

Abstract
The catabolism of ATP and other nucleotides participates partly in the important function of nucleotide salvage by activated cells and also in removal or de novo generation of compounds including ATP, ADP, and adenosine that stimulate purinergic signaling. Seven nucleotide pyrophosphatase/phosphodiesterase NPP family members have been identified to date. These isoenzymes, related by up conservation of catalytic domains and certain other modular domains, exert generally non-redundant functions via distinctions in substrates and/or cellular localization. But they share the capacity to hydrolyze phosphodiester or pyrophosphate bonds, though generally acting on distinct substrates that include nucleoside triphosphates, lysophospholipids and choline phosphate esters. PPi generation from nucleoside triphosphates, catalyzed by NPP1 in tissues including cartilage, bone, and artery media smooth muscle cells, supports normal tissue extracellular PPi levels. Balance in PPi generation relative to PPi degradation by pyrophosphatases holds extracellular PPi levels in check. Moreover, physiologic levels of extracellular PPi suppress hydroxyapatite crystal growth, but concurrently providing a reservoir for generation of pro-mineralizing Pi. Extracellular PPi levels must be supported by cells in mineralization-competent tissues to prevent pathologic calcification. This support mechanism becomes dysregulated in aging cartilage, where extracellular PPi excess, mediated in part by upregulated NPP1 expression stimulates calcification. PPi generated by NPP1modulates not only hydroxyapatite crystal growth but also chondrogenesis and expression of the mineralization regulator osteopontin. This review pays particular attention to the role of NPP1-catalyzed PPi generation in the pathogenesis of certain disorders associated with pathologic calcification.
Key words: ANK, ANKH, cartilage, inorganic phosphate, inorganic pyrophosphate, osteopontin
Introduction
The extracellular catabolism of ATP and other nucleotides by coordinated ecto-enzymes mediates nucleotide salvage by activated cells and also drives removal or de novo generation of compounds including ATP, ADP, and adenosine that stimulate purinergic signaling [1–3]. This subject is reviewed in depth by Stefan et al. in this special issue of the journal. Among the many enzymes participating in nucleotide catabolism are certain nucleotide pyrophosphatase/phosphodiesterase (NPP) family members, including NPP1, the principal subject of this review. Seven NPPs have been identified to date (Figure 1) [4]. These isoenzymes, related by 24%Y60% conservation in catalytic domains [4] and by conservation of certain other modular domains, exert generally non-redundant functions via distinctions in substrates and/or subcellular localization. For example, the type II transmembrane ecto-enzymes NPP1 (PC-1, npps) and NPP3 (B10, CD203c, PD-1β, gp130RB13-6), which exist as disulfide-bonded homodimers in membranes, and whose extracellular domains can be proteolytically liberated into secreted forms, exert nucleoside triphosphate pyrophosphohydrolase (NTPPPH) activity that generates PPi from ATP and other nucleoside triphosphates, as discussed below, NPP1 and NPP3 both subserve other functions by alkaline pH optimum nucleotide phosphodiesterase activities [5–8]. However, the dileucine motif in the cytosolic tail of NPP1 (but not NPP3) mediates differential subcellular localization to the basolateral and apical plasma membrane, respectively, in polarized cell types [5].
Figure 1.

General Structural Features of NPP family members. The schematic highlights related structural features of NPPs 1Y7, as discussed further in the text.
NPP2 (autotaxin, PD-1a), though very similar to NPP1 and NPP3 in structural organization (Figure 1), is synthesized as a pro-enzyme and further processed to be a secretory molecule (4.9). NPP2 lysophospholipase D specific activity is much higher than that of other NPP family members and specific activity as a nucleotide pyrophosphatase/phosphodiesterase much lower than that of NPP1 and NPP3 [10, 11]. Correspondingly, we have observed that direct expression of NPP2 did not increase extracellular PPi in chondrocytes, under conditions in which NPP2 did stimulate both alkaline phosphatase and increased calcification [12]. NPP6 and the intestinal enzyme NPP7 (Figure 1) exert lysophospholipase C or choline phosphate esterase activities [4]. The secretion of NPP2 by multiple tissues, and NPP2 accumulation in extracellular fluids, allows NPP2, in large part via lysophospholipase D activity, to exert a variety of biologically significant effects on cell growth, differentiation, adhesion, and migration, translated into functional effects in angiogenesis, tumor metastasis, and embryonic development [4, 13–15].
Comparative molecular structure-function of NPPs and their substrate specificities were recently reviewed in a thorough and lucid manner [4]. This review focuses on the functions of NPP1 in the regulation of physiologic and pathologic calcification, principally via PPi generation from nucleoside triphosphates in tissues (and cells) including cartilage (and chondrocytes), bone (and osteoblasts), and large arteries (and smooth muscle cells (SMCs)).
NPP1 and ANK in PPi metabolism and calcification
Subcellular trafficking mediated by the dileucine motif in the NPP1 (but not NPP3) cytosolic tail accounts for the observation that the majority of NPP activity in osteoblast plasma membranes and plasma membrane-derived mineralizing secretory vesicles (termed matrix vesicles) is accounted for by NPP1 [17]. Concordantly, cultured osteoblasts of NPP1 null mice demonstrate marked depletion (of up to 50%) in extracellular PPi [18].
PPi potently inhibits the nucleation and propagation of hydroxyapatite (HA) and other basic calcium phosphate crystals [19]. As such, maintenance of physiologic extracellular PPi levels by mineralization-competent cells suppresses spontaneous calcification. This has been strikingly illustrated in certain mouse models of deficient NPP1-catalyzed PPi generation [18, 20, 21], or alternately, ANK-mediated PPi transport [18, 22]. In humans, 18 of 23 kindreds demonstrated homozygosity or compound heterozygosity for mutations of NPP1 in association with generalized arterial calcification of infancy (GACI, IIAC, MIM# 208000) [23, 24]. This entity, described in approximately 180 individuals to date, is characterized by large artery media calcification and myointimal proliferation, commonly associated with periarticular calcification [23–25]. The disease is frequently lethal but may respond to treatment with bisphosphonates, which function in part as non-hydrolyzeable PPi analogues [25]. GACI is linked to systemic (blood, urine, tissue) extracellular PPi deficiency [25, 26] discovered by us to be caused by mutations widely spread through NPP1 extracellular domains [23]. Many of these NPP1 mutations, which are mostly in the nuclease-like and catalytic, domains, but also reported in the somatomedin B-like domain, have been established to impair NPP1 catalytic activity [23, 24].
Notably, PPi serves as reservoir for alkaline phosphatase-catalyzed Pi generation that is pro-mineralizing, as illustrated by osteopenia in long bones of NPP1 deficient mice [27, 28]. As such, PPi generation can both suppress and promote HA crystal deposition, depending on relative tissue levels of NPP1 and alkaline phosphatase (Figure 2) [16–21, 27, 28]. The capacity of chondrocytes to produce copious extracellular PPi is particularly double edged, as it is directly promotes calcium pyrophosphate dihydrate (CPPD) crystal deposition (Figure 2). Depending on cartilage ATP and PPi concentrations, and the level of activity of Pi-generating ATPases and pyrophosphatases, NPP1 excess promotes both HA and CPPD crystal formation by articular chondrocytes [12, 29–31], an event that commonly occurs in the joint in human aging and osteoarthritis (OA) [32].
Figure 2.
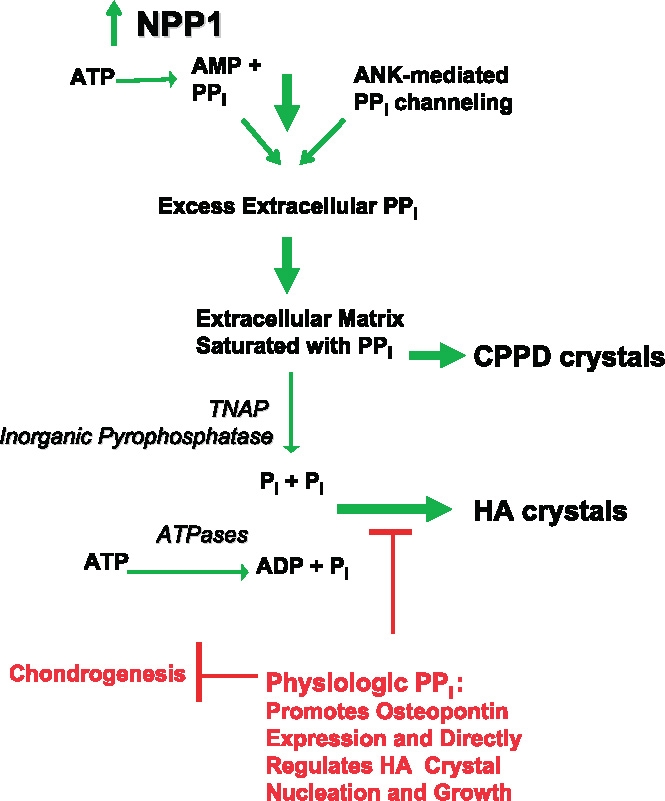
Proposed NPP1-mediated and PPi-dependent mechanisms stimulating CPPD and HA crystal deposition in aging and osteoarthritis (OA): Roles of ATP and PPi Metabolism and inorganic phosphate (Pi) generation in pathologic cartilage calcification. This model presents mechanisms underlying the common association of extracellular PPi excess with both CPPD and HA crystal deposition in OA and chondrocalcinosis cartilages, as well as the paradoxical association of extracellular PPi deficiency (from defective ANK or PC-1/NPP1 expression) with pathologic calcification of articular cartilage with HA crystals in vivo. Factors driving pathologic calcification are indicated in green and physiologic factors suppressing calcification in red. Excess PPi generation in aging cartilages in idiopathic CPPD deposition disease of aging, and in OA cartilages, is mediated in part by marked increases in NTPPPH activity, mediated in large part by the PC-1/NPP1 isoenzyme. In idiopathic chondrocalcinosis of aging and in OA, there are substantial increases in joint fluid PPi derived largely from cartilage. NPP1 not only directly induces elevated PPi but also matrix calcification by chondrocytes in vitro. Depending on extracellular availability of substrate PPi and the activity of pyrophosphatases, the availability of substrate ATP and the activity of ATPases, and other factors such as substantial local Mg++ concentrations, HA crystal deposition, as opposed to CPPD deposition, may be stimulated. In this model, excess extracellular PPi also may result from heightened release of intracellular PPi via increased ANK expression in OA and abnormal ANK function in familial chondrocalcinosis, as well as from deficient activity of pyrophosphatases (such as TNAP and possibly inorganic pyrophosphatase) in certain primary metabolic disorders. Also illustrated at the top of this schematic is the role in cartilage calcification in OA and aging of altered TGFβ expression and responsiveness, which drives PPi generation and release mediated via NPP1 and ANK, and diminished responsiveness to IGF-I, which normally suppresses elevation of chondrocyte extracellular PPi.
PPi appears to directly regulate expression of certain genes (including inductive effects first described by us for osteopontin and MMP-13 expression and suppressive effects on Sox9 expression)[18, 33–35]. PPi regulates certain cellular differentiation and functions including protein synthesis [19], chondrogenesis [35], and promineralizing chondrocyte maturation to terminal hypertophic differentiation transduced partly by Pit-1 mediated Pi uptake [36]. Such effects of PPi are analogous to effects of not only Pi [37, 38] but also bisphosphonate PPi analogues [39, 40]. It is not clear in which subcellular compartments PPi could act to carry out these effects and what contributions Pi derived from PPi makes in these activities of PPi.
Mammalian extramitochondrial mechanisms for PPi production, degradation, and transport were recently reviewed in depth [19]. In cells such as osteoblasts and chondrocytes that normally express NPP1 relatively robustly, NPP1 and NPP3 increase intracellular PPi, suspected to be in large part in the lumen of the ER and Golgi [12, 16, 31]. Critical to support of extracellular PPi is apparent direct PPi transport by the multiple-pass transmembrane protein ANK [41], which makes a major contribution to moving to the movement into the extracellular space of intracellular PPi, including the fraction of intracellular PPi generated by NPP1 [34].
NPP1 and PPi metabolism in cartilage and bone
Extracellular PPi rises markedly in articular cartilage in direct association with aging and OA, and resultant matrix supersaturation with PPi and cartilage matrix abnormalities that alter the solubility product of PPi and Ca2+ promote calcification [42]. Physiologic chondrocyte PPi metabolism is regulated in part by growth factor and cytokine regulatory effects on chondrocyte NPP1 expression. Interruption in regulatory checks and balances on articular cartilage PPi metabolism appears to occur in aging and diseases including OA. For example, the chondrocyte growth factor TGFβ induces both NPP1 expression and elevation of extracellular PPi [12, 31, 43]. The capacity of TGFβ to increase cartilage NPP activity and extracellular PPi levels directly correlates with donor age [12, 31, 44]. The TGFβ-stimulated cellular program for chondrocyte extracellular PPi elevation includes substantial increases in ATP generation [45] and stimulation of NPP1 movement to the plasma membrane [12, 31].
Osteoblasts and chondrocytes have particularly high levels of both NPP1 expression and NPP specific activity [19, 46, 47]. Moreover, chondrocyte NPP activity increases in direct condordance with cartilage PPi generation (to an average of double normal levels) in a donor age-dependent manner [47]. The age-dependent increases in NPP activity are directly linked to CPPD crystal deposition disease [47]. Upregulation of NPP1 but not NPP3 is associated with calcification by chondrocytic cells in situ and in vitro [12, 31]. Unlike NPP1, which regulates both intracellular and extracellular PPi in chondrocytes, NPP3 appears to principally regulate only intracellular PPi [12, 31].
Chondrocyte mitochondrial dysfunction associated with spontaneous OA in Hartley guinea pig knees promotes ATP depletion [48]. Significantly increased NPP activity and extracellular PPi develop concurrent with the ATPdepleted state [48]. Hence, increased ATP-scavenging by energy-depleted chondrocytes likely promotes extracellular PPi excess in human OA and aging cartilages.
A series of studies from one research group erroneously reported that cartilage intermediate layer protein (CILP), an interterritorial and pericellular matrix constituent in cartilage with a molecular weight similar to that of NPP1, was an NPP family member, even though there was no structural similarity of CILP to NPP family members [49–51]. We refuted this work [52], and in so doing, we demonstrated that increased expression of one of 2 CILP isoforms (CILP-1) in aging cartilage interferes with the regulatory effects of IGF-I on PPi metabolism, thereby promoting increased extracellular PPi and cartilage calcification.
NPP1 and PPi metabolism in pathologic soft tissue calcification syndromes and pivotal role of osteopontin depletion
Consistent with the apparent co-dependent function of ANK and NPP1 to raise extracellular PPi [34] is the remarkable similarity in the consequences of deficient ANK and PC-1 function in vivo. Both NPP1 deficient mice and mice homozygous for a natural C-terminal ANK mutant that appears to incapacitate ANK PPi transport function (ank/ank mice) spontaneously develop a progressive phenotype of pathologic soft tissue calcification that with increasing age comes to include perispinal ligament hyperostosis, periarticular calcification leading to ossific fusion of peripheral joints, extensive articular cartilage degeneration associated with HA deposits, and large artery calcification [22, 35]. The initial implication of NPP1 deficiency in spontaneous pathologic soft tissue calcification was in ‘tiptoe walking’ ttw/ttw mice, which are homozygous for a spontaneous nonsense mutation that encodes for a stop codon at tyrosine 568, a position 3′ of the NPP1 catalytic site [20]. It is not yet known if NPP1 expression is depressed or absent in ttw/ttw mice, or if the ttw mutation, like many of the NPP1 mutations seen in humans with GACI, critically impairs catalytic activity, putatively by interfering with substrate binding.
Human ossification of the posterior longitudinal ligament (OPLL), a form of spontaneous pathologic perispinal ligament calcification common in Japanese subjects, has been linked with certain SNPs in the NPP1 gene [53–55]. It will be of interest to see if the implicated NPP1 sequence variants affect NPP1 expression and function. Interestingly, the inflammatory cytokine IL-1 depresses NPP1 expression, NPP activity, and extracellular PPi in chondrocytes [43]. In this context, a ∼30% depression in serum NPP activity is seen in males with the chronic inflammatory disease ankylosing spondylitis [56], a condition that, like OPLL and spinal alterations in NPP1-deficient mice, associated with ankylosing intervertebral soft tissue calcification.
Interestingly, periarticular and bone abnormalities are far more substantial and progressive in NPP1-deficient mice than in NPP1-deficient humans with GACI. Conversely NPP1 deficient mice [35] do not demonstrate the severe myointimal proliferative changes seen in arteries in human GACI [24, 25]. We speculate that the relatively high level of normal serum Pi in mice compared to humans (≤8 vs. ≤4.5 mg/dL, respectively) [46] plays a major role in determining these phenotypic distinctions. In this context, high dietary Pi worsens pathologic calcification in NPP1 null mice [46]. Conversely, low serum Pi induced by crossbreeding with PHEX null mice is associated with correction of pathologic artery and soft tissue calcification in both NPP1 null and ank/ank mice [46].
Unlike cultured cells of ank/ank mice, NPP1-deficient cells demonstrate low intracellular as well as extracellular PPi levels [18]. Thus, the common basis for the remarkably similar hypermineralizing phenotypes seen in ank/ank mice and in NPP1 null mice (and the pathologic calcification seen in the human NPP1 deficiency state GACI) appears to rest in depression of extracellular PPi. Furthermore, the marked depletion of extracellular PPi and of osteopontin, the rapid, extensive calcification by both NPP1-/- and ank/ank osteoblasts in culture are corrected by soluble NPP1, reinforcing a central role of NPP1 in skeletal PPi and Pi metabolism and osteopontin expression [18], a notion strongly supported by in vivo studies [21, 33].
Pi, mediated by uptake through plasma membrane sodium-phosphate co-transport, stimulates expression of osteopontin, an inhibitor of HA crystal growth and promoter of mineral resorption [37, 38]. As cited above, exogenous PPi also induces osteopontin expression [18, 33]. It is not yet clear whether uptake of Pi derived from extracellular PPi is a major signaling intermediate in this process. Nevertheless, it is remarkable that one HA crystal growth inhibitor (PPi) promotes expression of a second in the form of osteopontin. Because osteopontin knockout mice have relatively mild changes in mineralization in contrast to the marked phenotypic abnormalities in extracellular PPi-deficient mice, PPi clearly higher than osteopontin in the physiologic hierarachy of HA crystal growth inhibitors.
As previously reviewed [19, 29], NPP1 plays a major role in regulating nucleation of mineral in chondrocyte-, osteoblast-, and apparently artery smooth muscle cellderived secretory bodies released by budding from the plasma membrane and termed matrix vesicles (MVs). The MVs, are enriched in NPP1 and TNAP, whose catalytic domains are predominantly exposed at the external face of MVs. The MVs provide a sheltered environment for initiation of mineral crystal formation in a manner modulated by the concentration of PPi, though mineral propagation is mediated by other factors, including availability of fibrillar collagen in ‘osteoid’ to serve as a nidus for calcification with HA [46]. NPP1 is clearly the principal NPP associated with chondrocyte-derived and osteoblast-derived MVs [16, 17, 21, 30, 57]. NPP1 and TNAP exert mutually antagonistic regulatory effects on crystal deposition in MVs, and activity not shared by NPP3 [16]. Cell differentiation and a variety of calciotropic hormones and cytokines (including 1,25 dihydroxyvitamin D3, TGFβ, and IL-1) can regulate the NPP1 content, NPP and alkaline phosphatase activities, PPi content, and other compositional features of MVs [29]. However, we have not seen concentrated ANK localization in MVs [33], likely contributing to the observation that correction of pathologic calcification by TNAP deficiency is less marked in ank/ank than NPP1-/- mice [33].
NPP1 and PPi deficiency states are linked to accelerated chondrogenesis
Taken together, it is clear that NPP1 and PPi physiologically function to prevent calcification of arteries and certain other soft tissues at the level of cell differentiation, and not simply at the level of mineral formation and resorption in the extracellular matrix. Most strikingly, we recently discovered that trans-differentiation of artery SMCs and accelerated intra-arterial chondrogenic differentiation mediated directly by PPi depletion promotes spontaneous artery media calcification in NPP1-/- and ank/ank mice [35]. Specifically, we observed that NPP1 deficiency promoted the spontaneous emergence of chondrogenesis from bone marrow stromal cells under noncalcifying conditions. Cultured NPP1-/- aortic SMC preparations and NPP1-/- aortic cells 023060 in situ expressed cbfa1, osteocalcin, and chondrocyte-specific collagens. Osteopontin expression was depressed and procalcifying alkaline phosphatase specific activity and calcification were markedly upregulated in cultured NPP1-/- SMCs [35]. In contrast, there was no gross alteration in expression of the physiologic artery calcification inhibitors matrix gla protein and osteoprotegerin in NPP1-/- mouse arterial cells [35]. The capacity of exogenous PPi to correct spontaneous chondrogenesis in NPP1-/- bone marrow stromal cells under non-calcifying conditions suggested that extracellular PPi deficiency directly promoted chondrogenesis and trans-differentiation to chondrocytes of the SMCs, a notion supported by aortic media calcification and changes in cultured SMC differentiation and calcification in ank/ank mice [35]. Therefore, acquired regional and systemic decrements in NPP1 and ANK expression and extracellular PPi could contribute to intra-arterial chondroosseous metaplasia and calcification in aging, diabetes mellitus, and atherosclerosis. In addition, it is noteworthy that systemic PPi deficiency is seen in hemodialysisdependent renal insufficiency, a condition associated with hyperphosphatemia and often extensive artery media and periarticular calcifications [58].
Conclusions and perspectives
Support of extracellular PPi levels by NPP1 and ANK inhibits pathologic soft tissue calcification but supports hard tissue mineralization in long bones and promotes calcification of articular cartilages in aging and OA. PPi is a central regulator of calcification in the extracellular matrix, but extracellular PPi regulates gene expression and cellular differentiation, including major physiologic effects on chondrogenesis and expression of osteopontin. The larger significance of mutants of NPP1 and ANK in disease continues to be elucidated. For example, mutants of ANKH, concentrated mainly at the N-terminal end of the molecule, have been linked with both autosomal dominant familial and ‘sporadic’ CPPD crystal deposition disease of articular cartilage [59, 60]. But other ANKH mutants clustered in putative cytosolic loops well-removed the N-and C-termini are linked with the distinct phenotype of craniometaphyseal dysplasia, a disease mediated by abnormal skeletal remodeling more than pathologic calcification [61, 62]. Polymorphisms in the human homologue of ANK (ANKH) also appear to contribute to differences in hand bone size and geometry that may influence bone fragility in a homogeneous Chuvasha population [63]. In the same population, NPP1 gene polymorphisms appeared to contribute to variance in severity of hand joint OA [64].
NPP1, in a catalytic activity-independent manner, inhibits ligand-induced insulin receptor signaling [65], an effect that appears linked to NPP1 mutations associated with type II diabetes mellitus in some but not all ethnic groups studied [66, 67]. Interestingly, the K173Q SNP of NPP1, which maps to the second somatomedin-B-like domain of NPP1 and has been linked to insulin resistance, does not modulate NPP1 dimerization or catalytic activity or affect physical interaction of NPP1 with the insulin receptor [68]. Inherited states of putative ‘gain-of-function’ of NPP1 also have been linked to obesity [69], also likely mediated primarily via effects on insulin receptor signaling. However, it is not likely that the numerous NPP1 catalytic site-independent mutants implicated as interfering with ligand-induced insulin receptor signaling directly affect mineralization.
Last, NPP1 not only generates PPi but also modulates N-glycosylation and secretion of glycoproteins, and proteoglycans sulfation [6–8], and NPP1 also scavenges ATP and thereby regulates purinergic receptor signaling. The potential roles in calcification of these alternative effects of NPP1, and of other NPP1 interactions with nucleotide-hydrolyzing ecto-ezymes, remain to be determined. Nevertheless, the remarkable phenotypic similarities between NPP1-deficient and ANK-deficient mice strongly support the central role of NPP1 catalyzed PPi generation in the regulation of calcification.
Acknowledgment
Our work has is supported by the Department of Veterans Affairs Research Service and by NIH grants HL077360, P01AGO7996, AR049366.
Abbreviations
- ANK
protein product of the murine ankylosis disease susceptibility gene
- CPPD
calcium pyrophosphate dihydrate
- CILP
cartilage intermediate layer protein
- HA
hydroxyapatite
- IIAC
Idiopathic Infantile Artery Calcification
- MV
matrix vesicles
- NPP
nucleotide pyrophosphatase/phosphodiesterase
- OPLL
ossification of the posterior longitudinal ligament
- SMC
smooth muscle cells
- SNP
single nucleotide polymorphism
- TNAP
tissue nonspecific alkaline phosphatase
References
- 1.Robson SC, Wu Y, Sun X et al. Ectonucleotidases of CD39 family modulate vascular inflammation and thrombosis in transplantation. Semin Thromb Hemost 2005; 31: 217–33. [DOI] [PubMed]
- 2.Vorhoff T, Zimmermann H, Pelletier J et al. Cloning and characterization of the ecto-nucleotidase NTPDase3 from rat brain: Predicted secondary structure and relation to other members of the E-NTPDase family and actin. Purinergic Signalling 2005; 1: 259–70. [DOI] [PMC free article] [PubMed]
- 3.Deterre P, Gelman L, Gary-Gouy H et al. Coordinated regulation in human T cells of nucleotide-hydrolyzing ecto-enzymatic activities, including CD38 and PC-1. Possible role in the recycling of nicotinamide adenine dinucleotide metabolites. J Immunol 1996; 157: 1381–8. [PubMed]
- 4.Stefan C, Jansen S, Bollen M. NPP-type ectophosphodiesterases: Unity in diversity. Trends Biochem Sci 2005; 30: 542–50. [DOI] [PubMed]
- 5.Bello V, Goding JW, Greengrass V et al. Characterization of a di-leucine-based signal in the cytoplasmic tail of the nucleotide-pyrophosphatase NPP1 that mediates basolateral targeting but not endocytosis. Mol Biol Cell 2001; 12: 3004–15. [DOI] [PMC free article] [PubMed]
- 6.Hickman S, Wong-Yip YP, Rebbe NF, Greco JM. Formation of lipid-linked oligosaccharides by MOPC 315 plasmacytoma cells. Decreased synthesis by a nonsecretory variant. J Biol Chem 1985; 260: 6098–106. [PubMed]
- 7.Goding JW, Grobben B, Slegers H. Physiological and pathophysiological functions of the ecto-nucleotide pyrophosphatase/phosphodiesterase family. Biochim Biophys Acta 2003; 1638: 1–19. [DOI] [PubMed]
- 8.Goding JW. Ecto-enzymes: Physiology meets pathology. J Leukoc Biol 2000; 67: 285–311. [DOI] [PMC free article] [PubMed]
- 9.Jansen S, Stefan C, Creemers JW et al. Proteolytic maturation and activation of autotaxin (NPP2), a secreted metastasis-enhancing lysophospholipase D. J Cell Sci 2005; 118: 3081–9. [DOI] [PubMed]
- 10.van Meeteren LA, Ruurs P, Christodoulou E et al. Inhibition of autotaxin by lysophosphatidic acid and sphingosine 1-phosphate. J Biol Chem 2005; 280: 21155–61. [DOI] [PubMed]
- 11.Cimpean A, Stefan C, Gijsbers R et al. Substrate-specifying determinants of the nucleotide pyrophosphatases/phosphodiesterases NPP1 and NPP2. Biochem J 2004; 381: 71–7. [DOI] [PMC free article] [PubMed]
- 12.Johnson K, Hashimoto S, Lotz M et al. Up-regulated expression of the phosphodiesterase nucleotide pyrophosphatase family member PC-1 is a marker and pathogenic factor for knee meniscal cartilage matrix calcification. Arthritis Rheum 2001; 44: 1071–81. [DOI] [PubMed]
- 13.Durgam GG, Virag T, Walker MD et al. Synthesis, structure-activity relationships, and biological evaluation of fatty alcohol phosphates as lysophosphatidic acid receptor ligands, activators of PPARgamma, and inhibitors of autotaxin. J Med Chem 2005; 48: 4919–30. [DOI] [PubMed]
- 14.Moolenaar WH. Lysophospholipids in the limelight: Autotaxin takes center stage. J Cell Biol 2002; 158: 197–9. [DOI] [PMC free article] [PubMed]
- 15.Koh E, Clair T, Woodhouse EC et al. Site-directed mutations in the tumor-associated cytokine, autotaxin, eliminate nucleotide phosphodiesterase, lysophospholipase D, and motogenic activities. Cancer Res 2003; 63: 2042–5. [PubMed]
- 16.Johnson KA, Hessle L, Vaingankar S et al. Osteoblast tissue-nonspecific alkaline phosphatase antagonizes and regulates PC-1. Am J Physiol Regul Integr Comp Physiol 2000; 279: R1365–77. [DOI] [PubMed]
- 17.Vaingankar SM, Fitzpatrick TA, Johnson K et al. Subcellular targeting and function of osteoblast nucleotide pyrophosphatase phosphodiesterase 1. Am J Physiol Cell Physiol 2004; 286: C1177–87. [DOI] [PubMed]
- 18.Johnson K, Goding J, Van Etten D et al. Linked deficiencies in extracellular PPi and osteopontin mediate pathologic calcification associated with defective PC-1 and ANK expression. J Bone Miner Res 2003; 18: 994–1004. [DOI] [PubMed]
- 19.Terkeltaub R. Inorganic pyrophosphate (PPi) generation and disposition in pathophysiology. Am J Physiol: Cell Physiol 2001; 281: C1–11. [DOI] [PubMed]
- 20.Okawa A, Nakamura I, Goto S et al. Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spine. Nat Genet 1998; 19: 271–3. [DOI] [PubMed]
- 21.Hessle L, Johnson KA, Anderson HC et al. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci USA 2002; 99: 9445–9. [DOI] [PMC free article] [PubMed]
- 22.Ho AM, Johnson MD, Kingsley DM. Role of the mouse ank gene in control of tissue calcification and arthritis. Science 2000; 289: 265–70. [DOI] [PubMed]
- 23.Ruf N, Uhlenberg B, Terkeltaub R et al. The mutational spectrum of ENPP1 as arising after the analysis of 23 unrelated patients with generalized arterial calcification of infancy (GACI). Human Mutat 2005; 25: 98–104. [DOI] [PubMed]
- 24.Rutsch F, Ruf N, Vaingankar S et al. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat Genet 2003; 34: 379–81. [DOI] [PubMed]
- 25.Rutsch F, Vaingankar S, Johnson K et al. PC-1 nucleoside triphosphate pyrophosphohydrolase deficiency in idiopathic infantile arterial calcification. Am J Pathol 2001; 158: 543–54. [DOI] [PMC free article] [PubMed]
- 26.Rutsch F, Schauerte P, Kalhoff H et al. Low levels of urinary inorganic pyrophosphate indicating systemic pyrophosphate deficiency in a boy with idiopathic infantile arterial calcification. Acta Paediatr 2000; 89: 1265–9. [DOI] [PubMed]
- 27.Anderson HC, Harmey D, Camacho NP et al. Sustained osteomalacia of long bones despite major improvement in other hypophosphatasia-related mineral deficits in tissue nonspecific alkaline phosphatase/nucleotide pyrophosphatase phosphodiesterase 1 double-deficient mice. Am J Pathol 2005; 166: 1711–20. [DOI] [PMC free article] [PubMed]
- 28.Kobayashi Y, Goto S, Tanno T et al. Regional variations in the progression of bone loss in two different mouse osteopenia models. Calcif Tissue Int 1998; 62: 426–36. [DOI] [PubMed]
- 29.Johnson K, Terkeltaub R. Inorganic pyrophosphate (PPi) in pathologic calcification of articular cartilage. Front Biosci 2005; 10: 98–97. [DOI] [PubMed]
- 30.Johnson K, Pritzker K, Goding J, Terkeltaub R. The nucleoside triphosphate pyrophosphohydrolase isozyme PC-1 directly promotes cartilage calcification through chondrocyte apoptosis and increased calcium precipitation by mineralizing vesicles. J Rheumatol 2001; 28: 2681–91. [PubMed]
- 31.Johnson K, Vaingankar S, Chen Y et al. Differential mechanisms of inorganic pyrophosphate production by plasma cell membrane glycoprotein-1 and B10 in chondrocytes. Arthritis Rheum 1999; 42: 1986–97. [DOI] [PubMed]
- 32.Derfus BA, Kurian JB, Butler JJ et al. The high prevalence of pathologic calcium crystals in pre-operative knees. J Rheumatol 2002; 29: 570–4. [PubMed]
- 33.Harmey D, Hessle L, Narisawa S et al. Concerted regulation of inorganic pyrophosphate and osteopontin by akp2, enpp1, and ank: An integrated model of the pathogenesis of mineralization disorders. Am J Pathol 2004; 164: 1199–209. [DOI] [PMC free article] [PubMed]
- 34.Johnson K, Terkeltaub R. Upregulated ank expression in osteoarthritis can promote both chondrocyte MMP-13 expression and calcification via chondrocyte extracellular PPi excess. Osteoarthritis Cartilage 2004; 12: 321–35. [DOI] [PubMed]
- 35.Johnson K, Polewski M, van Etten D, Terkeltaub R. Chondrogenesis mediated by PPi depletion promotes spontaneous aortic calcification in NPP1-/-mice. Arterioscler Thromb Vasc Biol 2005; 25: 686–91. [DOI] [PubMed]
- 36.Wang W, Xu J, Du B, Kirsch T. Role of the progressive ankylosis gene (ank) in cartilage mineralization. Mol Cell Biol 2005; 25: 312–23. [DOI] [PMC free article] [PubMed]
- 37.Beck GR Jr. Inorganic phosphate as a signaling molecule in osteoblast differentiation. J Cell Biochem 2003; 90: 234–43. [DOI] [PubMed]
- 38.Beck GR Jr, Zerler B, Moran E. Phosphate is a specific signal for induction of osteopontin gene expression. Proc Natl Acad Sci USA 2000; 97: 8352–7. [DOI] [PMC free article] [PubMed]
- 39.Rogers MJ. New insights into the molecular mechanisms of action of bisphosphonates. Curr Pharm Des 2003; 9: 2643–58. [DOI] [PubMed]
- 40.Fujita T, Izumo N, Fukuyama R et al. Incadronate and etidronate accelerate phosphate-primed mineralization of MC4 cells via ERK1/2-Cbfa1 signaling pathway in a Ras-independent manner: Further involvement of mevalonate-pathway blockade for incadronate. Jpn J Pharmacol 2001; 86: 86–96. [DOI] [PubMed]
- 41.Ho AM, Johnson DM. Kingsley: Role of the mouse ank gene in control of tissue calcification and arthritis. Science 2000; 289: 265–70. [DOI] [PubMed]
- 42.Kalya S, Rosenthal AK. Extracellular matrix changes regulate calcium crystal formation in articular cartilage. Curr Opin Rheumatol 2005; 17: 325–329. [DOI] [PubMed]
- 43.Lotz M, Rosen F, McCabe G et al. Interleukin 1 beta suppresses transforming growth factor-induced inorganic pyrophosphate (PPi) production and expression of the PPi-generating enzyme PC-1 in human chondrocytes. Proc Natl Acad Sci USA 1995; 92: 10364–8. [DOI] [PMC free article] [PubMed]
- 44.Rosen F, McCabe G, Quach J et al. Differential effects of aging on human chondrocyte responses to TGFβ: Increased pyrophosphate production and decreased cell proliferation. Arthritis Rheum 1997; 40: 1275–81. [DOI] [PubMed]
- 45.Johnson K, Jung AS, Andreyev A et al. Mitochondrial Oxidative Phosphorylation is a downstream regulator of nitric oxide effects on chondrocyte matrix synthesis and mineralization. Arthritis Rheum 2000; 43: 1560–70. [DOI] [PubMed]
- 46.Murshed M, Harmey D, Millan JL et al. Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev 2005; 19: 1093–104. [DOI] [PMC free article] [PubMed]
- 47.Costello JC, Ryan LM. Modulation of chondrocyte production of extracellular inorganic pyrophosphate. Curr Opin Rheumatol 2004; 16: 268–72. [DOI] [PubMed]
- 48.Johnson K, Svensson CI, Etten DV et al. Mediation of spontaneous knee osteoarthritis by progressive chondrocyte ATP depletion in Hartley guinea pigs. Arthritis Rheum 2004; 50: 1216–25. [DOI] [PubMed]
- 49.Masuda I, Halligan BD, Barbieri JT et al. Molecular cloning and expression of a porcine chondrocyte nucleotide pyrophosphohydrolase. Gene 1997; 197: 277–87. [DOI] [PubMed]
- 50.Masuda I, Hamada J, Haas AL et al. A unique ectonucleotide pyrophosphohydrolase associated with porcine chondrocyte-derived vesicles. J Clin Invest 1995; 95: 699–704. [DOI] [PMC free article] [PubMed]
- 51.Hirose J, Masuda I, Ryan LM. Expression of cartilage intermediate layer protein/nucleotide pyrophosphohydrolase parallels the production of extracellular inorganic pyrophosphate in response to growth factors and with aging. Arthritis Rheum 2000; 43: 2703–11. [DOI] [PubMed]
- 52.Johnson K, Farley D, Hu SI, Terkeltaub R. One of two chondrocyte-expressed isoforms of cartilage intermediate-layer protein functions as an insulin-like growth factor 1 antagonist. Arthritis Rheum 2003; 48: 1302–14. [DOI] [PubMed]
- 53.Nakamura I, Ikegawa S, Okawa A et al. Association of the human NPPS gene with ossification of the posterior longitudinal ligament of the spine (OPLL). Hum Genet 1999; 104: 492–7. [DOI] [PubMed]
- 54.Koshizuka Y, Kawaguchi H, Ogata N et al. Nucleotide pyrophosphatase gene polymorphism associated with ossification of the posterior longitudinal ligament of the spine. J Bone Miner Res 2002; 17: 138–44. [DOI] [PubMed]
- 55.Tahara M, Aiba A, Yamazaki M et al. The extent of ossification of posterior longitudinal ligament of the spine associated with nucleotide pyrophosphatase gene and leptin receptor gene polymorphisms. Spine 2005; 3: 877–80. [DOI] [PubMed]
- 56.Mori K, Chano T, Ikeda T et al. Decrease in serum nucleotide pyrophosphatase activity in ankylosing spondylitis. Rheumatology (Oxford) 2003; 42: 62–5. [DOI] [PubMed]
- 57.Johnson K, Moffa A, Chen Y et al. Matrix vesicle plasma cell membrane glycoprotein-1 regulates mineralization by murine osteoblastic MC3T3 cells. J Bone Miner Res 1999; 14: 883–92. [DOI] [PubMed]
- 58.Lomashvili KA, Khawandi W, O’Neill WC. Reduced plasma pyrophosphate levels in hemodialysis patients. J Am Soc Nephrol 2005; 16: 2495–500. [DOI] [PubMed]
- 59.Williams CJ. Familial calcium pyrophosphate dihydrate deposition disease and the ANKH gene. Curr Opin Rheumatol 2003; 15: 326–31. [DOI] [PubMed]
- 60.Zhang Y, Johnson K, Russell RG et al. Association of sporadic chondrocalcinosis with a —4-basepair G-to-A transition in the 5’-untranslated region of ANKH that promotes enhanced expression of ANKH protein and excess generation of extracellular inorganic pyrophosphate. Arthritis Rheum 2005; 52: 1110–7. [DOI] [PubMed]
- 61.Reichenberger E, Tiziani V, Watanabe S et al. Autosomal dominant craniometaphyseal dysplasia is caused by mutations in the transmembrane protein ANK. Am J Hum Genet 2001; 68: 1321–6. [DOI] [PMC free article] [PubMed]
- 62.Nurnberg P, Thiele H, Chandler D et al. Heterozygous mutations in ANKH, the human ortholog of the mouse progressive ankylosis gene, result in craniometaphyseal dysplasia. Nat Genet 2001; 28: 37–41. [DOI] [PubMed]
- 63.Malkin I, Dahm S, Suk A et al. Association of ANKH gene polymorphisms with radiographic hand bone size and geometry in a Chuvasha population. Bone 2005; 36: 365–73. [DOI] [PubMed]
- 64.Suk EK, Malkin I, Dahm S et al. Association of ENPP1 gene polymorphisms with hand osteoarthritis in a Chuvasha population. Arthritis Res Ther 2005; 7: R1082–90. [DOI] [PMC free article] [PubMed]
- 65.Dong H, Maddux BA, Altomonte J et al. Increased hepatic levels of the insulin receptor inhibitor, PC-1/NPP1, induce insulin resistance and glucose intolerance. Diabetes 2005; 54: 367–72. [DOI] [PubMed]
- 66.Morrison JA, Gruppo R, Glueck CJ et al. Population-specific alleles: The polymorphism (K121Q) of the human glycoprotein PC-1 gene is strongly associated with race but not with insulin resistance in black and white children. Metabolism 2004; 53: 465–8. [DOI] [PubMed]
- 67.Gijsbers R, Ceulemans H, Bollen M. Functional characterization of the non-catalytic ectodomains of the nucleotide pyrophosphatase/phosphodiesterase NPP1. Biochem J 2003; 371: 321–30. [DOI] [PMC free article] [PubMed]
- 68.Stefanovic V, Antic S. Plasma cell membrane glycoprotein 1 (PC-1): A marker of insulin resistance in obesity, uremia and diabetes mellitus. Clin Lab 2004; 50: 271–8. [PubMed]
- 69.Meyre D, Bouatia-Naji N, Tounian A et al. Variants of ENPP1 are associated with childhood and adult obesity and increase the risk of glucose intolerance and type 2 diabetes. Nat Genet 2005; 37: 863–7. [DOI] [PMC free article] [PubMed]