

Abstract
Diacylglycerol acyltransferase 2 (DGAT2) catalyses the final step of triacylglycerol (TG) synthesis. Despite the existence of an alternative acyltransferase (DGAT1), mice lacking DGAT2 have a severe deficiency of TG in adipose tissue, indicating a non redundant role for this enzyme in adipocyte TG synthesis. We have studied the regulation of DGAT2 expression during adipogenesis. In both isolated murine pre-adipocytes and 3T3-L1 cells the temporal pattern of DGAT2 expression closely mimicked that of genes whose expression is regulated by C/EBPβ. Inhibition of C/EBPβ expression in differentiating preadipocytes reduced DGAT2 expression and EMSA and chromatin immunopreciptation experiments identified a promoter element in the DGAT2 gene that is likely to mediate this effect. The importance of C/EBPβ in adipocyte expression of DGAT2 was confirmed by the finding of reduced DGAT2 expression in the adipose tissue of C/EBPβ null animals. However, DGAT2 expression is maintained at high levels during the later stages of adipogenesis, when C/EBPβ levels decline. We show that, at these later stages of differentiation, C/EBPα is capable of substituting for C/EBPβ at the same promoter element. These observations provide novel insight into the transcriptional regulation of DGAT2 expression. Moreover, they further refine the complex and serial roles of the C/EBP family of transcription factors in inducing and maintaining the metabolic properties of mature adipocytes.
The increased adipose tissue mass of obesity results from a combination of increased lipid storage in existing adipocytes and the generation of new adipocytes from precursors residing within the adipose tissue (1). The induction of genes responsible for the formation of triacylglycerol (TG) within developing or pre-existing adipocytes is therefore likely to make an important contribution to the enlargement of adipose mass. In contrast, pathologically decreased lipid accumulation or impaired adipogenesis in lipodystrophic subjects has deleterious metabolic consequences superficially like those seen in obesity, including insulin resistance and dyslipidemia with attendant increases in cardiovascular disease. Thus good metabolic control is likely to require the body to restrain adipose tissue mass whilst still maintaining the capacity to respond accurately to substrate availability. In this way, when necessary, lipids can be partitioned appropriately into adipose tissue and away from other insulin sensitive tissues where they may have detrimental effects. The demonstration that mutations of AGPAT2, a key enzyme in TG synthesis, can cause near total lipodystrophy demonstrates the importance of this pathway in such diseases of adipose development and function (2,3). Rational therapeutic strategies for both obesity and lipodystrophy will require a detailed knowledge of the regulatory pathways required for the formation of an appropriate mass of metabolically active adipocytes, capable of tightly controlling lipid synthesis and storage. The enzyme diacylglycerol acyltransferase (DGAT) catalyses the final step of mammalian TG synthesis and two isoforms, DGAT1 and DGAT2, exist, encoded by different genes. DGAT1 knockout mice have been shown to be resistant to high fat diet induced obesity due to increased metabolic rate with increased physical activity (4-6). Mice lacking DGAT2, however, show a more dramatic phenotype, with severe lipopenia and early post-natal death due to a lack of substrates for energy metabolism and defects of skin permeability (7). Thus, DGAT2 has a key role in TG synthesis and, in addition is highly expressed in adipocytes. However, the molecular mechanisms controlling its expression in adipose tissue have not been defined to date.
The transcriptional control of gene expression during adipogenesis involves the complex interplay of a multitude of transcription factors whose temporal expression must be precisely co-ordinated (8-10). Several factors have been shown to play central roles in this transcriptional cascade. The induction of C/EBPβ and C/EBPδ occurs rapidly following the initiation of adipogenesis and these factors, modulated by a plethora of co-factors, induce the expression of a second wave of genes including the so-called “master regulators” of adipogenesis, C/EBPα and PPARγ. The targets of these transcription factors include the genes encoding many genes of the mature lipogenic and insulin-sensitive adipocyte such as aP2, PEPCK, aquaporin 7, lipoprotein lipase, adiponectin and Glut-4 (8,11,12). Thus the C/EBP family of transcription factors has a critical role in adipogenesis and studies both of loss and gain of function in vitro and in vivo have demonstrated the importance of their activity in adipocyte development and lipid accumulation. Ablation of C/EBPα in mice leads to a loss of white adipose tissue and impaired adipogenesis in culture whilst the expression of C/EBPα in fibroblasts and preadipocytes facilitates adipogenesis (13-18). Studies examining the effects of the rapidly induced C/EBPβ and C/EBPδ have demonstrated that loss of function of one or both of these factors can lead to decreased adipose mass in mice and decreased adipogenesis in cellular models (19-22). Whilst C/EBPβ and C/EBPδ appear to have some compensatory, synergistic and overlapping functions, C/EBPβ appears to have the greater effect on adipocyte development and lipid accumulation (13-18). Here we examine the control of DGAT2 expression during adipogenesis and, for the first time, define a transcriptional mechanism for the regulation of its expression, demonstrating a key role for the C/EBP family of transcription factors.
Experimental Proceedures
Preadipocyte Isolation and Culture
Murine preadipocyte isolation, culture and differentiation were performed as described previously (23). Human preadipocytes were grown from the stromovascular fraction of collagenase digested abdominal subcutaneous adipose tissue as previously described (24). At various times following induction of differentiation, cells were harvested, and RNA extracted. 3T3-L1 preadipocytes were maintained and differentiated as described in (25). 3T3-L1 preadipocytes constitutively expressing the LIP (liver inhibitory protein) isoform of C/EBPβ were generated as follows: LIP cDNA was amplified by PCR using a forward primer immediately upstream of the LIP initiating ATG and using the pMT2-C/EBPβ expression vector (generously provided by Dr Q-Q Tang and Dr M.D. Lane). This was subcloned into pBabe retroviral vector which was then used to generate retrovirus in HEK293-BOSC cells and to subsequently infect 3T3-L1 preadipocytes. Mock transfected 3T3-L1 preadipocytes were generated with the same protocol using empty pBabe vector. Cells stably expressing ETO (eight-twenty one/MTG8) were as previously described (25). Differentiating 3T3-L1 cells were assessed for lipid content by staining with oil-red O as in (25).
siRNA knockdown
Synthetic double stranded si-RNA against C/EBPβ or C/EBPα mRNAs were purchased from Ambion. 3T3-L1 preadipocytes were plated at a density 1×105 cells per well in 12-well plates the day before siRNA transfection. siRNA/ liposome mixes containing 2 μg of Lipofectamine 2000 (Invitrogen) and 100nM of si-RNA/well were incubated with cells for 6 hours in the absence of serum. Medium was replaced with serum containing 3T3-L1 growth medium for 18 hours before the induction of differentiation.
RNA Isolation, cDNA Synthesis, and Real Time PCR
Total RNA was extracted from cell cultures using an RNeasy kit (Qiagen). Adipose tissue was isolated from C/EBPβ null mice or their wild type littermates as previously described (26). All procedures were approved by the UCHSC Animal Care and Use Committee. Frozen adipose tissue samples were first minced finely with scissors, then RNA isolated according to the RNeasy kit protocol (Qiagen). Samples were eluted in 50μl of RNase free H20 and RNA concentration determined by GeneQuant (Amersham Biosciences). The quality of extracted RNA was assessed by formaldehyde gel electrophoresis. Primer Express, version 1.0 software (Perkin Elmer Applied Biosystems) was used to design the probes and primers for real time quantitative PCR to determine DGAT2, DGAT1, C/EBPβ and 11βHSD1 mRNA expression. Primer/probe mix to assay C/EBPα was obtained from Applied Biosystems. RNA was reverse transcribed using Moloney murine leukemia virus-reverse transcriptase and random hexamer primers (Promega). The resulting cDNA was used in 12μl PCR reactions, in which 300nmol/l of forward and reverse primers and, where applicable, 150nmol/l of fluorogenic probe were used in combination with ABI Taqman or Sybr green master mix (Applied Biosystems). Reactions were carried out in duplicate for each sample on an ABI 7900 sequence detection system (Perkin Elmer Biosystems) according to the manufacturer's instructions. The relative quantities of amplified cDNAs were analyzed by the SDS software (Applied Biosystems) and target values were normalised to 18S rRNA (tissue samples) or cyclophilin A mRNA (cell culture samples).
EMSA
Post-confluent 3T3-L1 preadipocytes differentiated for various times were washed with phosphate-buffered saline containing 1mM phenylmethylsulfonyl fluoride before being scraped and centrifuged at 1,200 rpm at 4 °C. The cell pellet was subjected to crude nuclear protein preparation using a cytosolic and nuclear protein extraction kit (Pierce). EMSA was performed using LightShift chemiluminescent EMSA kit (Pierce). The probes were prepared by annealing complementary oligonucleotides with their 3'-end labeled with biotin. The oligonucleotides sequences were as follows: C/EBPα promoter C/EBP binding site, 5'-CAGTGGGCGTTGCGCCACGATCTCTCT; DGAT2 putative site 1 C/EBP site, 5'-ACACGTCTATTGGCCAATCTACCGT. The DNA-protein binding was performed at room temperature for 20 min in a final volume of 20μl containing 1x binding buffer (10mM Tris, pH 7.5, 50mM KCl, 1mM dithiothreitol), 2.5% (v/v) glycerol, 5mM MgCl2, 1μg of poly(dI-dC), 0.05% (v/v) Nonidet P-40, 8pmol of double-stranded biotinylated probe, and 10μg of nuclear extract. The DNA-protein complexes were separated by 5% PAGE in 0.5x TBE at 200 V at 4 °C for 2 hours. DNA-protein complexes in gel were transferred to Hybond N nylon membrane (Amersham Biosciences) by electroblotting with 0.5x TBE at 350 mA for 1.5 hours. DNA-protein complexes were fixed to the membrane by UV cross-linker and detected by a nonradioactive nucleic acid detection kit (Pierce). For the competition assay, 20x more concentrated double-stranded DNAs were included in the binding reaction. Where appropriate, 2μl of anti-C/EBPβ (C-19) or anti-C/EBPα antibody (14AA) (Santa Cruz Biotechnology) were preincubated in the binding reaction for 10 min before the probe was added.
ChIP Assay
3T3-L1 preadipocytes in 35-mm wells were differentiated for various times as indicated. The DNA and protein were cross-linked in situ with 0.5% (v/v) formaldehyde at 37 °C for 5 min. Soluble chromatin was prepared using a chromatin immunoprecipitation assay kit (Upstate Biotechnology, Inc.). The lysate was sonicated four times for 10 s at 4 °C. The lysates were precipitated with either 5μl of anti-C/EBPβ (C-19) or anti-C/EBPα antibody (Santa Cruz Biotechnology) overnight before protein A-agarose beads were added. The proteins were removed from DNA by digesting with 10μg/ml proteinase K at 45 °C for 30 min. The DNA was further purified by a QIAquick PCR purification kit (Qiagen). The DNA was eluted in 50μl of sterile water. Two microlitres of eluted DNA was used to assay the presence of DNA sequences associated with the immunoprecipitated proteins using specific primers amplifying DNA sequences including the binding sites being assayed and Sybr green master mix according to the manufacturer's instructions. Values obtained from immunoprecipitated samples were normalised to those from input samples.
Western Blotting
Protein samples were extracted by scraping in lysis buffer containing 1% NP40, followed by sonication as described previously (25). After centrifugation for 10 min at 13,000g, samples of supernatant containing 30μg of protein were denatured and analysed by western blotting. All antibodies were from Santa Cruz Biotechnology.
RESULTS
To determine the expression of DGAT1 and DGAT2 during adipogenesis, 3T3-L1 preadipocytes were induced to differentiate and DGAT1 and DGAT2 expression was assayed by real-time PCR. Both DGAT1 and DGAT2 mRNA were induced with a similar time course, with a minor increase in expression in the first few hours following induction and strong and sustained increase of mRNA occurring within 3 days. Of the two isoforms, DGAT2 showed a much greater increase, with an approximately 200-fold induction within 3 days, whilst DGAT1 expression increased by less than 10-fold (Fig. 1a). To determine whether the induction of DGAT2 also occurred when isolated mouse preadipocytes undergo differentiation, we next examined DGAT2 expression in cells isolated from the stromovascular fraction of mouse WAT, induced to differentiate in culture. In a manner similar to differentiating 3T3-L1 preadipocytes, DGAT2 expression increased significantly 2 days following induction of differentiation with maximal induction apparent after 4 to 6 days (Fig. 1b). To extend these observations we next determined DGAT2 expression in differentiating human preadipocytes. Cultures of cells isolated from the stromovascular fraction of abdominal subcutaneous adipose tissue were induced to differentiate for various times and the expression of DGAT2 mRNA determined (Fig. 1c). Again, a strong and significant upregulation of DGAT2 expression was observed within 3 days of the induction of differentiation. This demonstrates that the induction of DGAT2 expression in the 3T3-L1 model recapitulates not only murine, but also human adipogenesis and that DGAT2 may be an important enzyme of adipocyte triglyceride synthesis in humans as well as mice. Given the physiological importance of DGAT2, apparent from the phenotype of DGAT2 null mice, the strong induction of this isoform during adipogenesis and yet the lack of knowledge about the regulation of its expression, we decided to investigate its regulation in developing adipocytes further.
Figure 1. DGAT2 expression is strongly induced during adipocyte differentiation.

A, 3T3-L1 preadipocytes were induced to differentiate for various times up to 6 days (D6), RNA was isolated and DGAT1 (white bars) or DGAT2 (black bars) expression was determined by real-time PCR. Data shown are normalised to cyclophilin +/− SEM, n=4. B, DGAT2 expression was determined in RNA isolated from confluent isolated murine preadipocytes before or after induction of differentiation for various times between 4 hours (4h) and 8 days (D8). Data shown are normalised to cyclophilin +/− SD from 2 independent experiments performed with cells isolated and pooled from the adipose tissue of 4 mice in each case. C, confluent cultures of human isolated preadipocytes were induced to differentiate for various times, RNA isolated, reverse transcribed and DGAT2 expression determined by real-time PCR. Data shown are normalised to cyclophilin +/− SEM, n=5. In all cases * indicates statistically significant difference from expression at day 0 (p<0.05).
To identify potential transcriptional mechanisms by which DGAT2 is regulated during adipogenesis we first examined published microarray data of gene expression over an extended time course of 3T3-L1 preadipocyte differentiation. This revealed that the temporal regulation of DGAT2 very closely mimicked that of C/EBPα and 11βHSD1 (27), both well characterised targets of C/EBPβ (28,29). Despite the rapid induction of C/EBPβ during adipogenesis, the appearance of C/EBPα is characteristically delayed. Initially C/EBPβ is unable to bind the C/EBPα promoter due, at least in part, to the inhibitory actions of CHOP10 and/or ETO (25,30). Subsequently C/EBPβ binds promoter sequences in a repressed complex containing HDAC1 and Sin3a, before finally becoming an active complex 48-72 hours following induction (31,32). As the characteristically delayed time course of C/EBPα expression was shared by DGAT2 mRNA, this led us to speculate that C/EBPβ may also have a key role in the regulation of its expression. This is also consistent with the observation that inhibition of C/EBPβ in 3T3-L1 adipogenesis significantly inhibits lipid accumulation in these cells. To test this hypothesis we generated 3T3-L1 preadipocytes constitutively expressing the naturally occurring inhibitory form of C/EBPβ, LIP. LIP lacks the transactivating domain of the transcriptionally active LAP forms of C/EBPβ, but contains the leucine zipper and DNA binding motifs, allowing it to bind to C/EBPβ target sites and prevent binding by active forms of C/EBPβ (Fig. 2a) (33). Expression of C/EBPβ was assessed in mock or LIP transfected 3T3-L1 preadipocytes during differentiation by western blotting. In the LIP transfected cells, LIP expression was increased prior to the induction of differentiation to levels similar to the maximum level observed in mock transfected cells once differentiation had been induced (Fig. 2b). Consistent with this, following induction of differentiation, LIP expression in LIP overexpressing cells rose to levels approximately twice those in mock transfected cells.
Figure 2. Inhibtion of C/EBPβ activity by LIP expression inhibits the induction of DGAT2 during adipogenesis.

A, the inhibitory LIP isoform of C/EBPβ contains the DNA binding (DBD) and leucine zipper (LZ) domains but lacks the transcativation domain (TAD) present in the activating LAP isoform, and acts as a dominant negative. B, 3T3-L1 preadipocytes were stably transfected with LIP or empty vector (mock) and confluent cells were induced to differentiate for the times shown. Protein lysates were analysed for C/EBPβ expression by western blotting. C, lipid accumulation was assessed in mock or LIP transfected cells following differentiation for 8 days in the absence (MDI) or presence (MDI-BRL) of the PPARγ agonist BRL49653. Mock (black bars) or LIP (white bars) transfected cells were also differentiated for various times, RNA isolated and assayed for the expression of PPARγ1 (D), PPARγ2 (E) C/EBPα (F), 11-βHSD1 (G) or DGAT2 (H) by real-time PCR. Data shown are normalised to cyclophilin +/− SEM, n=4. * indicates statistically significant difference in expression compared with mock-transfected cells at the same time-point (p<0.05).
We next assessed the effect of LIP expression on lipid accumulation by staining cells with oil-red O 8 days following induction of differentiation in the presence or absence of the PPARγ agonist BRL. Consistent with previous studies, LIP expression almost completely inhibited the accumulation of lipid (Fig. 2c). Indeed, even BRL treatment, which enhanced lipid accumulation in mock transfected cells, was only able to induce very slight lipid accumulation in LIP cells. The expression of PPARγ1 and PPARγ2 was reduced by 57% and 60%, respectively, on day 3 of differentiation in these cells (Fig. 2d and 2e). Thus even pharmacological activation of the significant residual PPARγ expressed was unable to overcome the inhibitory effects of LIP expression.
We next assessed the expression of C/EBPα and 11βHSD1 in LIP cells. The induction of both these genes was significantly impaired in the LIP cells when compared with the mock transfected cells (Figs. 2f and 2g). Subsequent analysis of DGAT2 expression in these cells demonstrated that this was particularly strongly inhibited by LIP expression which caused a reduction in DGAT2 mRNA of 87% at day 3 and 77% at day 6 (Fig. 2f) (compared with reductions of 52% and 63% in C/EBPα expression at these time points). We also determined the expression levels of several other genes regulated during adipogenesis. At day 3 of differentiation, LIP expression led to reductions of 56% in aP2, and 56% in fatty acid synthase, only 14% in carnitine palmitoyltransferase 2, and no inhibition of the induction of the adiponectin receptor 2 (data not shown). The differences in the magnitude of inhibition observed for each of these genes probably reflects the complex cross-regulated nature of the adipogenic transcription cascade. However, the data also strongly suggest that C/EBPβ has an important role in the regulation of DGAT2 in addition to its more general effects on adipogenesis per se.
We have previously demonstrated that the transcriptional co-repressor ETO functions as an inhibitor of C/EBPβ (25). Thus, we next examined the effect of ETO expression on DGAT2 expression. As with the inhibition of C/EBPβ activity by LIP expression, constitutive ETO expression significantly inhibits lipid accumulation during 3T3-L1 adipogenesis (Fig. 3a). However, the expression of ETO does not affect the expression of C/EBPβ in these cells (Fig. 3b). Predictably, due to its inhibition of C/EBPβ activity, ETO expression inhibited both C/EBPα (Fig. 3c) and 11βHSD1 (Fig. 3d) mRNA expression. Moreover, the induction of DGAT2 mRNA was significantly impaired by ETO expression, again suggesting a role for C/EBPβ in DGAT2 expression (Fig. 3e).
Figure 3. Inhibtion of C/EBPβ activity by ETO expression inhibits the induction of DGAT2 during adipogenesis.

3T3-L1 preadipocytes were stably transfected with empty vector (mock) or ETO and confluent cells were induced to differentiate for the times shown. A, lipid accumulation was assessed in mock or ETO transfected cells following differentiation for 8 days. B, Mock transfected (m) or ETO expressing cells (E) were differentiated for various times and protein lysates analysed for C/EBPβ expression by western blotting. Mock (black bars) or ETO (white bars) transfected cells were also differentiated for various times, RNA isolated and assayed for the expression of C/EBPα (C), 11-βHSD1 (D) or DGAT2 (E) by real-time PCR. Data shown are normalised to cyclophilin +/− SEM, n=4. * indicates statistically significant difference in expression compared with mock-transfected cells at the same time-point (p<0.05).
The LIP isoform of C/EBPβ is known to affect the expression of genes regulated by other C/EBP isoforms by binding to DNA sequences that are also capable of binding C/EBPβ. In addition, whilst we have demonstrated that ETO affects C/EBPβ activity, the specificity of ETO is not yet completely defined. Thus we next sought to specifically interfere with C/EBPβ by using siRNA to inhibit its expression. Transfection of preadipocytes with C/EBPβ siRNA resulted in a significant and sustained inhibition of C/EBPβ, reducing expression levels at day 1 post-induction by greater than 60% and at day 3 by greater than 50% (Fig 4a). Analysis of C/EBPβ protein showed that this translated to a dramatic reduction of C/EBPβ protein in cells transfected with C/EBPβ siRNA (Fig. 4b). We next assessed the effect of C/EBPβ knockdown on lipid accumulation. Oil-red O staining demonstrated that specific loss of C/EBPβ significantly impairs the accumulation of lipid in a similar manner to LIP or ETO expression (Fig. 4c). Analysis of C/EBPα (Fig. 4d), 11βHSD1 (Fig. 4e) and DGAT2 (Fig. 4f) mRNA demonstrated that all showed significantly reduced induction in cells transfected with C/EBPβ siRNA.
Figure 4. siRNA knockdown of C/EBPβ activity inhibits DGAT2 induction during adipogenesis.
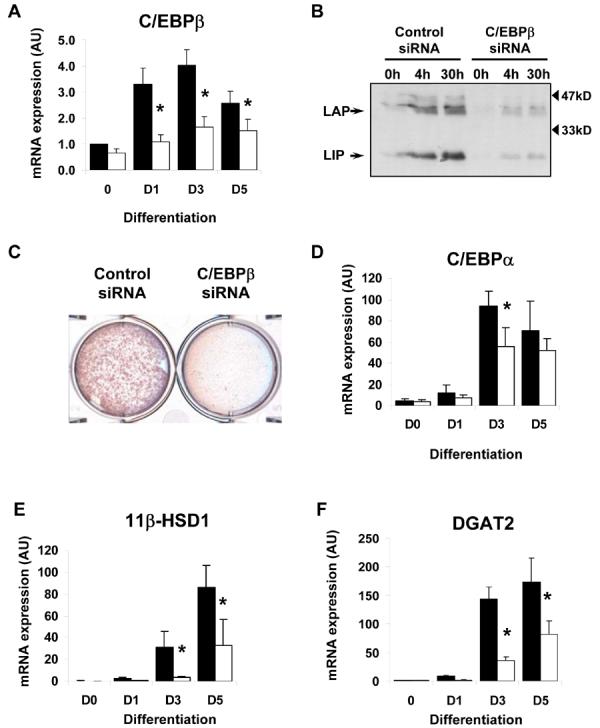

A, 3T3-L1 preadipocytes were transfected with control siRNA (black bars) or siRNA targeting C/EBPβ (white bars) and subsequently induced to differentiate for various times. C/EBPβ mRNA expression was assayed by real-time PCR. B, control or C/EBPβ siRNA transfected cells were also assayed for C/EBPβ protein expression by western blotting. C, lipid accumulation was assessed by oil-red O staining in control or C/EBPβ siRNA transfected cells following differentiation for 8 days. Control siRNA (black bars) or C/EBPβ siRNA (white bars) transfected cells were also differentiated for various times, RNA isolated and assayed for the expression of C/EBPα (D), 11-βHSD1 (E) DGAT2 (F), AGPAT2 (G), Lipin-α (H) or lipin-β (I) by real-time PCR. Data shown are normalised to cyclophilin +/− SEM, n=4. * indicates statistically significant difference in expression compared with control siRNA transfected cells at the same time-point (p<0.05).
Like DGAT2 both AGPAT2 and lipin have critical roles in triglyceride synthesis. However, in addition to their enzymatic activity, AGPAT2 and lipin also have roles in adipogenenic gene expression (34,35). The two splice variants of lipin, lipin-α and lipin-β have been shown to selectively enhance the expression of proteins of adipogenesis or lipogenesis, respectively. To assess the involvement of these enzymes in the observed reduction of lipid accumulation we measured their expression in C/EBPβ knockdown cells. Inhibition of C/EBPβ expression resulted in a reduction of approximately 50% in both AGPAT2 (Fig 4g) and lipin-α (Fig 4h) expression after 3 days of differentiation. However, no significant inhibition of these genes was observed after 5 days of differentiation, nor in the expression of more “lipogenic” lipin-β isoform at any of the time points tested (Fig. 4i). The reductions in DGAT2 expression were larger (70% and 45% at day 3 and day 5 of differentiation, respectively) (Fig. 4f). This suggests that the inhibition of DGAT2 expression probably makes a more significant contribution to the impaired lipid accumulation in these cells than reductions in AGPAT2, lipin-α or lipin-β. However it is possible that the concerted decrease in these enzymes compounds the effect of DGAT2 inhibition at the earlier stages of differentiation, either affecting lipogenesis directly or via effects on adipogenesis itself.
To determine whether our observations in cellular models of adipogenesis extended to adipocyte development in vivo, we examined gene expression in WAT isolated from C/EBPβ knockout mice. As expected C/EBPβ mRNA was undetectable in these samples (Fig. 5a). As predicted from previous studies the expression of C/EBPα was also significantly reduced (Fig. 5b), as was the expression of 11βHSD1 (Fig. 5c). In addition, we also found that the expression of DGAT2 was significantly decreased in the WAT of these mice (Fig. 5d), consistent with an important role for C/EBPβ in the expression of DGAT2 in adipocytes in vivo.
Figure 5. Gene expression analysis in adipose tissue of mice lacking C/EBPβ.

RNA was isolated from white adipose tissue of wild-type (WT) or C/EBPβ knockout (KO) mice, reverse transcribed and the expression of C/EBPβ (A), C/EBPα (B), 11βHSD1, (C) and DGAT2 (D), was assayed by real-time PCR. Results are the mean of 3 independent samples normalised to 18S +/− SEM, * indicates statistically significant expression compared with wild-type mice (p<0.05).
We next sought to determine whether C/EBPβ could directly regulate DGAT2 through binding to its promoter. Examination of the putative promoter of DGAT2 revealed four potential C/EBPβ consensus binding sites within 3kb upstream of the transcriptional start site. These were designated sites 1-4 and are schematically represented in figure 6a. To assess binding to these putative sites we performed ChIP analysis, immunoprecipitating C/EBPβ and using real-time PCR to quantify binding to specific DNA sequences. As a positive control we first assayed binding of C/EBPβ to its well-characterised target site in the C/EBPα promoter (Fig. 6b). Consistent with our previous findings and gel shift analyses by others (25,31), we detected increased binding of C/EBPβ to this site over the first 24 hours of differentiation. Subsequent analysis of binding to the four putative sites that we had identified demonstrated that significantly increased binding was only observed for site 1 (Fig. 6c), the most distal site identified. In contrast, we observed little change or consistent binding of C/EBPβ to the other putative sites (Fig. 6d, 6e, 6f).
Figure 6. Identification of C/EBPβ binding sites in the DGAT2 promoter.

A, analysis of 3kb upstream of the first exon of DGAT2 by TESS revealed four potential C/EBPβ binding sites, denoted site 1 to site 4. 3T3-L1 preadiocytes were induced to differentiate for various times and subjectd to ChIP analysis to assess binding of C/EBPβ to the well characterised site in the C/EBPα promoter (B) or the putative site 1 (C), site 2 (D), site 3 (E) or site 4 (F) in the DGAT2 promoter. C/EBPβ bound DNA in immunoprecipitates was quantified by real-time PCR. Values in each sample were normalised to total genomic DNA of the same sequence in the “input” starting sample prior to immunoprecipitation. Data are the average of 6 independent experiments +/− SEM, * indicates statistically significant difference from binding at time 0 (p<0.05).
To examine this further, we used gel shift assays with nuclear extracts from 3T3-L1 preadipocytes, differentiated for 0, 4, 8 or 24 hours. Once again, the well-characterised C/EBPβ binding site in the C/EBPα promoter was used as a positive control. A biotinylated DNA probe containing this site showed increased complex formation when incubated with nuclear extracts from differentiating cells (Fig. 7a). These complexes were capable of being supershifted with an antibody to C/EBPβ but only from cells that had been induced to differentiate for 4-48 hours. Similarly, nuclear extracts from differentiating preadipocytes formed complexes with probe containing site 1 of the DGAT2 promoter (Fig. 7b). Again, these showed increased binding as differentiation progressed and could be supershifted with C/EBPβ antibody. This data demonstrates that C/EBPβ can directly bind to site 1 of the DGAT2 promoter both in vitro and in vivo.
Figure 7. Gel shift analysis of C/EBPβ DNA binding activity in nuclear extracts.
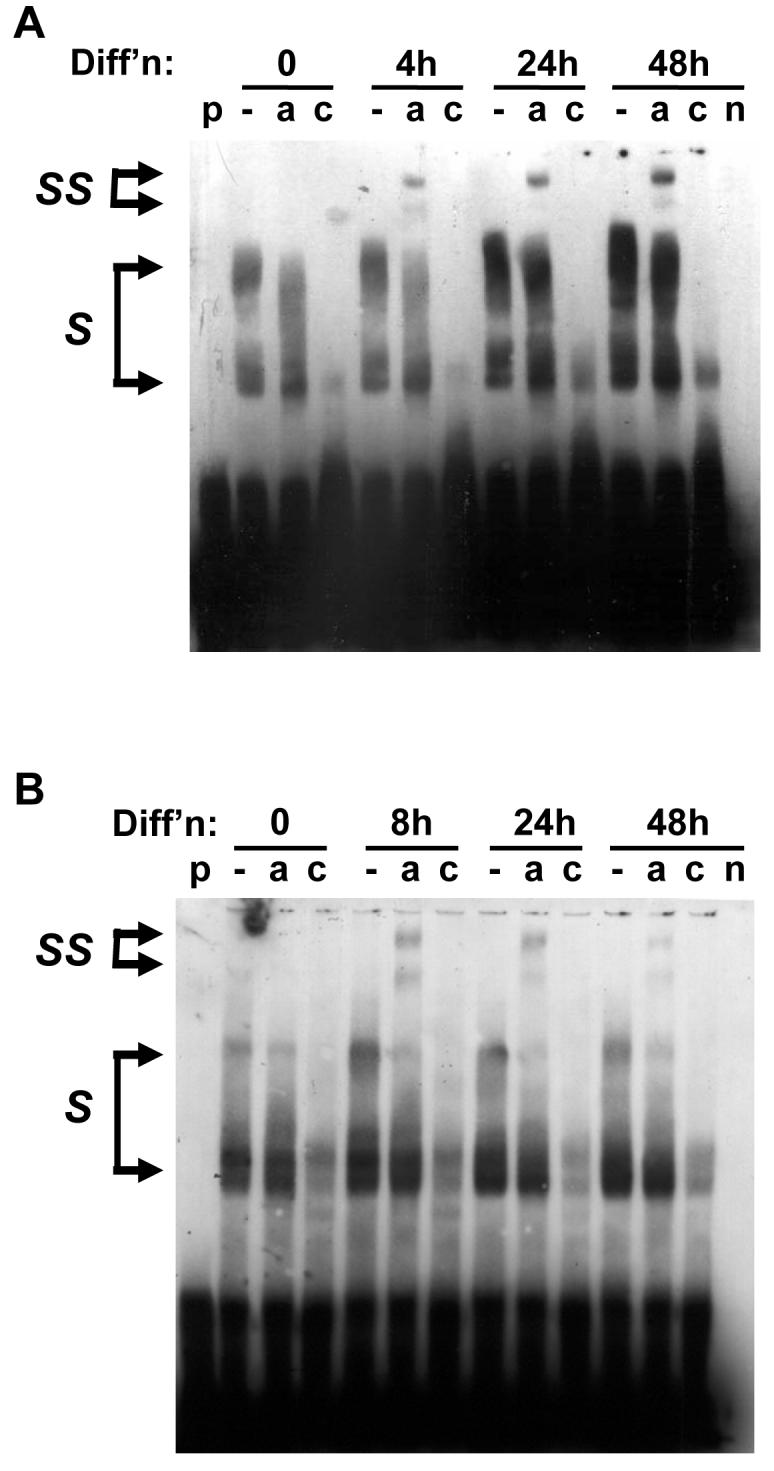
Nuclear extracts were prepared from 3T3-L1 preadiopocytes before or after differentiation for various times as indicated. These were incubated with biotinylated DNA probes corresponding to the well characterised C/EBPβ binding site in the C/EBPα promoter (A) or the site 1 identified in the DGAT2 promoter (B). Nuclear extracts were incubated with probe alone (−), or with probe in the presence of either anti-C/EBPβ antibody (a), or antibody and a 100-fold excess of unlabelled DNA probe (c). As controls, lanes were also run with probe alone (p) or 24h nuclear extract in the absence of probe (n). S indicates nuclear protein complexes binding probe, and SS indicates antibody-supershifted complexes containing C/EBPβ. Data are representative of 4 independent experiments.
DGAT2 expression persists in later stages of adipogenesis whilst C/EBPβ expression subsides in differentiating 3T3-L1 cells approximately 3 to 6 days after the induction of differentiation. Thus we investigated whether C/EBPα might take over the regulation of DGAT2 expression in the later stages of differentiation. We therefore used siRNAs to C/EBPα to inhibit its expression. Transfection of 3T3-L1 preadipocytes with C/EBPα siRNA led to a significant reduction in its induction at day 3 and day 6 of differentiation. However, as the transfections were performed prior to differentiation, the knockdown of C/EBPα was only 29% (Fig. 8a). Despite this, assay of DGAT2 mRNA expression revealed that this was sufficient to significantly impair DGAT2 expression in these cells (Fig. 8b). Given that C/EBP binding sites are often promiscuous for different C/EBP isoforms we examined whether the regulation of DGAT2 by C/EBPα may occur through the same site in its promoter bound by C/EBPβ. We therefore performed ChIP assays, immunoprecipitating C/EBPα from differentiating 3T3-L1 preadipocytes and assaying association with the proposed site 1 of the DGAT2 promoter. These assays revealed an increase in binding of C/EBPα to this site as differentiation progessed, with binding apparent after day 3 of differentiation when C/EBPα first appears (Fig. 8c). To further examine this we performed gel shift assays using a labelled DNA probe corresponding to site 1 and nuclear extracts from differentiating preadipocytes. In agreement with the ChIP assay data, complex formation involving proteins from the nuclear extracts and the DNA probe increased as differentiation progressed (Fig. 8d). Moreover, as differentiation progressed to later time points some complexes could be supershifted with an antibody to C/EBPα demonstrating binding of this transcription factor to site 1 of the DGAT2 promoter. These data indicate that during the later stages of adipogenesis, C/EBPα substitutes for C/EBPβ in the control of DGAT2 expression and that this occurs through the same regulatory element in the DGAT2 promoter.
Figure 8. Analysis DGAT2 regulation by C/EBPα during adipogenesis.

A, 3T3-L1 preadipocytes were transfected with control siRNA (black bars) or siRNA targeting C/EBPα (white bars) and subsequently induced to differentiate for various times. C/EBPα mRNA expression was assayed by real-time PCR. Data are +/− SEM, normalised to cyclophilin, n=4. B, cells transfected with control (black bars) or C/EBPα (white bars) siRNA were also assayed for DGAT2 mRNA expression by real time PCR. Data are +/− SEM normalised to cyclophilin, n=4. * indicates statistically significant difference in expression compared with control siRNA transfected cells at the same time-point (p<0.05). C, 3T3-L1 preadiocytes were induced to differentiate for various times and subjected to ChIP analysis to assess binding of C/EBPα to the putative site 1 in the DGAT2 promoter. C/EBPα bound DNA in immunoprecipitates was quantified by real-time PCR. Data are the average of 4 independent experiments +/− SEM, * indicates statistically significant difference from binding at time 0 (p<0.05). D, Nuclear extracts were prepared from 3T3-L1 preadiopocytes before or after differentiation for various times as indicated and incubated with biotinylated DNA probe corresponding to site 1 identified in the DGAT2 promoter. Nuclear extracts were incubated with probe alone (−) or with probe in the presence of either anti-C/EBPα antibody (a), or antibody and a 100-fold excess of unlabelled DNA probe (c). Control lanes were included containing probe alone (p) or 24h nuclear extract in the absence of probe (n). S indicates nuclear protein complexes binding probe, and SS indicates antibody-supershifted complexes containing C/EBPα. Data are representative of 3 independent experiments.
DISCUSSION
Dysregulation of TG synthesis is likely to play an important role in the development of disorders of adipose tissue mass. In obesity the occurrence of hypertrophic adipocytes correlates with features of the metabolic syndrome, consistent with the suggestion that excessive cellular lipid loading is part of a detrimental cellular phenotype of metabolic inflexibility and decreased insulin sensitivity (36). Evidently, inappropriately high TG synthesis may in principal cause, or exacerbate this situation. Conversely, the development of lipodystrophy due to mutations of AGPAT2, an upstream enzyme in the same pathway of lipid synthesis, demonstrates the potential for loss of function in the TG synthesis pathway to cause metabolic disease in humans (2,3). Thus, good metabolic control probably requires finely balanced control of the capacity of adipose tissue to store and metabolise lipids, responsive to the types and quantity of substrates available. Such a balance would allow an appropriate flow of fatty acids to tissues such as liver and skeletal muscle proportionate to their ability to use these as fuels for oxidation. However, when substrates are present in excess, the adipocytes must respond to prevent detrimental accumulation of lipids and resulting insulin resistance, in these other tissues. At a molecular level, the adipocyte's ability to take-up lipid substrates, and their fate once within the adipocyte, is likely to involve tight regulation of the expression of key enzymes of TG synthesis.
The DGAT enzymes catalyse the critical final step of TG synthesis, are expressed in adipocytes and so are well placed to function as a control point for lipid accumulation in these cells. Despite this little is known about how their expression is regulated, in this or any other tissue. In this study we chose to examine the regulation of DGAT2 because its expression is more highly induced than that of DGAT1 in differentiating preadipocytes and because studies in knockout animals demonstrate that loss of DGAT2 leads to a profound lack of lipid synthesis in the whole animal (4,7).
Using several means to inhibit C/EBPβ expression or activity we demonstrated that C/EBPβ plays a key role in the induction of DGAT2 during adipogenesis. The inhibition of C/EBPβ activity by LIP, CHOP10 or ETO, or the loss of synergistic factors such as KLF5 and Krox20 all significantly inhibit lipid accumulation (19,21,25,37-39). The reduced expression of downstream adipogenic factors such as PPARγ and C/EBPα, and subsequent impairment of the induction of the genes they control, will clearly make an important contribution to the lack of lipogenesis in these models. Similarly, it must be acknowledged that inhibition of many factors in the complex network of adipogenic gene expression downstream of C/EBPβ may have a role in the reduced induction of DGAT2 in our experiments. This complexity is aptly illustrated by the recent demonstration that PPARγ, which classically lies downstream of C/EBPβ in the adipogenic cascade, is also required to activate C/EBPβ at the C/EBPα promoter by counteracting HDAC1 (40). Thus reduced PPARγ, when C/EBPβ is suppressed, is likely to feed back to reduced C/EBPβ dependent DGAT2 expression through this mechanism. In addition, we cannot rule out that PPARγ makes a direct contribution to DGAT2 expression by binding to its promoter. However, our finding that DGAT2 is more strongly inhibited than PPARγ when C/EBPβ function is impaired argues against an important direct role for PPARγ. Importantly, the demonstration that C/EBPβ and C/EBPα physically bind the DGAT2 promoter in intact cells strongly supports the notion that direct regulation by these factors is also likely to make a significant contribution to its expression during adipogenesis.
It is particularly difficult to discriminate the relative importance of C/EBPβ activity directly from the effects of reduced expression of C/EBPα in the control of adipocyte DGAT2 expression in our experiments. Existing evidence implicating C/EBPα in lipogenesis includes data from fibroblasts from C/EBPα null mice that can be induced to differentiate by transfection with PPARγ in combination with thiazolidinedione treatment. Despite appropriate induction of adipocyte genes such as aP2, Glut4, adiponectin and FAS (41,42), these cells accumulate significantly less lipid than those from wild-type mice. Our data suggests that loss of C/EBPα driven DGAT2 expression may contribute to this phenomenon. Expression of C/EBPβ-LIP in our cells leads to a very dramatic reduction of DGAT2 and lipid accumulation, consistent with its ability to inhibit the activity of all C/EBP isoforms. The inhibition of DGAT2 expression by selective C/EBPβ knockdown was less dramatic, probably because the expression but not the activity of C/EBPα would be affected. The specificity of ETO for C/EBPs, or indeed other lipogenic factors, is not clear and may in part explain the intermediate effects on DGAT2 expression and lipid accumulation in ETO expressing cells. However, a caveat to these observations is that our data were obtained in independently derived cell populations each with respective control populations, and thus one should be cautious about directly comparing the effects of inhibiting C/EBPβ by each method.
Given the key role of DGAT2 in lipogenesis, the therapeutic potential of altering its expression has been investigated in vivo. Antisense knockdown of DGAT2 in mice led to significant improvement of hepatic steatosis, serum lipids and hyperinsulinaemia in ob/ob or diet-induced obese mice (43). Whilst body weight and epididymal or perirenal fat mass were not decreased in these mice, unlike controls DGAT2 knockdown mice failed to gain additional weight on a high fat diet during the study. This is consistent with DGAT2 playing an important role in promoting lipid accumulation in newly forming or expanding adipocytes. Recently, deletion of C/EBPβ has been shown to decrease adipocyte size and hepatic steatosis in db/db mice (44). It will be interesting to determine whether inhibition of DGAT2 expression contributes to these effects, and whether DGAT2 expression is also regulated by C/EBPβ in the liver.
In summary, we believe that this is the first study to address the regulation of DGAT2 transcription at the molecular level. We have shown that C/EBPβ and C/EBPα sequentially activate the expression of DGAT2 in developing adipocytes, for the first time demonstrating a means whereby C/EBPβ may directly affect the expression of genes controlling lipid accumulation, as well as through its already defined role in inducing other adipogenic transcription factors. In vivo knockdown of DGAT2 has demonstrated the potential therapeutic value of modulating DGAT2 activity. Thus, a greater understanding of the transcriptional mechanisms controlling DGAT2 expression is important in identifying further potential points of intervention, in addition to providing more general insight into the processes controlling lipid accumulation in adipose and other tissues.
Acknowledgments
This work was supported by the British Heart Foundation (JJR), Wellcome Trust (VAP, DH, SOR), The Dorothy Hodgkin Postgraduate Award Scheme (W-SA), the Natural Sciences and Engineering Research Council of Canada (SLG), the O.Arlotti Trust, Italy (EDN) and through NIH Grant DK059767 (JEF, SMH). JR and SOR are members of the EUGENE2 Consortium.
REFERENCES
- 1.Camp HS, Ren D, Leff T. Trends Mol Med. 2002;8(9):442–447. doi: 10.1016/s1471-4914(02)02396-1. [DOI] [PubMed] [Google Scholar]
- 2.Agarwal AK, Arioglu E, De Almeida S, Akkoc N, Taylor SI, Bowcock AM, Barnes RI, Garg A. Nat Genet. 2002;31(1):21–23. doi: 10.1038/ng880. [DOI] [PubMed] [Google Scholar]
- 3.Magre J, Delepine M, Van Maldergem L, Robert JJ, Maassen JA, Meier M, Panz VR, Kim CA, Tubiana-Rufi N, Czernichow P, Seemanova E, Buchanan CR, Lacombe D, Vigouroux C, Lascols O, Kahn CR, Capeau J, Lathrop M. Diabetes. 2003;52(6):1573–1578. doi: 10.2337/diabetes.52.6.1573. [DOI] [PubMed] [Google Scholar]
- 4.Smith SJ, Cases S, Jensen DR, Chen HC, Sande E, Tow B, Sanan DA, Raber J, Eckel RH, Farese RV., Jr. Nat Genet. 2000;25(1):87–90. doi: 10.1038/75651. [DOI] [PubMed] [Google Scholar]
- 5.Buhman KK, Smith SJ, Stone SJ, Repa JJ, Wong JS, Knapp FF, Jr., Burri BJ, Hamilton RL, Abumrad NA, Farese RV., Jr. J Biol Chem. 2002;277(28):25474–25479. doi: 10.1074/jbc.M202013200. [DOI] [PubMed] [Google Scholar]
- 6.Chen HC, Smith SJ, Ladha Z, Jensen DR, Ferreira LD, Pulawa LK, McGuire JG, Pitas RE, Eckel RH, Farese RV., Jr. J Clin Invest. 2002;109(8):1049–1055. doi: 10.1172/JCI14672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stone SJ, Myers HM, Watkins SM, Brown BE, Feingold KR, Elias PM, Farese RV., Jr. J Biol Chem. 2004;279(12):11767–11776. doi: 10.1074/jbc.M311000200. [DOI] [PubMed] [Google Scholar]
- 8.Rangwala SM, Lazar MA. Annu Rev Nutr. 2000;20:535–559. doi: 10.1146/annurev.nutr.20.1.535. [DOI] [PubMed] [Google Scholar]
- 9.Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM. Genes Dev. 2000;14(11):1293–1307. [PubMed] [Google Scholar]
- 10.MacDougald OA, Mandrup S. Trends Endocrinol Metab. 2002;13(1):5–11. doi: 10.1016/s1043-2760(01)00517-3. [DOI] [PubMed] [Google Scholar]
- 11.Rosen ED, Spiegelman BM. Annu Rev Cell Dev Biol. 2000;16:145–171. doi: 10.1146/annurev.cellbio.16.1.145. [DOI] [PubMed] [Google Scholar]
- 12.Semple RK, Chatterjee VK, O'Rahilly S. J Clin Invest. 2006;116(3):581–589. doi: 10.1172/JCI28003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin FT, Lane MD. Genes Dev. 1992;6(4):533–544. doi: 10.1101/gad.6.4.533. [DOI] [PubMed] [Google Scholar]
- 14.Freytag SO, Paielli DL, Gilbert JD. Genes Dev. 1994;8(14):1654–1663. doi: 10.1101/gad.8.14.1654. [DOI] [PubMed] [Google Scholar]
- 15.Lin FT, Lane MD. Proc Natl Acad Sci U S A. 1994;91(19):8757–8761. doi: 10.1073/pnas.91.19.8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang ND, Finegold MJ, Bradley A, Ou CN, Abdelsayed SV, Wilde MD, Taylor LR, Wilson DR, Darlington GJ. Science. 1995;269(5227):1108–1112. doi: 10.1126/science.7652557. [DOI] [PubMed] [Google Scholar]
- 17.Lane MD, Lin FT, MacDougald OA, Vasseur-Cognet M. Int J Obes Relat Metab Disord. 1996;20(Suppl 3):S91–96. [PubMed] [Google Scholar]
- 18.Linhart HG, Ishimura-Oka K, DeMayo F, Kibe T, Repka D, Poindexter B, Bick RJ, Darlington GJ. Proc Natl Acad Sci U S A. 2001;98(22):12532–12537. doi: 10.1073/pnas.211416898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tanaka T, Yoshida N, Kishimoto T, Akira S. Embo J. 1997;16(24):7432–7443. doi: 10.1093/emboj/16.24.7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen SS, Chen JF, Johnson PF, Muppala V, Lee YH. Mol Cell Biol. 2000;20(19):7292–7299. doi: 10.1128/mcb.20.19.7292-7299.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamm JK, Park BH, Farmer SR. J Biol Chem. 2001;276(21):18464–18471. doi: 10.1074/jbc.M100797200. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto H, Kurebayashi S, Hirose T, Kouhara H, Kasayama S. J Cell Sci. 2002;115(Pt 18):3601–3607. doi: 10.1242/jcs.00044. [DOI] [PubMed] [Google Scholar]
- 23.Jitrapakdee S, Slawik M, Medina-Gomez G, Campbell M, Wallace JC, Sethi JK, O'Rahilly S, Vidal-Puig AJ. J Biol Chem. 2005;280(29):27466–27476. doi: 10.1074/jbc.M503836200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laudes M, Christodoulides C, Sewter C, Rochford JJ, Considine RV, Sethi JK, Vidal-Puig A, O'Rahilly S. J Biol Chem. 2004;279(12):11711–11718. doi: 10.1074/jbc.M310240200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rochford JJ, Semple RK, Laudes M, Boyle KB, Christodoulides C, Mulligan C, Lelliott CJ, Schinner S, Hadaschik D, Mahadevan M, Sethi JK, Vidal-Puig A, O'Rahilly S. Mol Cell Biol. 2004;24(22):9863–9872. doi: 10.1128/MCB.24.22.9863-9872.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu S, Croniger C, Arizmendi C, Harada-Shiba M, Ren J, Poli V, Hanson RW, Friedman JE. J Clin Invest. 1999;103(2):207–213. doi: 10.1172/JCI4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soukas A, Socci ND, Saatkamp BD, Novelli S, Friedman JM. J Biol Chem. 2001;276(36):34167–34174. doi: 10.1074/jbc.M104421200. [DOI] [PubMed] [Google Scholar]
- 28.Tang QQ, Jiang MS, Lane MD. Mol Cell Biol. 1999;19(7):4855–4865. doi: 10.1128/mcb.19.7.4855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams LJ, Lyons V, MacLeod I, Rajan V, Darlington GJ, Poli V, Seckl JR, Chapman KE. J Biol Chem. 2000;275(39):30232–30239. doi: 10.1074/jbc.M001286200. [DOI] [PubMed] [Google Scholar]
- 30.Tang QQ, Lane MD. Proc Natl Acad Sci U S A. 2000;97(23):12446–12450. doi: 10.1073/pnas.220425597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang QQ, Lane MD. Genes Dev. 1999;13(17):2231–2241. doi: 10.1101/gad.13.17.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wiper-Bergeron N, Wu D, Pope L, Schild-Poulter C, Hache RJ. Embo J. 2003;22(9):2135–2145. doi: 10.1093/emboj/cdg218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Descombes P, Schibler U. Cell. 1991;67(3):569–579. doi: 10.1016/0092-8674(91)90531-3. [DOI] [PubMed] [Google Scholar]
- 34.Gale SE, Frolov A, Han X, Bickel PE, Cao L, Bowcock A, Schaffer JE, Ory DS. J Biol Chem. 2006;281(16):11082–11089. doi: 10.1074/jbc.M509612200. [DOI] [PubMed] [Google Scholar]
- 35.Peterfy M, Phan J, Reue K. J Biol Chem. 2005;280(38):32883–32889. doi: 10.1074/jbc.M503885200. [DOI] [PubMed] [Google Scholar]
- 36.Weyer C, Foley JE, Bogardus C, Tataranni PA, Pratley RE. Diabetologia. 2000;43(12):1498–1506. doi: 10.1007/s001250051560. [DOI] [PubMed] [Google Scholar]
- 37.Batchvarova N, Wang XZ, Ron D. Embo J. 1995;14(19):4654–4661. doi: 10.1002/j.1460-2075.1995.tb00147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Z, Torrens JI, Anand A, Spiegelman BM, Friedman JM. Cell Metab. 2005;1(2):93–106. doi: 10.1016/j.cmet.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 39.Oishi Y, Manabe I, Tobe K, Tsushima K, Shindo T, Fujiu K, Nishimura G, Maemura K, Yamauchi T, Kubota N, Suzuki R, Kitamura T, Akira S, Kadowaki T, Nagai R. Cell Metab. 2005;1(1):27–39. doi: 10.1016/j.cmet.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 40.Zuo Y, Qiang L, Farmer SR. J Biol Chem. 2006;281(12):7960–7967. doi: 10.1074/jbc.M510682200. [DOI] [PubMed] [Google Scholar]
- 41.Wu Z, Rosen ED, Brun R, Hauser S, Adelmant G, Troy AE, McKeon C, Darlington GJ, Spiegelman BM. Mol Cell. 1999;3(2):151–158. doi: 10.1016/s1097-2765(00)80306-8. [DOI] [PubMed] [Google Scholar]
- 42.Gustafson B, Jack MM, Cushman SW, Smith U. Biochem Biophys Res Commun. 2003;308(4):933–939. doi: 10.1016/s0006-291x(03)01518-3. [DOI] [PubMed] [Google Scholar]
- 43.Yu XX, Murray SF, Pandey SK, Booten SL, Bao D, Song XZ, Kelly S, Chen S, McKay R, Monia BP, Bhanot S. Hepatology. 2005;42(2):362–371. doi: 10.1002/hep.20783. [DOI] [PubMed] [Google Scholar]
- 44.Schroeder-Gloeckler JM, Rahman SM, Janssen RC, Qiao L, Shao J, Roper M, Fischer SJ, Lowe E, Orlicky DJ, McManaman JL, Palmer C, Gitomer WL, Huang W, O'Doherty R M, Becker TC, Klemm DJ, Jensen DR, Pulawa LK, Eckel RH, Friedman JE. J Biol Chem. 2007 doi: 10.1074/jbc.M701329200. [DOI] [PMC free article] [PubMed] [Google Scholar]