

Abstract
Interferon alpha (IFNα) is widely used in treatment of malignant melanoma patients. This cytokine acts on cells by engaging Type I IFN receptor consisting of two subunits, (IFNAR1 and IFNAR2) followed by activation of Janus kinases (Jak). Levels of IFNAR1 (regulated via degradation mediated by the βTrcp E3 ubiquitin ligase) and IFNα signaling were reduced in 1205Lu melanoma cell line that harbors activated BRAF and exhibits high levels of βTrcp ubiquitin ligase. Expression of stabilized IFNAR1 in melanoma cells decreased their tumorigenicity. Furthermore, RNAi-mediated BRAF knockdown and pharmacologic inhibition of either Raf or MEK1 decreased levels of βTrcp and stabilized IFNAR1. However, despite causing stabilization of IFNAR1, Raf inhibitor BAY 43-9006 interfered with cellular responses to IFNα most likely due to its ability to directly inhibit Jak activity. We discuss the implications of this result for combination therapy with BAY 43-9006 and IFNα in melanoma patients.
Keywords: BRAF, melanoma, interferon alpha, Raf inhibitor, β-Trcp, ubiquitin, BAY 43-9006
INTRODUCTION
Malignant melanoma is a lethal disease whose incidence is rapidly rising.1 Although surgery is effective in early melanoma stages, the high resistance of melanoma cells to chemo- and radiation therapy compromises post-surgical efforts to prevent recurrence and/or metastatic disease. Interferon alpha (IFNα) remains the adjuvant therapy of choice for high-risk patients with malignant melanoma.2 However, treatment with IFNα has a limited response rate and efficiency,3 and is very expensive.4,5 Overcoming these limitations and identification of novel therapeutic targets requires better understanding of mechanisms that restrict IFNα signaling.
INFα acts through its receptor consisting of IFNAR1 and IFNAR2 chains to induce the activation of Janus kinase (Jak) family members (Jak1 and Tyk2) who phosphorylate each other as well as the receptor subunits and the recruited members of signal transducers and activators of transcription (Stat) family at specific tyrosines. Tyrosine phosphorylation of Stat1 and Stat2 is required for their interaction with p48/IRF9 to form the IFN-stimulated transcription factor that binds to IFN-stimulated regulatory elements (ISRE) and activates transcription of IFN-stimulated genes (reviewed in Ref. 6–Ref. 8).
IFNAR1 is essential for anti-viral9 and anti-oncogenic10 effects of IFNα; levels of IFNAR1 play an important role in regulating the magnitude and duration of these effects.11 Previous work identified βTrcp2/HOS protein as an E3 ubiquitin ligase that negatively regulates IFNAR1 levels and signaling via targeting IFNAR1 for ubiquitination and degradation.12 Levels and activities of the βTrcp2 E3 ubiquitin ligase are maintained by mitogenic signaling via the MAPK pathway.13 Interestingly, we recently found that constitutively active MAPK signaling emanating from the oncogenic BRAF induces βTrcp2 E3 ubiquitin ligase levels in human malignant melanoma cells leading to accelerated degradation of IκB and constitutively high activity of NFκB transcription factor.14 Here we investigated the role of BRAF-MAPK-βTrcp signaling in regulating IFNAR1 stability and IFNα signaling in human melanoma cells.
MATERIALS AND METHODS
Cells, inhibitors and plasmids
Human melanoma cells were maintained as previously described.15 Transfections were performed with Lipofectamine Plus (Invitrogen) according to the manufacturer’s recommendations. MEK inhibitor PD 098059 (Calbiochem), and cycloheximide (Sigma) were purchased. BAY 43-9006 was a kind gift from Dr. Charles Smith (Hershey Medical Center, Pennsylvania State University). ShRNA against BRAFV600E was kindly provided by Dave Tuveson (Cambridge Research Institute, Cambridge UK). Vectors for the expression of Flag-tagged human and murine IFNAR1, and βTrcp2ΔN were described previously.12,16
Antibodies and Immunotechniques
Antibodies specific for Flag (M2, Sigma), HA (Covance), JAK1 (Santa Cruz), Erk, phospho-Erk, STAT1, phospho-STAT1 (Cell Signaling) and β-actin (Santa Cruz) were purchased. Antibodies recognizing endogenous βTrcp17 and IFNAR118 were described previously. Secondary antibodies conjugated to horseradish peroxidase were purchased from Chemicon. Immunoprecipitation and immunoblotting procedures are described elsewhere.19 Degradation of IFNAR1 was measured as previously described.12,20 Densitometry data were obtained and analyzed using Scion Image Software (version Beta 4.0.2) and the digital images were prepared using Adobe Photoshop 7.0 Software.
JAK in vitro kinase assay
Lu1205 cells were grown in 100mm plates and starved in serum free DMEM for 2 hr, followed by stimulation with IFN alpha 3000 u/ml for 30 minutes. Cells were rinsed with ice-cold PBS and lysed by adding lysis buffer (1% NP40, 50 mM Tris-HCl, 150 mM NaCl, protease inhibitor cocktail, 2 mM NaVO3, 0.2 µM okadaic acid, 1 mM PMSF, 50 mM NaF and 10% glycerol) directly onto the plates. Cell lysates were clarified by centrifugation and precleared with protein A beads for 1 hr at 4°C. 1 mg of total cell lysates were then incubated with JAK1 antibody for 1 hr and then incubated with protein A beads for an additional 2 hr. After centrifugation, beads were washed three times in JAK1 kinase assay buffer (10 mM HEPES pH 7.6, 50 mM NaCl, 5 mM MgCl2, 5 mM MnCl2, 0.1 mM Na3VO4) and preincubated with DMSO or different doses of BAY (1.25, 2.5, 5 and 10 µM) for ten minutes at 30°C and then the kinase reaction was initiated by adding 15 µM ATP followed by incubation at 30°C for 30 minutes. Reaction was stopped by adding SDS-PAGE loading buffer and analysed by immunoblotting with 4G10 phosphotyrosine antibody to detect tyrosine phosphorylation and JAK1 antibody to detect JAK1 levels.
RESULTS AND DISCUSSION
Given that βTrcp2 is also required for ubiquitination and degradation of IFNAR1, we analyzed the levels of IFNAR1 in human melanoma cell lines. As shown in Figure 1A, high levels of IFNAR1 and relatively low levels of βTrcp2 were detected in WM35 and SBcl2 cells that were shown to exhibit relatively low extent of MAPK activation.15 Conversely, 1205Lu and WM793 cell lines that are known to harbor BRAF mutations and exhibit high constitutive MAPK activity15 contained higher levels of βTrcp2 and lower levels of IFNAR1. Expression of other components of IFNα signaling pathway including Stat1, Stat2, Jak1 and Tyk2 were comparable in all tested cell lines (Fig. 1A). Consistent with the importance of IFNAR1 expression levels for IFNα signaling observed in fibroblasts,11,12,21 activation of Stat1 by IFNα was more robust in human melanoma cells that express higher levels of IFNAR1 (WM35 versus 1205Lu, Fig. 1B). This result indicates that melanoma cells that express high levels of βTrcp2 might be less sensitive to IFNα signaling due to a decrease in IFNAR1 levels. This, in turn, could be attributed to accelerated degradation of IFNAR1 mediated by βTrcp2. Indeed, similar to our previous results obtained in fibroblasts and epithelial cells,12 expression of a dominant negative βTrcp2ΔN mutant (known to inhibit the function of endogenous βTrcp proteins16,22) resulted in robust stabilization of IFNAR1 in 1205Lu cells (Fig. 1C). These results are consistent with the notion that βTrcp2 (which is induced by the BRAF-MAPK signaling,14) negatively regulates IFNAR1 stability and IFNα signaling in human melanoma cells.
Figure 1.
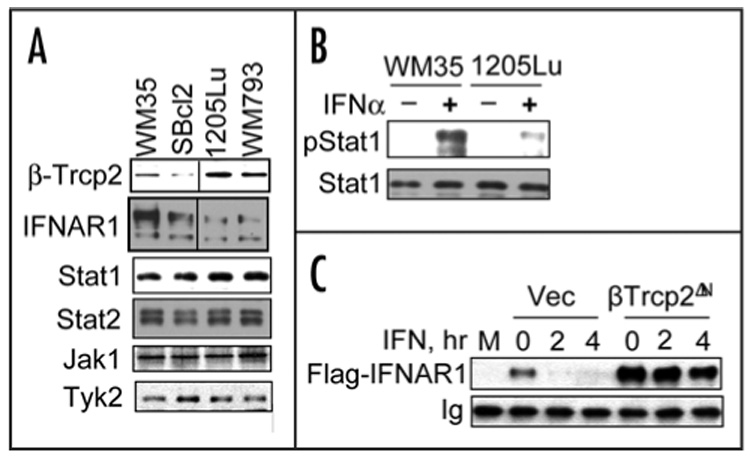
Degradation and levels of IFNAR1 in human melanoma cells are regulated by βTrcp2 E3 ubiquitin ligase. (A). Levels of expression of IFNAR1 and other components of Type I IFN signaling (Stat1, Stat2, Jak1 and Tyk2) as well as of βTrcp2 were analyzed by immunoblotting using indicated antibodies. (B). IFNα-induced tyrosine phosphorylation of Stat1 in human melanoma cells that express different levels of IFNAR1 analyzed by immunoblotting using indicated antibodies. (C). Activity of βTrcp2 is required for rapid IFNAR1 turnover in melanoma cells. Flag-tagged IFNAR1 was coexpressed in 1205Lu cells together with either empty vector (Vec) or with dominant negative βTrcp2ΔN mutant. Cells were treated with IFNα and cycloheximide as indicated. Levels of exogenous IFNAR1 were measured by immunoprecipitation-immunoblotting using anti- Flag antibody. M, mock-transfected cells. Ig, heavy chain immunoglobulin.
To determine the role of oncogenic BRAF and activated MAPK pathway in regulating IFNAR1 stability we next investigated the effects of BRAF and MAPK inhibitors on the rate of IFNAR1 turnover. Treatment of melanoma cells with MEK1 inhibitor PD098059 (which decreased the levels of βTrcp213,14 and data not shown) resulted in stabilization of endogenous IFNAR1 (Fig. 2A). Similar effects on the levels of βTrcp2 and the rate of IFNAR1 turnover were obtained when 1205Lu cells were treated with Raf inhibitor BAY43-9006 (Fig. 2B). These results indicate that BRAF-MAPK signaling might contribute to the destabilization of IFNAR1 and to a decrease in its levels in human melanoma cells.
Figure 2.

Maintenance of βTrcp2 levels via the BRAF signaling is required for degradation of IFNAR1 in human melanoma cells. (A) Efficient degradation of IFNAR1 in melanoma cells requires MAPK signaling. 1205Lu cells pretreated for 2 hr with either MEK1 inhibitor PD098059 or vehicle (DMSO) were incubated with IFNα and cycloheximide for indicated time. Levels of endogenous IFNAR1 were measured by immunoprecipitation-immunoblotting using anti-IFNAR1 antibodies. Ig, heavy chain immunoglobulin. NS, non-specific band. (B). BRAF activity mediates rapid degradation of IFNAR1 in melanoma cells. 1205Lu cells pretreated for 2 hr with either BAY43-9006 or vehicle (DMSO) were incubated with IFNα and cycloheximide for indicated time. Degradation of IFNAR1 was assessed as in (A). Levels of βTrcp2 and β-actin in whole cell extracts were also analyzed by immunoblotting. (C) Expression of BRAF is required for IFNAR1 turnover. Degradation of endogenous IFNAR1 in 1205Lu cells transfected with either irrelevant shRNA (shCon) or shRNA targeted against oncogenic BRAF (shBraf) was assessed as in (A). (C) Levels of βTrcp2 and activity of Erk in 1205Lu cells transfected with shRNA and βTrcp2-expressing vector (as indicated) were analyzed by immunoblotting using respective antibodies. Levels of β-actin are presented as a loading control. (E) Degradation of Flag-tagged IFNAR1 [coexpressed in 1205Lu cells with indicated shRNA and βTrcp2 constructs as described in (D)] were analyzed by cycloheximide chase followed by immunoblotting using anti-Flag antibody. Levels of β-actin are presented as a loading control. (F) The graph shows quantification of data presented in (E). Depicted is the percent of IFNAR1 remaining at each time point relative to time point “0” (100%). Symbols correspond to those shown in (D–E) and represent cells transfected with shCon (black diamonds), shBraf (gray squares) and shBraf together with βTrcp2 (white circles).
To further investigate the role of oncogenic BRAF in regulating IFNAR1 stability we used the RNAi approach in 1205Lu cells that are known to harbor activating V600E mutation in one allele of BRAF.15 Transfection of 1205Lu melanoma cells with an shRNA construct that specifically suppresses expression of the BRAFV600E mutant allele without affecting wild type BRAF14,23 led to a robust stabilization of IFNAR1 (Fig. 2C). This result provides genetic evidence in support of the role of oncogenic BRAF in accelerated degradation of IFNAR1.
In order to determine whether knockdown of oncogenic BRAF stabilizes IFNAR1 via downregulation of endogenous βTrcp2, we have cotransfected shRNA against oncogenic BRAFV600E together with recombinant βTrcp2 expression construct. The latter increased overall levels of βTrcp in 1205Lu human melanoma cells while having no effect on shBraf-mediated inhibition of the MAPK pathway (assessed by Erk phosphorylation, Fig. 2D). Under these conditions, expression of βTrcp2 noticeably reversed stabilization of IFNAR1 by shBraf (Figs. 2E–F). These results together suggest that oncogenic BRAF-MAPK signaling leads to acceleration of IFNAR1 degradation via inducing the expression of βTrcp2 in human melanoma cells.
Given that IFNAR1+/− heterozygous mice exhibit decreased responses to Type I IFN11,21 it is plausible that destabilization and downregulation of IFNAR1 in malignant melanomas might account for suboptimal antiproliferative effects of IFN. We next investigated whether IFNAR1 stability plays a role in growth of human melanoma cells. Inhibition of βTrcp function via retroviral introduction of the dominant negative βTrcp2ΔN mutant markedly slowed down both appearance and growth of tumors formed by 1205Lu cells transplanted into SCID mice (Fig. 3, compare open and closed circles). This effect should not be necessarily attributed to stabilization of IFNAR1 because βTrcp regulates degradation of many other proteins that might be important for tumor growth (such as IκB, Emi1, etc24). However, expression of a stabilized murine IFNAR1S526A mutant that cannot be regulated by βTrcp12 also delayed the appearance of tumors and inhibited their growth in mice (Fig. 3, closed squares). Together these results indicate that an increase in IFNAR1 levels that might result from impaired IFNAR1 degradation coincides with suppression of melanoma cell growth and suggest that targeting the mechanisms leading to IFNAR1 degradation might represent a promising strategy for treatment of melanoma.
Figure 3.

Stabilization of IFNAR1 inhibits growth of 1205Lu xenograft. Melanoma cells transduced either with empty retrovirus (open circles) or retroviruses expressing stable murine IFNAR1S526A mutant (closed squares) or dominant negative βTrcp2ΔN mutant (closed circles were transplanted subcutaneously in SCID mice. Percent of tumor-free mice (upper panel) and the volume of tumors (lower panel) at indicated days after the injection is shown. The inset depicts the size of representative tumors at day 30.
Such strategy could hypothetically combine treatment with Type I IFN together with available Raf or MAPK inhibitors to decrease BRAF signaling and βTrcp2 expression that should prevent rapid degradation of IFNAR1 and increase the extent of IFNα signaling. Indeed, knockdown of oncogenic BRAF via RNAi increased both basal and ligand-induced activity of luciferase driven by interferon stimulated response element (ISRE, Fig. 4A). Furthermore, pretreatment of 1205Lu cells with MEK1 inhibitor PD098059 noticeably enhanced the anti-proliferative effects of IFNα (Fig. 4B, open circles). These results are in line with observations of other groups who noted a similar contribution of MAPK to resistance to IFNα was shown in T cells,25 multiple myeloma26 and epidermoid carcinoma cells.27
Figure 4.
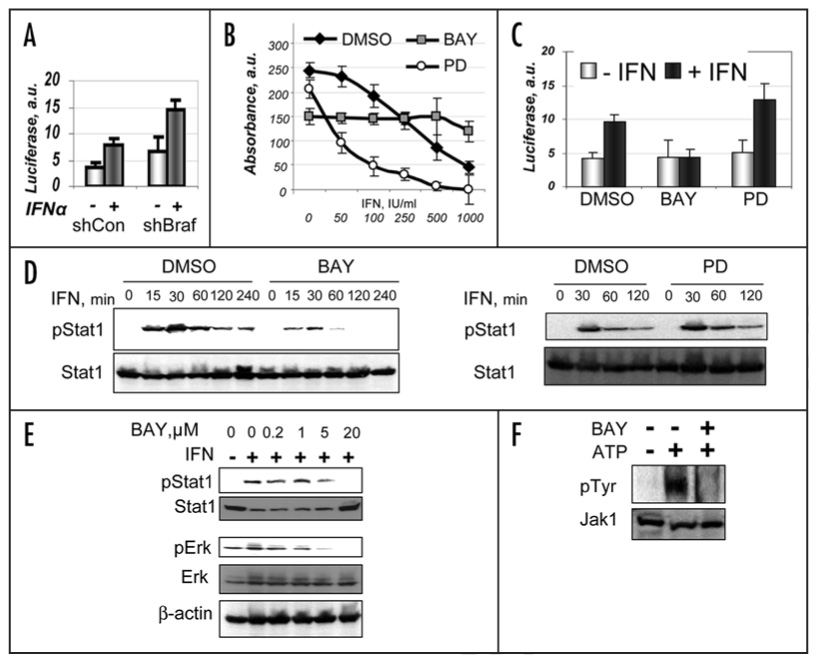
Raf inhibitor BAY43-9006 inhibits IFNα-induced signaling and anti-proliferative effects. (A) Activity of ISRE-driven luciferase (in arbitrary units) coexpressed with indicated shRNAs in either untreated (light gray bars) or IFNα-treated (dark gray bars) 1205Lu cells. Depicted data were normalized per activity of cotransfected Renilla luciferase. Average data from three independent experiments (in triplicates) are shown. (B) Effect of pretreatment (for 2 hr) with either BAY43-9006 (1 µM, gray squares) or PD098059 (1 µM, open squares) on suppression of growth of 1205Lu cells subsequently grown in the presence of indicated concentrations of IFNα for 48 hr was assessed by WST-1 colorimetric assay. Control cells (black diamonds) received DMSO. Absorbance (in arbitrary units, after subtraction of the background values) calculated from two independent experiments (each in five repetitions) is depicted. (C) Activity of ISRE-driven luciferase in 1205Lu cells pretreated with either DMSO or BAY43-9006 or PD098059 (as indicated) was measured as described in (A). (D) 1205Lu cells were pretreated or not with BAY43-9006 (10 µM for 3 hr, left panel) or with PD098059 (10 µM for 3 hr, right panel) followed by pulse treatment with IFNα (800 IU/ml for 15 min) and subsequent incubation in the absence of this cytokine for the indicated time before harvesting. Stat1 levels and its phosphorylation on tyrosine were analyzed as in Figure 1B. (E) 1205Lu cells were pretreated or not with indicated doses of BAY43-9006 for 2 hr before treatment with IFNα (500 IU/ml for 15 min). Phosphorylation and levels of Stat1 and Erk were analyzed and in Figure 1B and Figure 2D. (F) BAY43-9006 inhibits tyrosine kinase activity of JAK1 in vitro. JAK1 was immunoprecipitated (from Lu1205 cells lysate) and used for immunokinase assay in the presence of ATP and BAY43-9006 (10 µM) as indicated. JAK1 tyrosine phosphorylation and JAK1 levels were detected by immunoblotting with phosphor-Tyr specific 4G10 antibody and JAK1 antibody as indicated.
However, contrary to our expectations, Raf inhibitor BAY 43-9006, while inhibiting melanoma cell growth by itself (consistent with previous reports28), attenuated the anti-proliferative effects of IFNα (Fig. 4B) and inhibited IFNα signaling assessed by activity of ISRE-driven luciferase (Fig. 4C). Given that knock down of BRAF stimulates this luciferase reporter (Fig. 4A), we hypothesized that BAY 43-9006 may affect other components of IFNα signaling that are downstream from IFNAR1. Indeed, we observed that pretreatment of 1205Lu cells with BAY 43-9006 decreased the duration and magnitude of IFNα-stimulated phosphorylation of Stat1 (Fig. 4D). Further analysis demonstrated that the dose of BAY 43-9006 that was needed to inhibit Stat1 phosphorylation did not differ from the dose required to inhibit Erk phosphorylation by more than two orders of magnitude (Fig. 4E). These results indicate that, despite stabilizing IFNAR1 (Fig. 2B), BAY 43-9006 impairs IFNα signaling in 1205Lu human melanoma cells.
Signaling from IFNα through its receptor to Stat1 requires the activation of Jak1 and Tyk2 via tyrosine phosphorylation (including autophosphorylation). Autophosphorylation of Jak1 (immunoprecipitated from 1205Lu cells) incubated with ATP in vitro was inhibited by addition of BAY 43-9006 (10 µM, Fig. 4F and 1.25–5.0 µM, data not shown). This data suggests that of BAY 43-9006 might decrease the extent of cellular responses to IFNα via directly inhibiting the catalytic activity of Janus kinases.
In addition to Raf, BAY 43-9006 is capable of inhibiting many other targets including tyrosine kinases such as c-Kit, Flt-2 and PDGF and VEGF receptors (reviewed in Ref. 29). This agent (produced under the names Sorafenib and Nexavar) is under active clinical investigation for its use in malignant melanoma, renal cell carcinoma and some other malignancies alone or in combinations with other anti-cancer agents.30,31 While initial studies reported that IFN does not affect pharmacokinetics of Sorafenib,32 it is yet not know how Sorafenib would affect the responses to IFN. Our studies in vitro presented here indicate that BAY 43-9006 can also inhibit Jak activity that might contribute to its anti-oncogenic potential (for example, via decreasing the activation of pro-oncogenic Stat3) but also might decrease the effects of Type I IFN. Given that concentration of BAY 43-9006 (4 mg/L = 9.88 µM,32) that is achieved in plasma of patients that receive standard doses (400 mg twice daily) is similar to one capable of decreasing Stat1 phosphorylation in cells in vitro (~5–10µM) it is plausible that inhibitory effect of this agent on IFNα responses could occur in patients as well. Additional preclinical studies are, therefore, warranted to provide a rationale for combining BAY 43-9006 with IFNα in treatment of malignant melanoma.
ACKNOWLEDGEMENTS
We thank Drs. C. Horvath and D. Tuveson for the reagents and Dr. V. Spiegelman for critical comments and discussion. The work was supported in part by NIH grants CA 092900 to Serge Y. Fuchs, CA102709 to Andrei Thomas-Tikhonenko, and CA25874 and CA93372 to Meenhard Herlyn.
References
- 1.Noonan FP, Recio JA, Takayama H, Duray P, Anver MR, Rush WL, De Fabo EC, Merlino G. Neonatal sunburn and melanoma in mice. Nature. 2001;413:271–272. doi: 10.1038/35095108. [DOI] [PubMed] [Google Scholar]
- 2.Pawlik TM, Sondak VK. Malignant melanoma: Current state of primary and adjuvant treatment. Crit Rev Oncol Hematol. 2003;45:245–264. doi: 10.1016/s1040-8428(02)00080-x. [DOI] [PubMed] [Google Scholar]
- 3.Eggermont AM. Critical appraisal of IFN-alpha-based adjuvant therapy in stage II–III malignant melanoma. Expert Rev Anticancer Ther. 2002;2:563–569. doi: 10.1586/14737140.2.5.563. [DOI] [PubMed] [Google Scholar]
- 4.Lafuma A, Grob JJ. Cost-effectiveness of interferon-alpha2 as adjuvant therapy in malignant melanoma. Expert Opin Pharmacother. 2003;4:343–349. doi: 10.1517/14656566.4.3.343. [DOI] [PubMed] [Google Scholar]
- 5.Lafuma A, Dreno B, Delaunay M, Emery C, Fagnani F, Hieke K, Bonerandi JJ, Grob JJ. Economic analysis of adjuvant therapy with interferon alpha-2a in stage II malignant melanoma. Eur J Cancer. 2001;37:369–375. doi: 10.1016/s0959-8049(00)00411-1. [DOI] [PubMed] [Google Scholar]
- 6.Taniguchi T, Takaoka A. A weak signal for strong responses: Interferon-alpha/beta revisited. Nat Rev Mol Cell Biol. 2001;2:378–386. doi: 10.1038/35073080. [DOI] [PubMed] [Google Scholar]
- 7.Aaronson DS, Horvath CM. A road map for those who know JAK-STAT. Science. 2002;296:1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- 8.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 9.Colamonici OR, Porterfield B, Domanski P, Constantinescu S, Pfeffer LM. Complementation of the interferon alpha response in resistant cells by expression of the cloned subunit of the interferon alpha receptor: A central role of this subunit in interferon alpha signaling. J Biol Chem. 1994;269:9598–9602. [PubMed] [Google Scholar]
- 10.Colamonici OR, Porterfield B, Domanski P, Handa RK, Flex S, Samuel CE, Pine R, Diaz MO. Ligand-independent anti-oncogenic activity of the alpha subunit of the type I interferon receptor. J Biol Chem. 1994;269:27275–27279. [PubMed] [Google Scholar]
- 11.Hwang SY, Hertzog PJ, Holland KA, Sumarsono SH, Tymms MJ, Hamilton JA, Whitty G, Bertoncello I, Kola I. A null mutation in the gene encoding a type I interferon receptor component eliminates antiproliferative and antiviral responses to interferons alpha and beta and alters macrophage responses. Proc Natl Acad Sci USA. 1995;92:11284–11288. doi: 10.1073/pnas.92.24.11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar KG, Tang W, Ravindranath AK, Clark WA, Croze E, Fuchs SY. SCF(HOS) ubiquitin ligase mediates the ligand-induced down-regulation of the interferon-alpha receptor. Embo J. 2003;22:5480–5490. doi: 10.1093/emboj/cdg524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spiegelman VS, Tang W, Chan AM, Igarashi M, Aaronson SA, Sassoon DA, Katoh M, Slaga TJ, Fuchs SY. Induction of homologue of Slimb ubiquitin ligase receptor by mitogen signaling. J Biol Chem. 2002;277:36624–36630. doi: 10.1074/jbc.M204524200. [DOI] [PubMed] [Google Scholar]
- 14.Liu J, Suresh Kumar KG, Yu D, Molton SA, McMahon M, Herlyn M, Thomas-Tikhonenko A, Fuchs SY. Oncogenic BRAF regulates beta-Trcp expression and NF-kappaB activity in human melanoma cells. Oncogene. 2007;26:1954–1958. doi: 10.1038/sj.onc.1209994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Satyamoorthy K, Li G, Gerrero MR, Brose MS, Volpe P, Weber BL, Van Belle P, Elder DE, Herlyn M. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res. 2003;63:756–759. [PubMed] [Google Scholar]
- 16.Fuchs SY, Chen A, Xiong Y, Pan ZQ, Ronai Z. HOS, a human homolog of Slimb, forms an SCF complex with Skp1 and Cullin1 and targets the phosphorylation-dependent degradation of IkappaB and beta-catenin. Oncogene. 1999;18:2039–2046. doi: 10.1038/sj.onc.1202760. [DOI] [PubMed] [Google Scholar]
- 17.Spiegelman VS, Tang W, Katoh M, Slaga TJ, Fuchs SY. Inhibition of HOS expression and activities by Wnt pathway. Oncogene. 2002;21:856–860. doi: 10.1038/sj.onc.1205132. [DOI] [PubMed] [Google Scholar]
- 18.Goldman LA, Zafari M, Cutrone EC, Dang A, Brickelmeier M, Runkel L, Benjamin CD, Ling LE, Langer JA. Characterization of antihuman IFNAR-1 monoclonal antibodies: Epitope localization and functional analysis. J Interferon Cytokine Res. 1999;19:15–26. doi: 10.1089/107999099314379. [DOI] [PubMed] [Google Scholar]
- 19.Fuchs SY, Chen A, Xiong Y, Pan ZQ, Ronai Z. HOS, a human homolog of Slimb, forms an SCF complex with Skp1 and Cullin1 and targets the phosphorylation-dependent degradation of IkappaB and beta-catenin. Oncogene. 1999;18:2039–2046. doi: 10.1038/sj.onc.1202760. [DOI] [PubMed] [Google Scholar]
- 20.Kumar KG, Krolewski JJ, Fuchs SY. Phosphorylation and specific ubiquitin acceptor sites are required for ubiquitination and degradation of the IFNAR1 subunit of type I interferon receptor. J Biol Chem. 2004;279:46614–46620. doi: 10.1074/jbc.M407082200. [DOI] [PubMed] [Google Scholar]
- 21.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 22.Soldatenkov VA, Dritschilo A, Ronai Z, Fuchs SY. Inhibition of homologue of Slimb (HOS) function sensitizes human melanoma cells for apoptosis. Cancer Res. 1999;59:5085–5088. [PubMed] [Google Scholar]
- 23.Hingorani SR, Jacobetz MA, Robertson GP, Herlyn M, Tuveson DA. Suppression of BRAF(V599E) in human melanoma abrogates transformation. Cancer Res. 2003;63:5198–5202. [PubMed] [Google Scholar]
- 24.Fuchs SY, Spiegelman VS, Kumar KG. The many faces of beta-TrCP E3 ubiquitin ligases: Reflections in the magic mirror of cancer. Oncogene. 2004;23:2028–2036. doi: 10.1038/sj.onc.1207389. [DOI] [PubMed] [Google Scholar]
- 25.Romerio F, Zella D. MEK and ERK inhibitors enhance the anti-proliferative effect of interferon-alpha2b. Faseb J. 2002;16:1680–1682. doi: 10.1096/fj.02-0120fje. [DOI] [PubMed] [Google Scholar]
- 26.Walters DK, French JD, Arendt BK, Jelinek DF. Atypical expression of ErbB3 in myeloma cells: Cross-talk between ErbB3 and the interferon-alpha signaling complex. Oncogene. 2003;22:3598–3607. doi: 10.1038/sj.onc.1206512. [DOI] [PubMed] [Google Scholar]
- 27.Caraglia M, Abbruzzese A, Leardi A, Pepe S, Budillon A, Baldassare G, Selleri C, Lorenzo SD, Fabbrocini A, Giuberti G, Vitale G, Lupoli G, Bianco AR, Tagliaferri P. Interferon-alpha induces apoptosis in human KB cells through a stress-dependent mitogen activated protein kinase pathway that is antagonized by epidermal growth factor. Cell Death Differ. 1999;6:773–780. doi: 10.1038/sj.cdd.4400550. [DOI] [PubMed] [Google Scholar]
- 28.Sharma A, Trivedi NR, Zimmerman MA, Tuveson DA, Smith CD, Robertson GP. Mutant V599EB-Raf regulates growth and vascular development of malignant melanoma tumors. Cancer Res. 2005;65:2412–2421. doi: 10.1158/0008-5472.CAN-04-2423. [DOI] [PubMed] [Google Scholar]
- 29.Sridhar SS, Hedley D, Siu LL. Raf kinase as a target for anticancer therapeutics. Mol Cancer Ther. 2005;4:677–685. doi: 10.1158/1535-7163.MCT-04-0297. [DOI] [PubMed] [Google Scholar]
- 30.Sosman JA, Puzanov I. Molecular targets in melanoma from angiogenesis to apoptosis. Clin Cancer Res. 2006;12:2376s–2383s. doi: 10.1158/1078-0432.CCR-05-2558. [DOI] [PubMed] [Google Scholar]
- 31.Flaherty KT. Sorafenib in renal cell carcinoma. Clin Cancer Res. 2007;13:747s–752s. doi: 10.1158/1078-0432.CCR-06-2063. [DOI] [PubMed] [Google Scholar]
- 32.Escudier B, Lassau N, Angevin E, Soria JC, Chami L, Lamuraglia M, Zafarana E, Landreau V, Schwartz B, Brendel E, Armand JP, Robert C. Phase I trial of sorafenib in combination with IFN alpha-2a in patients with unresectable and/or metastatic renal cell carcinoma or malignant melanoma. Clin Cancer Res. 2007;13:1801–1809. doi: 10.1158/1078-0432.CCR-06-1432. [DOI] [PubMed] [Google Scholar]