

Abstract
Our knowledge of the structure and function of alkaline phosphatases has increased greatly in recent years. The crystal structure of the human placental isozyme has enabled us to probe salient features of the mammalian enzymes that differ from those of the bacterial enzymes. The availability of knockout mice deficient in each of the murine alkaline phosphatase isozymes has also given deep insights into their in vivo role. This has been particularly true for probing the biological role of bone alkaline phosphatase during skeletal mineralization. Due to space constraints this mini-review focuses exclusively on structural and functional features of mammalian alkaline phosphatases as identified by crystallography and probed by site-directed mutagenesis and kinetic analysis. An emphasis is also placed on the substrate specificity of alkaline phosphatases, their catalytic properties as phosphohydrolases as well as phosphodiesterases and their structural and functional relatedness to a large superfamily of enzymes that includes nucleotide pyrophosphatase/phosphodiesterase.
Key words: alkaline phosphatases, crystal structures, hydrolases, hypophosphatasia, isozymes, modeling, physiological substrates, pyrophosphatases, site-directed mutagenesis, structure/function studies
Introduction
Alkaline phosphatases (APs; EC 3.1.3.1) occur widely in nature, and are found in many organisms from bacteria to man [1]. With few exceptions, APs are homodimeric enzymes and each catalytic site contains three metal ions, i.e., two Zn and one Mg, necessary for enzymatic activity. The enzymes catalyze the hydrolysis of monoesters of phosphoric acid and also catalyze a transphosphorylation reaction in the presence of large concentrations of phosphate acceptors. While the main features of the catalytic mechanism are conserved comparing mammalian and bacterial APs, mammalian APs have higher specific activity and Km values; have a more alkaline pH optimum; display lower heat stability; are membrane-bound and are inhibited by l-amino acids and peptides through an uncompetitive mechanism. These properties, however, differ noticeably among the different mammalian AP isozymes and are likely to reflect very different in vivo functions. Table 1 summarizes the nomenclature, tissue distribution and some of the functions of human and mouse AP isozymes. Importantly, APs have homology to a large number of other enzymes and are, thus, part of a superfamily of enzymes with some overlapping catalytic properties and substrate specificities. This mini-review will briefly point out some of the most salient structural features of mammalian APs and refer to their substrate specificity and homology to other members of the nucleoside pyrophosphatase/phosphodiesterase family of isozymes with which APs synergize and overlap in function, particularly during skeletal mineralization. For more information on any aspect of APs, including their in vivo roles as defined by gene knockout studies, the reader is referred to a recent book covering in depth 25 years of progress in understanding the structure and function of mammalian APs [2].
Table 1.
Summary of the gene nomenclature, accession numbers, common names, tissue distribution and function, if known, for the human and mouse alkaline phosphatase isozymes.
| Accession numbers | Protein names | Common names, synonyms | Tissue distribution | Function | |
|---|---|---|---|---|---|
| Human genes | |||||
| ALPL | NM_000478 | TNAP | Tissue-nonspecific alkaline phosphatase; TNSALP; “Bliver-bone-kidney type” AP | Developing nervous system, skeletal tissues, liver, kidney | Bone mineralization |
| ALPP | NM_001632 | PLAP | Placental alkaline phosphatase; PLALP | Syncytiotrophoblast, a variety of tumors | Unknown |
| ALPP2 | NM_031313 | GCAP | Germ cell alkaline phosphatase, GCALP | Testis, malignant trophoblasts, testicular cancer | Unkown |
| ALPI | NM_001631 | IAP | Intestinal alkaline phosphatase, IALP | gut, influenced by fat feeding and ABO status | Intestinal absorption? |
| Mouse genes | |||||
| Akp2 | NM_007431 | TNAP | Tissue-nonspecific alkaline phosphatase; TNSALP; “liver-bone-kidney type” AP | Developing nervous system, skeletal tissues, liver, kidney | Bone mineralization |
| Akp3 | NM_007432 | IAP | Intestinal alkaline phosphatase, IALP | Gut | Fat absorption |
| Akp5 | NM_007433 | EAP | Embryonic alkaline phosphatase | preimplantation embryo, testis, gut | Early embryogenesis |
Protein structure and functional domains
For many years the crystallographic coordinates of the Escherichia coli AP (ECAP) [3] molecule provided the only source of structural information on APs, but now the three-dimensional structure of the first mammalian AP, i.e., human placental AP (PLAP; 1EW2; http://pdbbeta.rcsb.org/pdb/) has been solved [4]. As had been predicted from sequence comparisons, the central core of PLAP, consisting of an extended β-sheet and flanking α-helices, is very similar to that of ECAP. The overall structure of PLAP is a dimer and each monomer contains 484 residues, four metal atoms, one phosphate ion, and 603 water molecules. The two monomers are related by a two-fold crystallographic axis (Figure 1). The surface of PLAP is poorly conserved with that of ECAP with only 8% residues in common. PLAP possesses additional secondary structure elements, comprising an N-terminal α-helix (residues 9 to 25), an α-helix and a β-strand in a highly divergent region (residues 208–280) and a different organization of the small β-sheet in domain 365–430. In the active site, only the residues that are essential for catalysis are preserved, i.e., the catalytic Ser, the three metal ion sites, M1 (occupied by Zn2+, also called Zn1), M2 (occupied by Zn2+; also called Zn2) and M3 (occupied by Mg2+) as well as their ligands, while most of the surrounding residues are different. Half of the enzyme surface corresponds to three clearly identifiable regions whose sequences largely vary among human APs, and are lacking in non-mammalian enzymes, i.e., the long N-terminal α-helix, an interfacial flexible loop known as the ‘crown domain’ and a fourth metal binding domain (M4). The availability of the PLAP structure facilitated modeling the human germ cell AP (GCAP), intestinal AP (IAP) and tissue-nonspecific AP (TNAP, a.k.a. liver/bone/kidney type AP) isozymes, revealing that all the novel features discovered in PLAP are conserved in those human isozymes as well [5]. The active site Ser is conserved in all species where an AP has been sequenced to-date.
Figure 1.
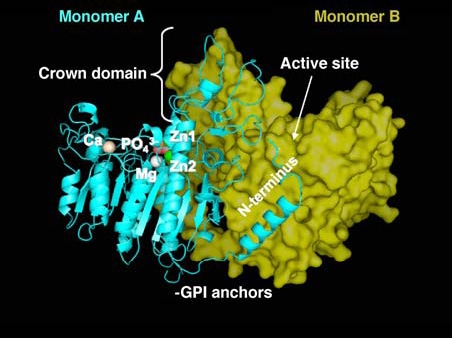
Three-dimensional structure of PLAP. Overview of the structure of human PLAP from the crystallographic coordinates determined by Le Du et al. [4]. Monomer A is shown in ribbon representation and in cyan, while monomer B is shown in surface representation in yellow. Indicated are the active site metals, Zn1, Zn2 and Mg, the novel fourth metal site occupied by Ca, the crown domain and the amino terminal arm. The relative location of the GPI anchor on the processed enzyme is also indicated.
Structure/function studies comparing PLAP and the ECAP structure have found a conserved function for those residues that stabilize the active site Zn1, Zn2 and Mg metal ions, i.e., Asp42, His153, Ser155, Glu311, Asp316, His320, Asp357, His358, His360 and His432 [6] (Figure 2). However, while mutations at Zn2 or Mg sites have similar effects in PLAP and ECAP, the environment of the Zn1 ion in PLAP is less affected by substitutions than in ECAP. This reflects the fact that, as shown on Figure 2, residue Glu429 is strategically positioned at the roof of the active site cleft where it provides additional stabilization to the active site environment, so that Zn2+ cannot easily diffuse in or out of the PLAP molecule [6]. An additional non-catalytic metal-binding site M4 that appears to be occupied by calcium and that is not present in ECAP was revealed upon solving the PLAP 3D structure [4, 7]. This fourth metal is coordinated by carboxylates from Glu216, Glu270 and Asp285, by the carbonyl of Phe269 and a water molecule and this architecture is conserved in all human and mouse APs and presumably represents a novel feature common to all mammalian APs. However, the structural and functional significance of this new metal site remains to be established.
Figure 2.
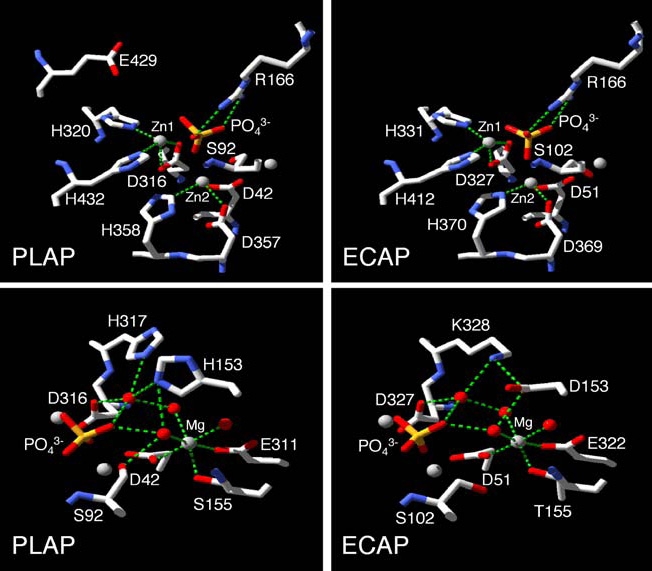
Comparison of the residues coordinating to the active site metals in PLAP and ECAP. The upper panels focus on the environment of the Zn1 and Zn2 metal sites and their ligands while the lower panels display the environment of the Mg metal site and its ligands. Water molecules are shown as red spheres. Green dotted lines denote metal-ligand interactions and hydrogen bonds. The figure is taken from Kozlenkov et al. [6] and is reproduced with permission from the Journal of Biological Chemistry.
All mammalian APs have five cysteine residues (Cys101, Cys121, Cys183, Cys467 and Cys474 in PLAP) per subunit, not homologous to any of the four cysteines in ECAP. They form two disulfide bonds, Cys121-Cys183 and Cys467-Cys474, whereas the Cys101 residue remains in free form [6]. The N-terminus of PLAP has an additional α-helix not present in the ECAP structure and its position is removed from the rest of the monomer, but it interacts with the second monomer with a buried surface area of 555 Å2, suggesting an involvement in enzyme dimerization. Recently, Hoylaerts et al. [8] have shown that the integrity of the N-terminal arm, and in particular of the α-helix comprising residues 10–25, represents a structural requirement for the active site to execute the intramolecular transition required during enzyme catalysis in both PLAP and TNAP.
The top flexible loop that constitutes the crown domain of PLAP is formed by the insertion of a 60-residue segment (366–430) from each monomer. It consists of two small interacting β-sheets, each composed of three parallel strands, and surrounded by six large and flexible loops containing a short α-helix [4]. This region displays the least degree of sequence conservation among mammalian APs and isozyme-specific properties, such as the characteristic uncompetitive inhibition [6, 9–12], their variable heat-stability [13], and their allosteric behavior [14] have all been attributed to residue 429 located on this crown domain (Figures 2 and 3). This domain may also mediate the interaction between APs and extracellular matrix proteins, such as collagens [13, 15–17]. Another interesting structural feature of PLAP, with no counterpart in the ECAP structure, is Y367 (Figure 3). This residue is part of the subunit interface in the PLAP dimer, where it protrudes from one subunit and its hydroxyl group is located 6.1 Å from the phosphate and 3.1 Å from His432, which in turn chelates the zinc atom Zn1 in the active site of the other subunit. Kozlenkov et al. [6, 12] has shown that Y367, similar to residue 429, also plays a crucial role in determining the uncompetitive inhibition properties to the mammalian APs but very likely also the allosteric character of mammalian APs.
Figure 3.
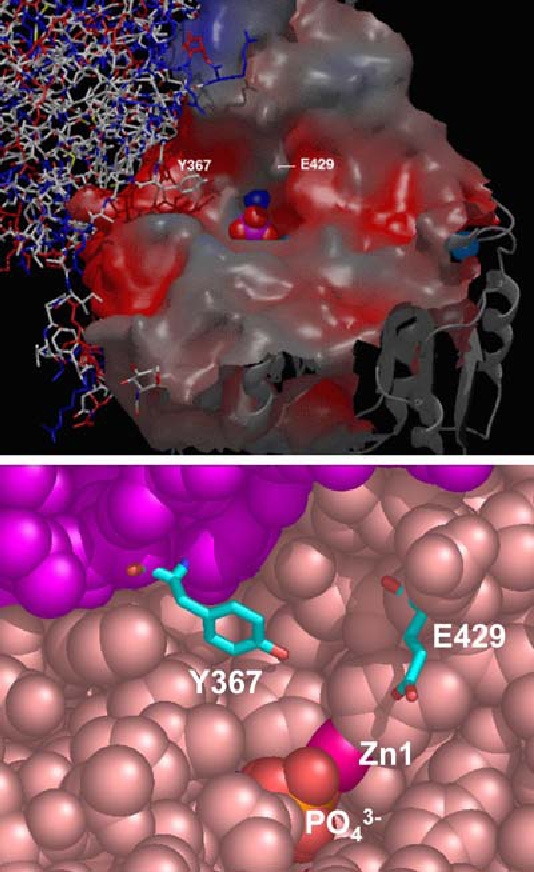
Location and environment of the Glu429 and Tyr367 residue. Upper panel ) Top view of the entrance to the active site, showing the position of the Y367 residue from one subunit (wireframe representation) in the immediate vicinity of the Glu429 residue of the other subunit (spacefill representation). Lower panel ) Detail of the active site entrance in spacefill representation showing the location of Glu429 relative to Zn1 and the active site PO3−4 in one subunit and the relative location of Tyr367 of the other subunit. The upper panel was taken from Kozlenkov et al. [6] and is reproduced with permission from the Journal of Biological Chemistry.
The monomer-monomer interface in PLAP has a strong hydrophobic character in contrast to that of ECAP. Less than 30% of the residues are involved in hydrogen-bonding interactions, which confer flexibility on the interface. Both the amino terminal arm and the crown domain take part in stabilizing the AP dimeric structure [4, 5]. Hoylaerts et al. [14] investigated whether the monomers in a given PLAP or GCAP dimer are subject to cooperativity during catalysis following an allosteric model or act via a half-of-sites model, in which at any time only a single monomer is operative. The authors used wt, single and double mutant PLAP homodimers and heterodimers for the analysis and found that mammalian APs are non-cooperative allosteric enzymes, but the stability and catalytic properties of each monomer are controlled by the conformation of the second AP subunit. The 3D structure of PLAP determined by Le Du et al. [4] contributed the visualization of the residues postulated to be involved in conferring the allosteric character to PLAP. The allosteric behavior is probably further favored by the quality of the dimer interface, by the long N-terminal α-helix from one monomer that embraces the other subunit, and by the protrusion of Tyr367 from one monomer into the active site of the other.
Recently, Llinas et al. [18] identified a novel, non-catalytic peripheral site on the surface of PLAP. This newly identified peripheral site is 28 Å from the active site but only 13 Å from the calcium binding site. The residues that constitute this site are located on two α-helices, 250–257 and 287–297. The loop between Ser257 and Ser287 contains the three residues that coordinate the Ca2+ ion, namely Phe269, Glu270, and Asp285 located only two residues from Ser287. Arg250 is located two residues away from Trp248 that interacts with the Ca2+ through a water molecule. Therefore, the large loop, comprising residues 250–297, that belongs to the peripheral domain of PLAP includes both the M4 site, and the peripheral binding site. The location of the Ca2+ ion in this loop suggests that the function of this novel M4 site could be related to the conformational stabilization of the two α-helices that form the peripheral site.
Finally, while ECAP is located in the periplasmic space of the bacterium, mammalian APs are ectoenzymes bound to the plasma membrane via a glycosyl-phosphatidylinositol (GPI) anchor [19]. The GPI-glycan moiety, with general structure Protein-(ethanolamine-PO4-6Manα1-2Manα1-6Manα1-4 GlcN1-6myo-inositol-1-P-lipid, is added post-translationally to the nascent PLAP peptide chain in a transamidation reaction that involves the removal of a stretch of 29 hydrophobic amino acids from the COOH-terminus and the concomitant transfer of the preassembled GPI anchor to the new carboxyterminal amino acid, Asp484 in PLAP [20]. To-date, all mammalian APs are thought to be anchored to the plasma membrane with each monomer carrying one unit of the GPI anchor.
Substrate specificities
Mammalian APs have broad substrate specificity and are able to hydrolyze or transphosphorylate a wide variety of phosphated compounds in vitro. However only a select few of those compounds have to-date been confirmed to serve as natural substrates for some of the AP isozymes. It has been proposed that the role of TNAP in the bone matrix is to generate the Pi needed for hydroxyapatite crystallization [1, 21, 22]. However, TNAP has also been hypothesized to hydrolyze the mineralization inhibitor PPi to facilitate mineral precipitation and growth [1, 23–25]. In fact, an inbornerror-of metabolism affecting the function of TNAP, hypophosphatasia, leads to severe rickets and osteomalacia [26]. Recent studies have provided compelling proof that a major function of TNAP in bone tissue consists in hydrolyzing PPi to maintain a proper concentration of this mineralization inhibitor to ensure normal bone mineralization. The nucleosidetriphosphate pyrophosphohydrolase activity of NPP1 and the transmembrane PPi-channeling protein ANK are responsible for supplying the larger amount of PPi to the extracellular spaces. Since mice deficient in TNAP (Akp2−/− mice) display high extracellular PPi levels, which lead to osteomalacia, Millán and colleagues [27, 28] tested the hypothesis that affecting the function of either NPP1 or ANK would have beneficial consequences on hypophosphatasia by reducing the amounts of extracellular PPi in the Akp2−/− mice and conversely that affecting the function of TNAP should also ameliorate the soft tissue ossification in the Enpp1−/− and ank/ank mutant animals. Indeed, experiments to rescue these abnormalities by combining two mutations, i.e., [Akp2−/−; Enpp1−/−] and [Akp2−/−; ank/ank] revealed that both double mutants led to improvement of hypophosphatasia as well as of the ossification of the vertebral apophyses confirming a major role of TNAP in regulating extracellular PPi levels [27, 28].
Pyridoxal-5′-phosphate (PLP) has also been shown to be a physiological substrate for the TNAP present in leukocytes [29, 30]. TNAP isolated from human SAOS-2 osteosarcoma cells hydrolyses phosphoethanolamine and PLP at physiologic pH [31]. Abnormalities in PLP metabolism also explain the epileptic seizures experienced by some patients with hypophosphatasia [26, 32]. Other described substrates include monofluorophosphate that can also be hydrolyzed by TNAP [33]. Human AP isozymes are able to hydrolyze phosphatidates with various fatty acyl chains (egg phosphatidate and dioleoyl, distearoyl, dipalmitoyl, dimyristoyl and dilauroyl phosphatidates). PLAP and IAP were capable of hydrolyzing all the phosphatidates examined with maximal activity in the presence of 10 g/l sodium deoxycholate and dilauroyl phosphatidate being the best substrate. The phosphatidate hydrolytic activity of PLAP was two-three times higher than that of the IAP enzyme, while TNAP did not hydrolyze phosphatidates with long fatty acyl chains (C16–18) even in the presence of sodium deoxycholate [34]. Inorganic polyphosphates (polyP), being energy-rich linear polymers of orthophosphate residues known from bacteria and yeast, also exist in higher eukaryotes. Calf IAP is able to cleave polyP molecules up to a chain length of about 800, acting as an exopolyphosphatase progressively degrading polyP. The pH optimum is in the alkaline range. TNAP was not able to hydrolyze polyP under the conditions tested while PLAP and ECAP displayed polyP-degrading activity [35].
APs appear also to be involved in the metabolism of nucleotides. Say et al. [36] reported that purified osseous plate TNAP displayed broad substrate specificity and was able to hydrolyze ATP, ADP, AMP, PPi, glucose-1-phosphate, glucose-6-phosphate, fructose-6-phosphate, β-glycerophosphate, bis-(p-nitrophenyl)-phosphate in addition to pNPP. However, ATP, bis-(p-nitrophenyl)-phosphate and PPi were among the less hydrolyzed substrates assayed. Nevertheless, TNAP appears to be able to hydrolyze ATP both at pH 7.5 and pH 9.4 [37]. However, Pizauro et al. [38] concluded that the membrane-specific ATPase activity present in osseous plate membranes and TNAP are different proteins. Several reports have indicated that TNAP is involved in AMP hydrolysis [39, 40]. Extracellular adenine nucleotides induce cAMP elevation through local adenosine production at the membrane surface and subsequent activation of adenosine A (2A) receptors in NG108-15 neuronal cells. NG108-15 cells hydrolyzed AMP to adenosine and this activity was suppressed at pH 6.5, but markedly increased at pH 8.5. The AMP hydrolysis was also blocked by levamisole, an uncompetitive inhibitor of TNAP [12], and the cells expressed TNAP mRNA. These results indicated that AMP phosphohydrolase activity in NG108-15 cells is due to TNAP, and suggest that this enzyme plays an essential role for the P1 antagonist-sensitive ATP-induced cAMP accumulation in NG108-15 cells [39]. Similarly, Picher et al. [40] found that two ectonucleotidases mediated the conversion of AMP to adenosine on the mucosal surface of human airway epithelia, i.e., ecto 5′-nucleotidase (CD73) and TNAP. While both mucosal and serosal epithelial surfaces displayed ecto 5′-nucleotidase activity,TNAP activity was restricted to the mucosal surface. These experiments support a major role for extracellular nucleotide catalysis and for the involvement of TNAP in the regulation of adenosine concentrations on airway surfaces.
Other catalytic properties of APs are also worth mentioning. AP isolated from the mouse intestine is able to catalyze the synthesis of PPi from Pi during hydrolysis of glucose 6-phosphate, ATP, ADP, PPi or pNPP [41]. While the rate of PPi synthesis is 1,000-fold lower than the rate of substrate hydrolysis, PPi synthesis increased by the addition of Mg2+ and also by decreasing the pH from 8.5 to 6.0. The data indicated that at the catalytic site of APs the energies of hydrolysis of the phosphoserine residue and of PPi are different from those measured in aqueous solutions. In another study, calf IAP was found to be able to effectively transphosphorylate thiamin to thiamin monophosphate using β-glycerophosphate or creatine phosphate as phosphate donors at pH 8.5. TMP production in the brush border membrane however was very small and corresponded to 0.001–0.01% of the total Pi simultaneously released by hydrolytic activity [42].
Rezende et al. [43] reported phosphodiesterase activity as a novel property of osseous plate TNAP. Bis-(p-nitrophenyl)-phosphate was hydrolyzed at both pH 7.5 and 9.4 with an apparent dissociation constant of 1.9 and 3.9 mM, respectively. The hydrolysis of p-nitrophenyl-5′-thymidinephosphate followed hyberbolic kinetics with a K0.5 of 500 µM. The hydrolysis of cAMP by the enzyme followed more complex kinetics, showing site-site interactions (h = 1.7) and K0.5 = 300 µM for high-affinity sites. ATP and cAMP were competitive inhibitors of bis-(p-nitrophenyl)-phosphatase activity of the enzyme and Ki values (25 and 0.6 mM for cAMP and ATP, respectively) very close to those of the K0.5 (22 and 0.7 mM for cAMP and ATP, respectively), determined by direct assay, indicated that a single catalytic site was responsible for the hydrolysis of both substrates. The alkaline apparent pH optima, the requirement for bivalent metal ions and the inhibition by methylxanthines, amrinone and amiloride demonstrated that rat osseous plate TNAP was a type I phosphodiesterase [43]. More recently, Zhang et al. [44] suggested that TNAP, together with NPP1, could contribute to the pool of extracellular PPi via its phosphodiesterase activity. So, it is possible that TNAP both degrades and produces PPi but the relative significance for skeletal mineralization of this hypothetical dual role still remains to be clarified. Various in vitro studies have also led to the suggestion that APs may function as a plasma membrane phosphoprotein phosphatase. Sarrouilhe et al. [45] used purified rat liver plasma membranes to study endogenous phosphorylation and dephosphorylation events and detected an 18-kDa phosphoprotein as a potential substrate for TNAP. Fedde et al. [46], however, compared the phosphorylation of plasma membrane proteins from control fibroblasts to those from profoundly TNAP-deficient fibroblasts of hypophosphatasia patients. They found no consistent different among all identifiable plasma membrane phosphoproteins in the control and hypophosphatasia samples and concluded that TNAP does not modulate the phosphorylation of plasma membrane proteins. Scheibe et al. [47] also showed that a 98-kDa membrane protein, autophosphorylated via its endogenous ectokinase activity, was dephosphorylated by TNAP in both murine P19 teratocarcinoma and HL-60 human myelopblastic leukemic cells. So, a putative role for APs as phosphoprotein phosphatases remains an open question at this point in time.
APs as members of a superfamily of enzymes
Of interest is the fact that APs appear to have structural similarity to a large number of other enzymes. Cofactorindependent phosphoglycerate mutase (iPGM) (EC 5.4.2.1) has been previously identified as a member of the AP superfamily of enzymes, based on the conservation of the predicted metal-binding residues [48]. Iterative homology searches resulted in the identification of similarly conserved regions in phosphopentomutases (EC 5.4.2.7), APs (EC 3.1.3.1) and in several previously uncharacterized proteins. All the amino acid residues that interact with Zn1 (Asp327, His331, and His412) and Zn2 (Asp51, Asp369, and His370) in ECAP [3] (Figure 2) are absolutely conserved in phosphocarbohydrate-binding proteins of the AP superfamily [48]. On the other hand, the ligands to the Mg site of ECAP are much less conserved, since an E322N substitution is found in phosphopentomutases and iPGMs, while Asp153 and Thr155 do not seem to be conserved at all. The strong conservation of the metal-binding residues in both phosphopentomutase and iPGM indicates that both these enzymes are metal dependent. Structural alignment of iPGM with ECAP and cerebroside sulfatase confirmed that all these enzymes have a common core structure and revealed similarly located conserved Ser (in iPGM, ECAP and mammalian APs) or Cys (in sulfatases) residues in their active sites. In ECAP and mammalian APs, this Ser residue is phosphorylated during catalysis, whereas in sulfatases the active site Cys residue is modified to formylglycine and sulfatated. Similarly located Thr residue forms a phosphoenzyme intermediate in phosphodiesterase/nucleotide pyrophosphatase-1 (NPP1). In fact, APs are known to have phosphotransferase and also phosphodiesterase [43] activities, while iPGM and NPP1 can also function as phosphatases [49, 50]. Using structure-based sequence alignment, Galperin and Jedrezjas [51] identified homologous Ser, Thr, or Cys residues in other enzymes of the AP superfamily, such as phosphopentomutase, phosphoglycerol transferase, phosphonoacetate hydrolase, and GPI-anchoring enzymes (GPI-phosphoethanolamine transferases). Thus, this AP superfamily includes enzymes with substantially different activities (isomerases, hydrolases, and a putative lyase), which, however, all act on similar phosphocarbohydrate (or sulfocarbohydrate) substrates.
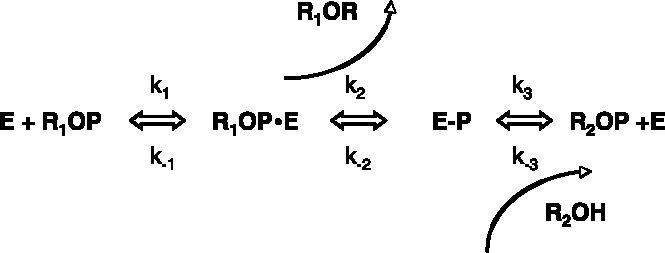
While these bioinformatics studies were done using the ECAP structure as paradigm, the same structural similarity is bound to exist with the mammalian APs since as discussed the active site of mammalian APs is well-conserved with that of ECAP. The authors predict that the catalytic cycles of all the members of this superfamily involve phosphorylation, or sulfatation, or phosphonation of these conserved Ser/Thr/Cys residues. This would imply that all enzymes of the AP superfamily would have the same reaction scheme (Scheme 1) where phosphate (sulphate or phosphonate) acceptor can be either water (r2 = H, in phosphatases, sulfatases, phosphonate hydrolases) or a second substrate (in phosphoglycerol and phosphoethanolamine transferases) or just a different hydroxyl group of the same original substrate r1 (in iPGM and phosphopentomutase).
Acknowledgement
This work was supported by grants DE12889 and AR47908 from the National Institutes of Health, USA.
Abbreviations
- ADP
adenosine diphosphate
- AMP
adenosine monophosphate
- AP
alkaline phosphatase
- ATP
adenosine triphosphate
- cAMP
cyclic AMP
- ECAP
Escherichia coli AP
- GCAP
germ cell alkaline phosphatase
- GPI
glycosylphosphatidylinositol
- IAP
intestinal alkaline phosphatase
- iPGM
cofactor-independent phosphoglycerate mutase
- kcat
catalytic rate constant
- Ki
inhibition constant
- Km
Michaelis constant
- NPP1
nucleosidetriphosphate pyrophosphohydrolase-1
- Pi
inorganic phosphate
- PLAP
placental alkaline phosphatase
- PLP
pyridoxal-5-phosphate
- pNPP
p-nitrophenylphosphate
- PPi
inorganic pyrophosphate
- TNAP
tissue-nonspecific alkaline phosphatase
- Vmax
maximal velocity
- Wt
wild-type
References
- 1.McComb RB, Bowers GN Jr, Posen S. Alkaline Phosphatase. New York: Plenum 1979.
- 2.Millán JL. Mammalian Alkaline Phosphatases. From Biology to Applications in Medicine and Biotechnology. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co, 2006; 1–322.
- 3.Stec B, Holtz KM, Kantrowitz ER. A revised mechanism for the alkaline phosphatase reaction involving three metal ions. J Mol Biol 2000; 299: 1303–11. [DOI] [PubMed]
- 4.Le Du MH, Stigbrand T, Taussig MJ et al. Crystal structure of alkaline phosphatase from human placenta at 1.8 A resolution. Implication for substrate specificity. J Biol Chem 2001; 276: 9158–65. [DOI] [PubMed]
- 5.Le Du MH, Millán JL. Structural evidence of functional divergence in human alkaline phosphatases. J Biol Chem 2002; 277: 49808–14. [DOI] [PubMed]
- 6.Kozlenkov A, Manes T, Hoylaerts MF, Millán JL. Function assignment to conserved residues in mammalian alkaline phosphatases. J Biol Chem 2002; 277: 22992–9. [DOI] [PubMed]
- 7.Mornet E, Stura E, Lia-Baldini AS et al. Structural evidence for a functional role of human tissue nonspecific alkaline phosphatase in bone mineralization. J Biol Chem 2001; 276: 31171–8. [DOI] [PubMed]
- 8.Hoylaerts MF, Ding L, Narisawa S et al. Mammalian alkaline phosphatase catalysis requires active site structure stabilization via the N-terminal amino acid microenvironment. Biochemistry 2006; in press. [DOI] [PubMed]
- 9.Hoylaerts MF, Millán JL. Site-directed mutagenesis and epitope-mapped monoclonal antibodies define a catalytically important conformational difference between human placental and germ cell alkaline phosphatase. Eur J Biochem 1991; 202: 605–16. [DOI] [PubMed]
- 10.Hummer C, Millán JL. Gly429 is the major determinant of uncompetitive inhibition of human germ cell alkaline phosphatase by l-leucine. Biochem J 1991; 274: 91–5. [DOI] [PMC free article] [PubMed]
- 11.Hoylaerts MF, Manes T, Millán JL. Molecular mechanism of uncompetitive inhibition of human placental and germ-cell alkaline phosphatase. Biochem J 1992; 286: 23–30. [DOI] [PMC free article] [PubMed]
- 12.Kozlenkov A, Le Du MH, Cuniasse P et al. Residues determining the binding specificity of uncompetitive inhibitors to tissue-nonspecific alkaline phosphatase. J Bone Miner Res 2004; 19: 1862–72. [DOI] [PubMed]
- 13.Bossi M, Hoylaerts MF, Millán JL. Modifications in a flexible surface loop modulate the isozyme-specific properties of mammalian alkaline phosphatases. J Biol Chem 1993; 268: 25409–16. [PubMed]
- 14.Hoylaerts MF, Manes T, Millán JL. Mammalian alkaline phosphatases are allosteric enzymes. J Biol Chem 1997; 272: 22781–7. [DOI] [PubMed]
- 15.Tsonis PA, Argraves WS, Millán JL. A putative functional domain of human placental alkaline phosphatase predicted from sequence comparisons. Biochem J 1988; 254: 623–4. [DOI] [PMC free article] [PubMed]
- 16.Vittur F, Stagni N, Moro L, de Bernard B. Alkaline phosphatase binds to collagen; a hypothesis on the mechanism of extravesicular mineralization in epiphyseal cartilage. Experientia 1984; 40: 836–7. [DOI] [PubMed]
- 17.Wu LN, Genge BR, Lloyd GC, Wuthier RE. Collagen-binding proteins in collagenase-released matrix vesicles from cartilage. Interaction between matrix vesicle proteins and different types of collagen. J Biol Chem 1991; 266: 1195–203. [PubMed]
- 18.Llinas P, Stura E, Menez A et al. Structural studies of human placental alkaline phosphatase in complex with functional ligands. J Mol Biol 2005; 350: 441–51. [DOI] [PubMed]
- 19.Low MG, Saltiel AR. Structural and functional roles of glycosyl-phosphatidylinositol in membranes. Science 1988; 239: 268–75. [DOI] [PubMed]
- 20.Micanovic R, Bailey CA, Brink L et al. Aspartic acid-484 of nascent placental alkaline phosphatase condenses with a phosphatidylinositol glycan to become the carboxyl terminus of the mature enzyme. Proc Natl Acad Sci USA 1988; 85: 1398–402. [DOI] [PMC free article] [PubMed]
- 21.Majeska RJ, Wuthier RE. Studies on matrix vesicles isolated from chick epiphyseal cartilage. Association of pyrophosphatase and ATPase activities with alkaline phosphatase. Biochim Biophys Acta 1975; 391: 51–60. [DOI] [PubMed]
- 22.Fallon MD, Whyte MP, Teitelbaum SL. Stereospecific inhibition of alkaline phosphatase by l-tetramisole prevents in vitro cartilage calcification. Lab Invest 1980; 43: 489–94. [PubMed]
- 23.Moss DW, Eaton RH, Smith JK, Whitby LG. Association of inorganic-pyrophosphatase activity with human alkaline-phosphatase preparations. Biochem J 1967; 102: 53–7. [DOI] [PMC free article] [PubMed]
- 24.Whyte MP. Hypophosphatasia and the role of alkaline phosphatase in skeletal mineralization. Endocr Rev 1994; 15: 439–61. [DOI] [PubMed]
- 25.Rezende LA, Ciancaglini P, Pizauro JM, Leone FA. Inorganic pyrophosphate-phosphohydrolytic activity associated with rat osseous plate alkaline phosphatase. Cell Mol Biol (Noisy-le-grand). 1998; 44: 293–302. [PubMed]
- 26.Whyte MP. Hypophosphatasia. In Scriver CR, Beaudet AL, Sly WS, Valle D, (eds): The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill 1995; 4095–112.
- 27.Hessle L, Johnson KA, Anderson HC et al. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci USA 2002; 99: 9445–9. [DOI] [PMC free article] [PubMed]
- 28.Harmey D, Hessle L, Narisawa S et al. Concerted regulation of inorganic pyrophosphate and osteopontin by Akp2, Enpp1, and Ank: An integrated model of the pathogenesis of mineralization disorders. Am J Pathol 2004; 164: 1199–209. [DOI] [PMC free article] [PubMed]
- 29.Smith GP, Peters TJ. Subcellular localization and properties of pyridoxal phosphate phosphatases of human polymorphonuclear leukocytes and their relationship to acid and alkaline phosphatase. Biochim Biophys Acta 1981; 661: 287–94. [DOI] [PubMed]
- 30.Wilson PD, Smith GP, Peters TJ. Pyridoxal 5’-phosphate: A possible physiological substrate for alkaline phosphatase in human neutrophils. Histochem J 1983; 15: 257–64. [DOI] [PubMed]
- 31.Fedde KN, Lane CC, Whyte MP. Alkaline phosphatase is an ectoenzyme that acts on micromolar concentrations of natural substrates at physiologic pH in human osteosarcoma (SAOS-2) cells. Arch Biochem Biophys 1988; 264: 400–9. [DOI] [PubMed]
- 32.Di Mauro S, Manes T, Hessle H et al. Kinetic characterization of hypophosphatasia mutations with physiological substrates. J Bone Miner Res 2002; 17: 1383–91. [DOI] [PubMed]
- 33.Farley JR, Tarbaux NM, Lau KH, Baylink DJ. Monofluorophosphate is hydrolyzed by alkaline phosphatase and mimics the actions of NaF on skeletal tissues, in vitro. Calcif Tissue Int 1987; 40: 35–42. [DOI] [PubMed]
- 34.Sumikawa K, Okochi T, Adachi K. Differences in phosphatidate hydrolytic activity of human alkaline phosphatase isozymes. Biochim Biophys Acta 1990; 1046: 27–31. [DOI] [PubMed]
- 35.Lorenz B, Schroder HC. Mammalian intestinal alkaline phosphatase acts as highly active exopolyphosphatase. Biochim Biophys Acta 2001; 1547: 254–61. [DOI] [PubMed]
- 36.Say JC, Ciuffi K, Furriel RP et al. Alkaline phosphatase from rat osseous plates: Purification and biochemical characterization of a soluble form. Biochim Biophys Acta 1991; 1074: 256–62. [DOI] [PubMed]
- 37.Demenis MA, Leone FA. Kinetic characteristics of ATP hydrolysis by a detergent-solubilized alkaline phosphatase from rat osseous plate. IUBMB. Life 2000; 49: 113–9. [DOI] [PubMed]
- 38.Pizauro JM, Demenis MA, Ciancaglini P, Leone FA. Kinetic characterization of a membrane-specific ATPase from rat osseous plate and its possible significance on endochodral ossification. Biochim Biophys Acta 1998; 1368: 108–14. [DOI] [PubMed]
- 39.Ohkubo S, Kimura J, Matsuoka I. Ecto-alkaline phosphatase in NG108-15 cells: A key enzyme mediating P1 antagonist-sensitive ATP response. Br J Pharmacol 2000; 131: 1667–72. [DOI] [PMC free article] [PubMed]
- 40.Picher M, Burch LH, Hirsh AJ et al. Ecto 5’-nucleotidase and nonspecific alkaline phosphatase. Two AMP-hydrolyzing ectoenzymes with distinct roles in human airways. J Biol Chem 2003; 278: 13468–79. [DOI] [PubMed]
- 41.Nayudu RV, de Meis L. Energy transduction at the catalytic site of enzymes: Hydrolysis of phosphoester bonds and synthesis of pyrophosphate by alkaline phosphatase. FEBS Lett 1989; 255: 163–6. [DOI] [PubMed]
- 42.Rindi G, Ricci V, Gastaldi G, Patrini C. Intestinal alkaline phosphatase can transphosphorylate thiamin to thiamin monophosphate during intestinal transport in the rat. Arch Physiol Biochem 1995; 103: 33–8. [DOI] [PubMed]
- 43.Rezende AA, Pizauro JM, Ciancaglini P, Leone FA. Phosphodiesterase activity is a novel property of alkaline phosphatase from osseous plate. Biochem J 1994; 301: 517–22. [DOI] [PMC free article] [PubMed]
- 44.Zhang L, Balcerzak M, Radisson J et al. Phosphodiesterase activity of alkaline phosphatase in ATP-initiated Ca2+ and phosphate deposition in isolated chicken matrix vesicles. J Biol Chem 2005; doi:10.1074/jbc.M504260200. [DOI] [PubMed]
- 45.Sarrouilhe D, Lalegerie P, Baudry M. Endogenous phosphorylation and dephosphorylation of rat liver plasma membrane proteins, suggesting a 18 kDa phosphoprotein as a potential substrate for alkaline phosphatase. Biochim Biophys Acta 1992; 1118: 116–22. [DOI] [PubMed]
- 46.Fedde KN, Michel MP, Whyte MP. Evidence against a role for alkaline phosphatase in the dephosphorylation of plasma membrane proteins: Hypophosphatasia fibroblast study. J Cell Biochem 1993; 53: 43–50. [DOI] [PubMed]
- 47.Scheibe RJ, Moeller-Runge I, Mueller WH. Retinoic acid induces the expression of alkaline phosphatase in P19 teratocarcinoma cells. J Biol Chem 1991; 266: 21300–5. [PubMed]
- 48.Galperin MY, Bairoch A, Koonin EV. A superfamily of metalloenzymes unifies phosphopentomutase and cofactor-independent phosphoglycerate mutase with alkaline phosphatases and sulfatases. Protein Sci 1998; 7: 1829–35. [DOI] [PMC free article] [PubMed]
- 49.Breathnach R, Knowles JR. Phosphoglycerate mutase from wheat germ: Studies with 18O-labeled substrate, investigations of the phosphatase and phosphoryl transfer activities, and evidence for a phosphoryl-enzyme intermediate. Biochemistry 1977; 16: 3054–60. [DOI] [PubMed]
- 50.Gijsbers R, Ceulemans H, Stalmans W, Bollen M. Structural and catalytic similarities between nucleotide pyrophosphatases/phosphodiesterases and alkaline phosphatases. J Biol Chem 2001; 276: 1361–8. [DOI] [PubMed]
- 51.Galperin MY, Jedrzejas MJ. Conserved core structure and active site residues in alkaline phosphatase superfamily enzymes. Proteins 2001; 45: 318–24 [DOI] [PubMed]