transformable functional group for each molecule was processed independently. If the high-energy structure was chiral, all stereoisomers were calculated. Reactions catalysed by the amidohydrolases cytosine deaminase (CDA), adenosine deaminase (ADA), dihydroorotase (DHO), d-hydantoinase (HYD), isoaspartyl-d-dipeptidase (IAD), N-acetyl-d-glucosamine-6-phosphate deacetylase (NaGA) and phosphotriesterase (PTE) are shown.
These retrospective results encouraged us to prospectively predict the substrates of Tm0936 from T. maritima. The X-ray structure of the enzyme had been determined as part of a broad structural genomics effort (PDB codes 1p1m and 1j6p), and it can be assigned to the AHS by fold classification and the identity of certain active site groups. Despite this, its substrate preference is anything but clear. By sequence similarity, Tm0936 most resembles the large chlorohydrolase and cytosine deaminase subgroup, which is often used to annotate amidohydrolases of unknown function17. Consistent with the view that this reflects an assignment to a broad subfamily and not a functional annotation, we tested 14 cytosine derivatives as Tm0936 substrates; no turnover was detected for any of them (see Methods). In an effort to find the true substrate, we therefore docked the database of high-energy intermediates into the structure of Tm0936, sampling thousands of configurations and conformations of each molecule. Each of these was scored by electrostatic and van der Waals complementarity, corrected for ligand desolvation energy, and ranked accordingly (see Methods)22,23.
The molecules best-ranked computationally were dominated by adenine and adenosine analogues, which make up 9 of the 10 top-scoring docking hits (Table 1, Supplementary Fig. 1). For all of these, an exocyclic nitrogen has been transformed into a tetrahedral, high energy centre, as would occur in a deamination reaction. The dominance of adenine and adenosine analogues, in this form, is due to nearly ideal interactions with the active site. An example is the docked structure of the high-energy intermediate for the deamination of 5-methylthioadenosine (MTA), the 6th ranked molecule (Fig. 2).
Table 1. The occurrence of adenine analogues among the top-ranked docking hits.
| Analogues in docking hit list | Top 10 ranked hits | Top 20 ranked hits | Top 100 ranked hits | Top 300 ranked hits |
|---|---|---|---|---|
| Adenine analogues | ||||
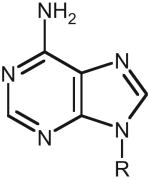
|
9 | 17 | 32 | 44 |
| Enrichment factor | 34 | 32 | 12 | 6 |
The enrichment factor is measured relative to the abundance of the analogues among the 4,207 potential substrates docked.
Figure 2. Binding and conversion of MTA by Tm0936.
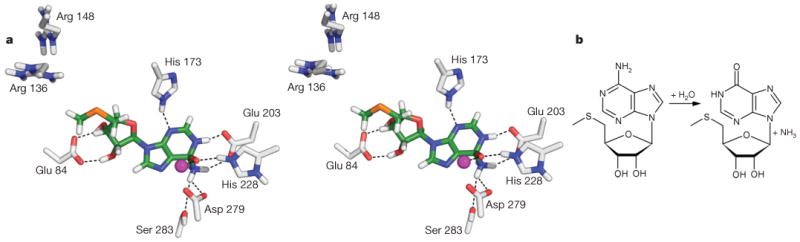
a, Stereoview of MTA in its high-energy intermediate form docked into the active site of Tm0936. Oxygen atoms are coloured red; enzyme carbons, grey; ligand carbons, green; hydrogens, white; nitrogens, blue; sulphur, orange; and the metal ion, purple. The oxyanion, representing the nucleophilic hydroxyl, ion-pairs with the metal ion and His 228. The ammonia leaving group is placed between Glu 203 and Asp 279, at 3.2 Å and 2.9 Å, respectively, also interacting with Ser 283 (3.2 Å). The N1-nitrogen donates a hydrogen bond to Glu 203, whereas N3 accepts one from His 173 (2.5 Å and 2.9 Å). Ribose hydroxyls hydrogen bond to Glu 84 (2.8 Å and 2.9 Å). Adenosines larger than MTA, such as SAH, make additional interactions with more distal residues, such as Arg 136 and Arg 148. All figures were rendered using PyMOL (http://pymol.sourceforge.net). b, The deamination of MTA to MTI, a reaction catalysed by Tm0936.
Experimental testing of the predicted substrates
On the basis of the docking ranks and compound availability, we selected four potential substrates for deamination by Tm0936: MTA, SAH, adenosine and adenosine monophosphate (AMP), all of which scored well (5th, 6th, 14th, 80th out of 4,207 docked metabolites), underwent the same reaction, and chemically resembled one another (Table 2). Although there were other high-ranking molecules in the docking hit list, most were single representatives of a chemotype and lacked the virtue of consistency of the adenines in general and the adenosines in particular. By extension, we also investigated the well-known metabolite S-adenosyl-l-methionine (SAM), a close analogue of SAH, even though its docking rank, at 511th, was poor.
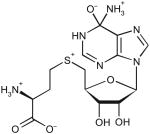
Table 2. Docking ranks and Tm0936 catalytic constants for five predicted substrates.
| Substrate tested | Docked high-energy intermediate form | Dock rank | Relative docking scores (kcal mol−1)* | Km (μM) | kcat (s−1) | kcat/Km (M−1s−1) |
|---|---|---|---|---|---|---|
| S-adenosyl-l-homocysteine | ||||||
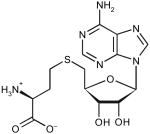
|
|
5 | 0 | 210 ± 40 | 12.2 ± 0.8 | 5.8 × 104 |
|
| ||||||
| 5-Methyl-thioadenosine | ||||||
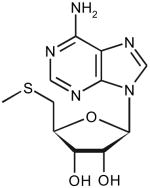
|
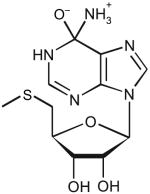
|
6 | 4.4 | 44 ± 4 | 7.2 ± 0.2 | 1.4 × 105 |
|
| ||||||
| Adenosine | ||||||
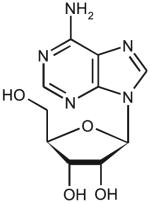
|
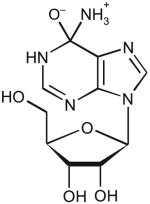
|
14 | 9.5 | 250 ± 40 | 2.3 ± 0.2 | 9.2 × 103 |
|
| ||||||
| Adenosine-5-monophosphate | ||||||
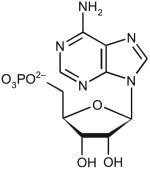
|
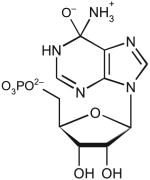
|
80 | 20.2 | ND | <10−3 | ND |
|
| ||||||
| S-adenosyl-l-methionine | ||||||
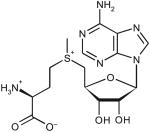
|
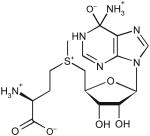
|
511 | 35.2 | ND | <10−3 | ND |
Deamination was measured by the production of ammonia. The standard deviations are given.
Docking energies relative to the best-ranked compound shown, SAH. Higher energies indicate worse scores. ND, not determined.
Of these five molecules, three had substantial activity as substrates, with MTA and SAH reaching kcat/Km values of 1.4 × 105 and 5.8 × 104 M−1s−1 respectively, and adenosine close to 104 M−1s−1 (Table 2 and Supplementary Information). The first order rate constant for the spontaneous deamination of adenosine in water is 1.8 × 10−10s−1, making this enzyme proficient for these substrates. Tm0936 is relatively active compared to other adenosine deaminases24, especially because the optimal temperature for this thermophilic enzyme is almost certainly higher than the 30 °C at which it was assayed. Consistent with the docking predictions, SAM was not deaminated by Tm0936, despite its close similarity to SAH. Conversely, AMP, which did rank relatively well (80th of 4,207), was also not an enzyme substrate. The inability of the docking programme to fully de-prioritize AMP reflects some of the well-known problems in docking scoring functions, in this case balancing ionic interactions and desolvation penalties for the highly charged phosphate group of AMP.
To investigate the mechanism further, we determined the structure of Tm0936 in complex with the purified product of the SAH deamination reaction, S-inosylhomocysteine (SIH), to 2.1 Å resolution by X-ray crystallography (Fig. 3, Methods). The differences between the docked prediction and the crystallographic result are minor, with every key polar and non-polar interaction represented in both structures (except that we docked the tetrahedral intermediate and the X-ray structure is of the ground state product). Indeed, the correspondence between the docked and crystallographic structures is closer than one might expect for inhibitor predictions, where docking has been more commonly used25–28. This may reflect the advantages of docking substrates in high-energy intermediate geometries, which encode more of the information necessary to specify fit.
Figure 3. Comparing the docking prediction and the crystallographic result.

Superposition of the crystal structure of Tm0936 in complex with SIH (red) and the docking predicted structure of the high-energy intermediate of SAH (carbons in green). Enzyme carbons are coloured light blue, SAH and enzyme oxygen atoms are coloured red, nitrogens blue and sulphurs orange. The purple sphere represents the divalent metal ion. An FO–FC omit electron density map for SIH is shown, contoured at 4.1 σ. The structure was determined at 2.1 Å resolution.
Metabolic pathway of a family of MTA/SAH deaminases
It is tempting to speculate that Tm0936 is not simply an isolated enzyme acting on particular substrates, but is involved in the deamination of metabolites in a previously uncharacterized MTA/SAH pathway. The deamination of adenosine itself is well known in all kingdoms of life, and the deamination of SAH to SIH has been reported in one organism, Streptomyces flocculus29. Very recently it was shown that MTA is deaminated in Plasmodium falciparum in an alternative degradation pathway of adenosine analogues30. To investigate whether the products of the deamination reactions, catalysed by Tm0936, SIH and MTI, could be further metabolized by other enzymes in T. maritima, we measured the activity of S-adenosyl homocysteinase (Tm0172), which hydrolyses SAH to homocysteine and adenosine, using SIH as a potential substrate. We found that Tm0172 catalyses the formation of homocysteine from either SIH or SAH about equally well (Supplementary Table 1 and Supplementary Information). This is consistent with Tm0172 and Tm0936 participating in a degradation pathway, though it does not confirm it. We cannot exclude the possibility that Tm0936 functions as an adenosine deaminase in T. maritima, because no other enzyme in the organism has been identified that serves this role.
What is clear is that Tm0936 has orthologues across multiple species. On the basis of the conservation of characteristic residues that interact with the substrate and product in the docked and X-ray structures, respectively, 78 other previously unannotated AHS enzymes from different species may now be classified as MTA/SAH/adenosine deaminases (Supplementary Fig. 2 and Supplementary Information). In all of these sequences, the metal-ligating residues (His 55, His 57, His 200 and Asp 279, Tm0936 numbering) are conserved, as are the residues recognizing the reactive centre (His 228, Ser 259, Ser 283 and Glu 203). Specificity is conferred by interactions between the substrate and Trp 75, Glu 84 and His 173, all of which are also conserved among the 78 amidohydrolases. Active site residues that vary include Arg 136 and Arg 148, which in Tm0936 interact with the α-carboxylate of the homocysteine moiety of SAH. These latter interactions are not critical to the activity of the enzyme, because these arginines do not seem to interact with MTA or adenosine, but they may be important for the recognition of SAH.
Many of the Tm0936 orthologues cluster with other genes that can now be associated with the metabolism of SAM, SAH or MTA. For example, in T. maritima Tm0936 is closely associated with Tm0938, which is currently annotated as a SAM-dependent methyl transferase. In Bacillus cereus, the Tm0936 orthologue is Bc1793, which is also closely associated with a SAM-dependent methyl transferase, Bc1797. In Pseudomonas aeruginosa, the Tm0936 orthologue, Pa3170, is adjacent to UbiG-methyltransferase, Pa3171. Other orthologues are adjacent or close to adenosyl homocysteinase, 5′-methylthioadenosine phosphorylase, MTA/SAH nucleosidase and other SAM-dependent methyl transferases.
Predicting function from form
This work describes one case of successful function prediction by structure-based docking, and it is appropriate to consider caveats. Our recognition of Tm0936 as an amidohydrolase limited the number of possible reactions to be considered. When even the gross mechanistic details of an enzyme cannot be inferred, this will not be possible. Restricting ourselves to metabolites was also helpful, but this too will not always be appropriate. Finally, we were fortunate that Tm0936 experienced little conformational change between the apo structure and that of the product complex. Enzymes that undergo large conformational changes along their reaction coordinates will be more challenging for docking.
If prudence warns against over-generalization, it is also unlikely that Tm0936 represents an isolated case. Other enzyme structures will be broadly classifiable by mechanism, and whereas conformational change remains a serious challenge, retrospective studies suggest that it is not insurmountable. Indeed, the most important technical innovation adopted here, modelling substrates as high-energy intermediates, was particularly useful when docking to apo structures in those studies (Supplementary Table 2 and Supplementary Information)20. Thus, the prediction and determination that Tm0936 acts as an MTA/SAH deaminase illustrates the possibilities of this and related structure-based approaches, at least for a subset of targets. The enzyme has no obvious sequence similarity to any known adenosine deaminase and exploits interactions not previously identified in the active sites of these enzymes. The very pathway in which Tm0936 participates seems novel. Structure-based docking of high-energy intermediates should be a useful tool to decrypt the activity of enzymes of unknown function, and will be especially interesting for those targets where bioinformatics inference is unreliable.
METHODS
Molecular docking
The 1.5 Å X-ray structure of Tm0936 (Protein Data Bank (PDB) code 1P1M) was used in docking calculations. High-energy intermediates of potential substrates were calculated20 and docked into the enzyme structure using the program DOCK3.5.54. Poses were scored for electrostatic and van der Waals complementarity and penalized for ligand desolvation31,32.
Enzymology
Tm0936 and Tm0172 from T. maritima were cloned, expressed and purified using standard techniques. The deamination reaction was measured by coupling the production of ammonia to the oxidation of NADH catalysed by glutamate dehydrogenase. The decrease in the concentration of NADH was followed spectrophotometrically at 340 nm. The chemical identities of the deaminated products were confirmed by mass spectrometry and specific changes in the ultraviolet absorption (UV) spectra for the deamination of adenosine derivatives. The SAH hydrolase activity by Tm0172 was determined by reaction of the free thiol group of the homocysteine product with dithio-bis(2-nitrobenzoic acid), monitoring the absorbance at 412 nm.
X-ray crystallography
Tm0936 was co-crystallized with ZnCl2 and SIH. X-ray diffraction data were collected at the NSLS X4A beamline (Brookhaven National Laboratory). The structure of the Tm0936-SIH complex was determined by molecular replacement, using apo Tm0936 (PDB code 1J6P) as the search model. The structure has been deposited in the protein data bank (PDB code 2PLM).
Supplementary Material
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Acknowledgments
This work was supported by grants from the National Institutes of Health, supporting docking analyses (to B.K.S.), large scale structural analysis (to S.C.A.), and function prediction (to F.M.R., B.K.S. and S.C.A.). F.M.R. thanks the Robert A. Welch Foundation for support. J.C.H. thanks the Deutsche Akademie der Naturforscher Leopoldina for a fellowship. We thank J. Irwin, V. Thomas and K. Babaoglu for reading this manuscript. The clone for Tm0172 was kindly supplied by the Joint Center for Structural Genomics.
Footnotes
J.C.H designed the docking database, performed the docking runs, and analysed the docking results. F.M.R. and R.M.-A. performed the enzymatic characterization of Tm0936 and Tm0172, including cloning and purification of the proteins. S.C.A., E.F. and A.A.F. determined the X-ray structure of Tm0936 with S-inosyl-homocysteine. J.C.H. and B.K.S. largely wrote the paper. All authors discussed the results and commented on the manuscript.
Author Information The complex structure of Tm0936 with SIH has been deposited in the PDB (accession code 2PLM). Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests.
References
- 1.Whisstock JC, Lesk AM. Prediction of protein function from protein sequence and structure. Q Rev Biophys. 2003;36:307–340. doi: 10.1017/s0033583503003901. [DOI] [PubMed] [Google Scholar]
- 2.Gerlt JA, Babbitt PC. Can sequence determine function? Genome Biol. 2000;1:REVIEWS0005. doi: 10.1186/gb-2000-1-5-reviews0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brenner SE. Errors in genome annotation. Trends Genet. 1999;15:132–133. doi: 10.1016/s0168-9525(99)01706-0. [DOI] [PubMed] [Google Scholar]
- 4.Devos D, Valencia A. Intrinsic errors in genome annotation. Trends Genet. 2001;17:429–431. doi: 10.1016/s0168-9525(01)02348-4. [DOI] [PubMed] [Google Scholar]
- 5.Schapira M, Abagyan R, Totrov M. Nuclear hormone receptor targeted virtual screening. J Med Chem. 2003;46:3045–3059. doi: 10.1021/jm0300173. [DOI] [PubMed] [Google Scholar]
- 6.Rao MS, Olson AJ. Modelling of factor Xa-inhibitor complexes: a computational flexible docking approach. Proteins. 1999;34:173–183. doi: 10.1002/(sici)1097-0134(19990201)34:2<173::aid-prot3>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 7.Sukuru SC, et al. Discovering new classes of Brugia malayi asparaginyl-tRNA synthetase inhibitors and relating specificity to conformational change. J Comput Aided Mol Des. 2006;20:159–178. doi: 10.1007/s10822-006-9043-5. [DOI] [PubMed] [Google Scholar]
- 8.Shoichet BK. Virtual screening of chemical libraries. Nature. 2004;432:862–865. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Macchiarulo A, Nobeli I, Thornton JM. Ligand selectivity and competition between enzymes in silico. Nature Biotechnol. 2004;22:1039–1045. doi: 10.1038/nbt999. [DOI] [PubMed] [Google Scholar]
- 10.Kalyanaraman C, Bernacki K, Jacobson MP. Virtual screening against highly charged active Sites: identifying substrates of α–β barrel enzymes. Biochemistry. 2005;44:2059–2071. doi: 10.1021/bi0481186. [DOI] [PubMed] [Google Scholar]
- 11.Irwin JJ, Shoichet BK. ZINC—a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schramm VL. Enzymatic transition states and transition state analogues. Curr Opin Struct Biol. 2005;15:604–613. doi: 10.1016/j.sbi.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 13.Hermann JC, Ridder L, Holtje HD, Mulholland AJ. Molecular mechanisms of antibiotic resistance: QM/MM modelling of deacylation in a class A β-lactamase. Org Biomol Chem. 2006;4:206–210. doi: 10.1039/b512969a. [DOI] [PubMed] [Google Scholar]
- 14.Warshel A, Florian J. Computer simulations of enzyme catalysis: finding out what has been optimized by evolution. Proc Natl Acad Sci USA. 1998;95:5950–5955. doi: 10.1073/pnas.95.11.5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holm L, Sander C. An evolutionary treasure: unification of a broad set of amidohydrolases related to urease. Proteins. 1997;28:72–82. [PubMed] [Google Scholar]
- 16.Seibert CM, Raushel FM. Structural and catalytic diversity within the amidohydrolase superfamily. Biochemistry. 2005;44:6383–6391. doi: 10.1021/bi047326v. [DOI] [PubMed] [Google Scholar]
- 17.Pegg SC, et al. Leveraging enzyme structure–function relationships for functional inference and experimental design: the structure–function linkage database. Biochemistry. 2006;45:2545–2555. doi: 10.1021/bi052101l. [DOI] [PubMed] [Google Scholar]
- 18.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tantillo DJ, Houk KN. Transition state docking: a probe for noncovalent catalysis in biological systems. Application to antibody-catalyzed ester hydrolysis. J Comput Chem. 2002;23:84–95. doi: 10.1002/jcc.10019. [DOI] [PubMed] [Google Scholar]
- 20.Hermann JC, et al. Predicting substrates by docking high-energy intermediates to enzyme structures. J Am Chem Soc. 2006;128:15882–15891. doi: 10.1021/ja065860f. [DOI] [PubMed] [Google Scholar]
- 21.Nowlan C, et al. Resolution of chiral phosphate, phosphonate, and phosphinate esters by an enantioselective enzyme library. J Am Chem Soc. 2006;128:15892–15902. doi: 10.1021/ja0658618. [DOI] [PubMed] [Google Scholar]
- 22.Wei BQ, Baase WA, Weaver LH, Matthews BW, Shoichet BK. A model binding site for testing scoring functions in molecular docking. J Mol Biol. 2002;322:339–355. doi: 10.1016/s0022-2836(02)00777-5. [DOI] [PubMed] [Google Scholar]
- 23.Lorber DM, Shoichet BK. Hierarchical docking of databases of multiple ligand conformations. Curr Top Med Chem. 2005;5:739–749. doi: 10.2174/1568026054637683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Radzicka A, Wolfenden R. A proficient enzyme. Science. 1995;267:90–93. doi: 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- 25.Mohan V, Gibbs AC, Cummings MD, Jaeger EP, DesJarlais RL. Docking: successes and challenges. Curr Pharm Des. 2005;11:323–333. doi: 10.2174/1381612053382106. [DOI] [PubMed] [Google Scholar]
- 26.Jorgensen WL. The many roles of computation in drug discovery. Science. 2004;303:1813–1818. doi: 10.1126/science.1096361. [DOI] [PubMed] [Google Scholar]
- 27.Kairys V, Fernandes MX, Gilson MK. Screening drug-like compounds by docking to homology models: a systematic study. J Chem Inf Model. 2006;46:365–379. doi: 10.1021/ci050238c. [DOI] [PubMed] [Google Scholar]
- 28.Klebe G. Virtual ligand screening: strategies, perspectives and limitations. Drug Discov Today. 2006;11:580–594. doi: 10.1016/j.drudis.2006.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Speedie MK, Zulty JJ, Brothers P. S-adenosylhomocysteine metabolism in Streptomyces flocculus. J Bacteriol. 1988;170:4376–4378. doi: 10.1128/jb.170.9.4376-4378.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tyler PC, Taylor EA, Fröhlich RFG, Schramm VL. Synthesis of 5′-methylthio coformycins: specific inhibitors for malarial adenosine deaminase. J Am Chem Soc. 2007;129:6872–6879. doi: 10.1021/ja0708363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meng EC, Shoichet B, Kuntz ID. Automated docking with grid-based energy evaluation. J Comp Chem. 1992;13:505–524. [Google Scholar]
- 32.Gschwend DA, Kuntz ID. Orientational sampling and rigid-body minimization in molecular docking revisited: on-the-fly optimization and degeneracy removal. J Comput Aided Mol Des. 1996;10:123–132. doi: 10.1007/BF00402820. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.