

Abstract
Protective antigen (PA) is a central component of anthrax toxin and a major antigen in anthrax vaccines. However, the use of native PA as a vaccine is not optimal. If administered to people who have been freshly exposed to anthrax, PA may actually aid in anthrax toxin formation and thus may pose a serious safety concern for postexposure vaccination applications. A non-functional PA mutant may be much safer alternative. To identify an improved anthrax vaccine antigen, we examined four non-functional mutants of PA, each being impaired in a critical step of the cellular intoxication pathway of PA. These mutants were Rec- (unable to bind PA-receptors), SSSR (resistant to activation by furin), Oligo- (unable to form oligomers), and DNI (unable to form endosomal transmembrane pores). When tested in mice and after three doses of immunization, all four mutants were highly potent in eliciting PA-specific, toxin-neutralizing antibodies, with immunogenicity increasing in the order of PA < Rec- < SSSR < Oligo- < DNI. While the differences between Rec- or SSSR and PA were small and not statistically significant, DNI and Oligo- were significantly more immunogenic than wild-type PA. One year after immunization and compared with PA-immunized mice, DNI-immunized mice maintained significantly higher levels of anti-PA IgG with correspondingly higher titers of toxin-neutralizing activity. In contrast, Oligo--immunized mice had high levels of anti-PA IgG but lower titers of toxin-neutralizing activity, suggesting that Oligo- mutation sites may overlap with critical protective epitopes of PA. Our study demonstrates that PA-based vaccines could be improved both in terms of safety and efficacy by strategic mutations that not only render PA non-functional but simultaneously enhance its immunogenic potency. Recombinant PA mutants, particularly DNI, hold great promise as better and safer antigens than wild-type PA for use in postexposure vaccination.
Introduction
High lethality and ease of production and dissemination make anthrax spores a likely candidate for use as a biological weapon. A recent example is the intentional delivery of anthrax spores through the US mail in 2001 that killed several people and caused a mass panic. However, anthrax vaccination is not mandated for civilians, and most people have not been vaccinated against anthrax because of the natural rarity of inhalational anthrax, the most lethal form of anthrax. The uncertainty about whether and when an anthrax attack will occur also makes it less likely that an entire population will be vaccinated prophylactically against anthrax. Immediately following any future anthrax attack, however, there may be an urgent demand for large-scale postexposure protection. The development of a safe and potent postexposure anthrax vaccine is therefore of significant importance.
The currently licensed anthrax vaccine in the US is based on PA (protective antigen), a central component of anthrax toxin [1]. Anthrax toxin and capsule are the two major virulence factors of Bacillus anthracis, the etiologic agent of anthrax [2-4]. Anthrax toxin is a tripartite complex formed by three nontoxic monomeric proteins secreted by B. anthracis, namely PA, lethal factor (LF), and edema factor (EF) [5]. LF is a zinc-dependent metalloprotease that inactivates the mitogen-activated protein kinase kinase signaling pathway [6, 7]. EF is a calcium- and calmodulin-dependent adenylate cyclase that catalyzes the production of intracellular cyclic AMP from host ATP [8]. PA is the common carrier, responsible for transporting LF and EF from the extracellular space into the host cytosol where LF and EF exert their enzymatic toxicities [5]. The binary complexes of PA/LF and PA/EF are also called lethal and edema toxin, respectively. PA-specific antibodies can prevent PA-mediated transportation of LF and EF and neutralize both lethal and edema toxins. Hence, PA has served as a major antigen in most anthrax vaccine formulations.
We sought to design an improved anthrax vaccine based on two considerations. First, as a component of the anthrax toxin, PA in its native form may not be ideal for postexposure vaccination. In a person freshly exposed to anthrax, administration of native PA could actually aid in anthrax toxin formation and thus be dangerous. A non-functional PA mutant that is not active in anthrax toxin formation would likely be much safer and cause much less concern. Second, the natural function of PA is the targeted delivery of toxic LF and EF enzymes that compromise the host to ensure the survival of its producer, while minimizing the induction of the host immunity against itself. In other words, native PA molecules have evolved to exploit a cellular pathway that is optimal for cytosolic translocation and to avoid a pathway that will stimulate the host defense, e.g., by being efficiently processed by antigen presenting cells leading to antibody production. Hence, we hypothesized that native PA is not the most potent molecule for inducing a protective antibody response and thus not the best antigen for use in vaccines. This hypothesis is supported by the fact that the PA-based AVA vaccine requires multiple immunizations and yearly boosters, primarily due to lack of potency [1]. In order to improve protective immunity, we examined the use of specific PA mutants carrying mutations that not only impair the critical intoxication steps of PA but also result in altered cellular trafficking.
We selected four non-functional mutants from a library of PA mutants, including a receptor binding-deficient mutant (Rec-) [9, 10], a furin cleavage-deficient mutant (SSSR) [11, 12], an oligomerization-deficient mutant (Oligo-) [13, 14], and a dominant negative inhibitory (DNI) mutant that does not form endosomal pores [3, 15]. Each mutant is impaired in one of the following steps of PA-mediated anthrax intoxication action: (i) binding of PA to a cell surface receptor [16, 17], (ii) cleavage of PA by a furin-type enzyme into two fragments (PA63 and PA20) [12], (iii) oligomerization of cell-bound PA63 to (PA63)7 heptamers [14], and (iv) conformational change of (PA63)7 upon endocytosis and in acidic endosomes [18] to form transmembrane pores for LF/EF translocation [19, 20]. Using these different non-functional mutants, we desired to identify PA mutants that are devoid of toxicity, yet highly efficient in inducing protective antibodies for use as future anthrax vaccine candidates.
Materials and Methods
Preparation of recombinant wild-type PA and mutant proteins
Plasmids for wild-type PA and mutant Rec- [9], SSSR [11], Oligo- [13], and DNI [21] were obtained from Dr. Collier’s group at Harvard Medical School. The construction and verification of specific mutations of these plasmids have been described previously [9, 11, 13, 21]. For preparation of recombinant proteins, plasmids were cloned into the pET-22b(+) expression vector and transformed into Escherichia coli BL21(DE3) (Novagen, Madison, WI) as previously described [13]. All proteins, i.e., recombinant wild-type PA and the four PA mutants, were prepared and purified using the same procedures. Typically, 1 L of freshly inoculated E. coli BL21(DE3) culture was grown at 37 °C for 4 h until the OD600 reached 0.8-1.0. Protein expression was induced by the addition of 1 mM IPTG to the bacterial culture, and the cells were cultured at 30 °C for 4 h. Cells were harvested by centrifugation at 8,000 g for 10 min at 4 °C, and cell pellets were resuspended in 200 ml sucrose shock buffer containing 20% (w/v) sucrose in 20 mM Tris (pH 8.0) and 1 mM EDTA and stirred for 15 min at 4 °C. After centrifugation for 15 min at 4 °C, cell pellets were collected and resuspended in 200 ml of ice-chilled 5 mM MgSO4 solution and stirred at for 15 min 4 °C. After centrifugation at 10,000 g for 15 min at 4 °C, the supernatant was obtained and purified on a Q Sepharose anion exchange column (GE Healthcare, Piscataway, NJ), eluting with 10 mM Tris buffer (pH 8.0) and a linear 0-0.5 M NaCl gradient at a flow rate of 1.5 ml/min. Wild-type PA or mutant protein was eluted at approximately 0.18 M NaCl. Protein fractions containing PA or mutant were concentrated with a 50-kDa cut-off Ultrafree-15 centrifugal filter device (Millipore, Billerica, MA) and further purified on a Superdex S-200 gel filtration column (GE HealthCare) in 10 mM Tris buffer (pH 8.0). All protein preparations were analyzed by SDS-PAGE and identified with Coomassie blue stain.
Mouse immunization
Groups of eight six-week-old female BALB/c mice (Jackson Laboratory, Bar Harbor, ME) were immunized with PA or mutant by intraperitoneal injection. Each mouse received three doses of PA, DNI, SSSR, Oligo-, or Rec- at two-week intervals. Each dose contained 10 μg protein dissolved in 50 μl of PBS and emulsified with 50 μl of adjuvant gel containing 0.18 mg Al(OH)3 (Sigma-Aldrich, St. Louis, MO). As a negative control, a group of eight mice were each injected with a mixture of 50 μl of PBS and 50 μl of Al(OH)3 gel in the same manner. Ten drops of tail vein blood samples were collected one day before the first immunization and 7 days after each immunization from every mouse. Mice immunized with PA, DNI, or Oligo- were kept for one year for assessment of a sustained antibody and memory response. All other mice were euthanized with CO2 two weeks after the last immunization.
Antibody measurement
Anti-PA IgG and IgM levels in all serum samples were determined by quantitative ELISA. 96-well flat-bottom Nunc-Immuno plates (Nalge Nunc International, Rochester, NY) were coated with 100 μl of 2.5 μg/ml PA in 0.1 M sodium carbonate buffer (pH 9.6) at 4 °C for 16 h. Mouse serum samples were diluted in Tris-buffered saline (TBS) (50 mM Tris-HCl, pH 7.4, 0.15 M NaCl, and 0.05% (v/v) Brij-35) supplemented with 5% (v/v) fetal calf serum and added to assay wells. The plates were incubated at 37 °C for 2 h. After three washes with a wash buffer (10 mM Tris-HCl, pH 7.4, 0.15 M NaCl, and 0.1% (v/v) Brij-35), each well received 100 μl of 0.5 μg/ml alkaline phosphatase-labeled goat anti-mouse IgG (Sigma-Aldrich) and was incubated at 37 °C for 1.5 h. Color was developed by adding 100 μl of 1 mg/ml p-nitrophenylphosphate (Southern Biotechnology, Birmingham, AL) in 1 M Tris buffer (pH 9.8) with 0.3 mM MgSO4. Plates were read in a μQuant microplate reader (Bio-Tek Instruments, Winooski, VT) at 405 nm with 650 nm as reference.
For calculation of the IgG concentrations, a standard IgG curve was generated for each assay plate in parallel to the anti-PA IgG measurement as described above. Instead of coating with PA, the top 2 rows of each plate were coated with 100 μl of 2.5 μg/ml of F(ab’)2 of goat anti-mouse IgG (Southern Biotechnology) in 0.1 M sodium carbonate buffer (pH 9.6) at 4 °C for 16 h. Instead of adding mouse serum, standard mouse IgG (Sigma-Aldrich) at known concentrations was added after washing to duplicate wells in serial 2-fold dilutions. The amount of bound IgG was determined by alkaline phosphate-labeled goat anti-mouse IgG and p-nitrophenylphosphate as described above. A curve of IgG concentrations vs. OD readings (405 nm) was plotted and served as the standard curve. IgG isotypes (including IgG1, IgG2a, IgG2b, and IgG3) and IgM in each mouse serum sample were quantified similarly by ELISA.
Cell cytotoxicity assay
Murine macrophage J774A.1 cells (ATCC, Manassas, VA, USA) were seeded into flat-bottom 96-well tissue culture plates at a density of 2.5×104/well. Cells were grown in cDMEM-10 medium containing Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum, 100 U/ml of penicillin, 100 μg/ml of streptomycin sulfate, and 0.292 mg/ml of L-glutamine (Invitrogen, Carlsbad, CA). After cells were cultured at 37 °C in a 5% (v/v) CO2 incubator for 16 h, culture medium was removed by aspiration and replaced with 100 μl of fresh cDMEM-10 medium. 2 nM LF and various concentrations of PA or mutant (ranging from 100 nM to 0.001 nM) were added to wells. After cells were cultured at 37 °C in a 5% (v/v) CO2 incubator for 18 h, 10 μl of 5 mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added to each well. Cells were cultured for another 1 h at 37 °C, and the colorimetric reaction was stopped by addition of 150 μl of a stop solution containing 0.04 M HCl in isopropanol. Cell viability in each well was determined by its absorbance at 570 nm (reference at 630 nm). Cells cultured in cDMEM-10 medium alone were used as positive controls and as the baseline (i.e., 100% cell survival) for calculating the percentage of cell death. The potency of PA or mutants as transporters of the cytotoxic LF was assumed as proportional to the percentage of cell death.
Toxin neutralization and cell protection assay
Murine J774A.1 macrophage cells were cultured in cDMEM-10 medium and 96-well tissue culture plates at 37 °C in a 5% (v/v) CO2 incubator for 16 h as described above. The medium was removed and replaced with 100 μl of fresh cDMEM-10 medium. Lethal doses of lethal toxin (20 nM PA and 10 nM LF) that had been premixed with mouse test sera at various dilutions (ranging from 1:200 to 1:25,600 in 2-fold dilutions) were added in duplicates. Cells were then cultured for 12 h at 37 °C in a 5% (v/v) CO2 incubator. Cell viability was detected by the MTT assay as described above. The percentage of cell survival in each well was calculated as described above. The toxin-neutralizing activity of each serum was assessed as the percentage of cell survival.
Statistical analysis
The Student’s t-test was employed to assess the statistical significance of differences between independent sample groups. A two-sided probability value of p <0.05 was considered statistically significant.
Results
Preparation of PA and mutants and verification of reduced mutant toxicity
We successfully expressed wild-type PA and the Rec-, SSSR, Oligo-, and DNI mutants in E. coli and purified these proteins by Q Sepharose anion exchange and Superdex S-200 gel filtration column chromatography. The average yields for PA, Rec-, SSSR, Oligo-, and DNI were 10, 7, 7.5, 2.5, and 10 mg per liter of culture, respectively. The purity of these protein preparations was confirmed by SDS-PAGE analysis as single bands with an expected molecular size of ∼83 kDa (Fig. 1).
FIG. 1.
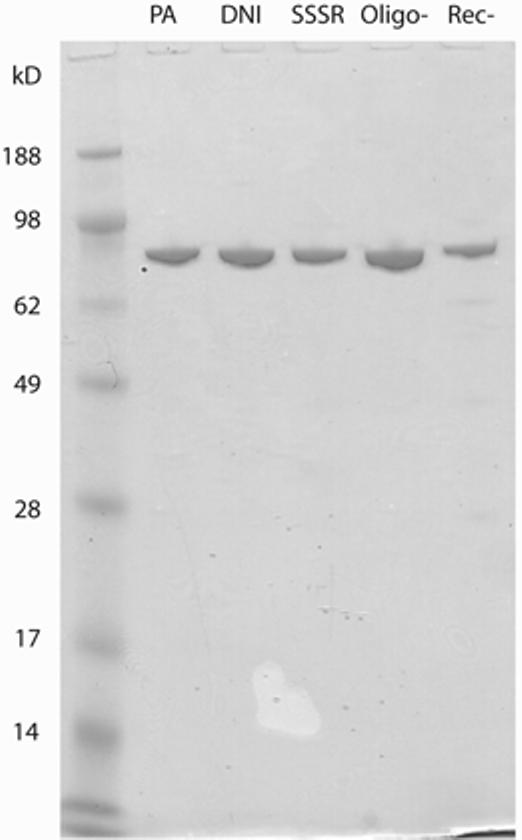
Purified recombinant wild-type PA and PA mutants analyzed by SDS-PAGE and Coomassie blue staining.
To determine whether the Rec-, SSSR, Oligo-, and DNI mutants are functionally impaired in anthrax toxin action, we tested whether they can combine with LF and exert cell cytotoxicity in vitro. Murine macrophage J774A.1 cells were treated with 2 nM LF and 0.01-100 nM recombinant wild-type PA or PA mutant for 16 h, and cell viability was determined by the MTT assay. As shown in Fig. 2, treating macrophage cells with 10 nM PA and 2 nM LF resulted in >95% cell death. However, virtually no killing was observed when cells were incubated with LF and up to 100 nM DNI, SSSR, Oligo-, or Rec-. The results show that these mutants of PA do not form functional lethal toxins with LF. It may thus be reasonable to expect that these mutants would not contribute to functional anthrax toxin formation in people who have been freshly exposed to anthrax and whose body may already contain circulating LF and EF proteins. Consequently, these mutants would likely be significantly safer than wild-type PA for use in postexposure vaccination.
FIG. 2.
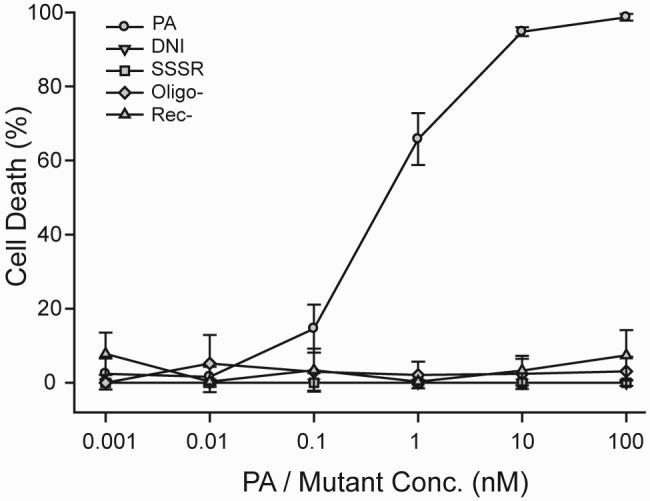
Cell cytotoxicity assays demonstrating diminished activity of PA mutants in PA-mediated anthrax lethal toxin action. Murine macrophage J774A.1 cells were treated with 2 nM LF and serial dilutions of wild-type PA or mutants. Only wild-type PA was active in forming cytotoxic lethal toxins. All four mutants were nonfunctional and did not form lethal toxins with LF.
Potency of PA and mutants in antibody induction
To evaluate the potential of the PA mutants as anthrax vaccine candidates, we examined their ability to induce specific antibodies in mice. Wild-type PA and each mutant were tested at 10-μg doses in groups of eight mice each. Immune serum samples were obtained from each mouse one week after each immunization. Serum levels of PA-specific IgG and IgM were determined by quantitative ELISA.
All four PA mutants were highly immunogenic in BALB/c mice (Fig. 3). After the first immunization, DNI, Oligo-, Rec-, and SSSR induced 0.06 ± 0.05, 0.17 ± 0.08, 0.05 ± 0.01, and 0.26 ± 0.24 μg/ml (mean ± standard error) of PA-specific IgG, respectively. Wild-type PA induced 0.09 ± 0.04 μg/ml of specific IgG. After the second immunization, anti-PA IgG increased to 21.5 ± 3.1, 31.9 ± 2.5, 26.9 ± 4.9, and 18.0 ± 4.6 μg/ml in mice immunized with DNI, Oligo-, Rec-, and SSSR, respectively. Mice immunized with PA produced 20.2 ± 5.3 μg/ml of specific IgG. While PA-immunized mice produced 79.7 ± 12.8 μg/ml of anti-PA IgG after the third immunization, anti-PA IgG levels increased to 142.8±9.1, 131.9±15.9, 100.7±17.9, and 93.7±17.4 μg/ml in mice immunized with DNI, Oligo-, Rec-, and SSSR, respectively. After three immunizations, DNI and Oligo- mutants had induced significantly higher PA-specific IgG than wild-type PA, with p values of 0.0013 and 0.023, respectively. Although the Rec- and SSSR mutants also appeared to be slightly more potent than PA, the differences were not statistically significant (with p values of 0.36 and 0.53, respectively). IgG isotype analysis revealed that anti-PA IgG produced by the wild-type PA and all four mutants were predominantly (>96 %) IgG1. Amounts of IgG2a, IgG2b, and IgG 3 were low (each isotype <3% of total IgG; data not shown). IgM responses to PA and all four mutants were minimal (near detection limit; data not shown).
FIG. 3.
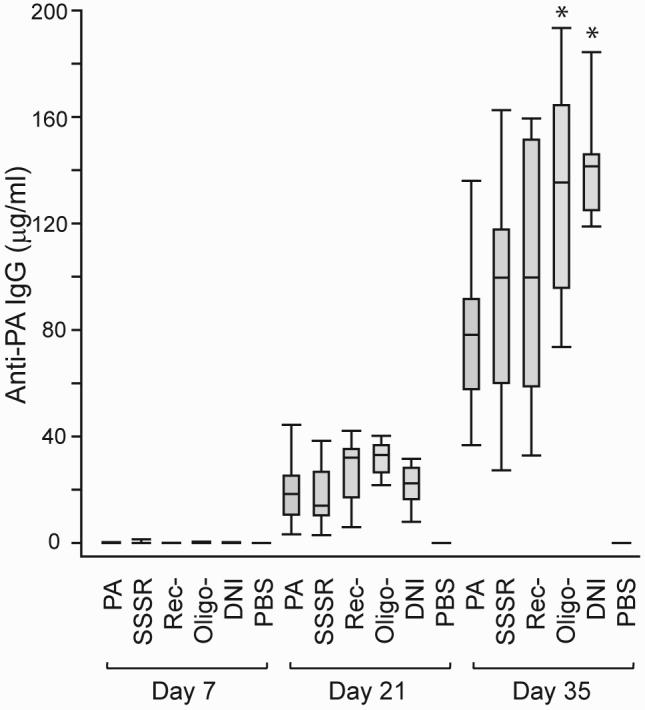
Serum levels of anti-PA IgG in mice immunized with wild-type PA or PA mutants. Each group includes eight mice immunized with three 10-μg doses of PA or mutant at 2-week intervals. Sera were obtained one week after each immunization. Boxes represent 25th to 75th percentiles of eight data points, and bars indicate minimum, median, and maximum values. After the 3rd immunization, the mean values (± standard deviation) of serum anti-PA IgG were 79.7 ± 12.8, 93.7±17.4, 100.7±17.9, 131.9±15.9, and 142.8±9.1 μg/ml in mice immunized with wild-type PA, SSSR, Rec-, Oligo-, and DNI, respectively. The pairwise p values (mutant vs. PA) are 0.53, 0.36, 0.023*, and 0.0013* for SSSR, Rec-, Oligo-, and DNI, respectively (* denotes statistically significant [p < 0.05]).
Toxin-neutralizing activity
In addition to inducing a strong antibody response, it is critical that the induced antibodies be capable of functionally neutralizing anthrax toxins. Thus, we carried out in vitro toxin protection assays using macrophages to evaluate the functional potency of antibodies induced by the various PA mutants. Sera obtained from each group of mice after three immunizations were pooled, and serial dilutions of immune sera were mixed with a lethal dose of anthrax lethal toxin. Protection conferred by the immune sera against lethal toxin was assessed by cell survival using the MTT colorimetric assay.
As shown in Fig. 4, antisera from all mutant groups protected macrophages with >95% survival at 1:200 dilution. The toxin-neutralizing titers for 50% protection (IC50) were 1:12,800, 1:6,400, 1:6,400, 1:3,200, and 1:3,200 for DNI, PA, Oligo-, Rec-, and SSSR, respectively. Although mean total anti-PA IgG levels from mice immunized with the four mutants were all higher than those from wild-type PA immunization (see above), only the sera from DNI-immunized mice showed consistently higher levels of functional protection against lethal toxin than those from PA-immunized mice. These findings suggest that the Oligo-, Rec-, and SSSR mutants may have lost at least some of the critical protective epitopes of wild-type PA.
FIG. 4.
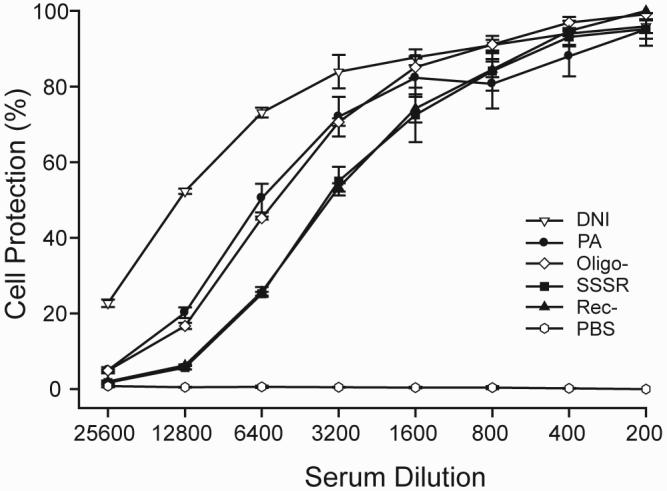
Toxin-neutralizing potency of immune sera against anthrax lethal toxin. Immune sera from groups of eight mice that had been immunized with three doses of PA or mutant were pooled and used in the test. Sera from PBS-immunized mice were used as a negative control. Sera were serially diluted and mixed with a constant amount of anthrax lethal toxin. Note that only sera from DNI-immunized mice showed higher levels of protection that those from PA-immunized mice at all dilutions. At high dilutions, sera from mice immunized with Oligo-, SSSR, and Rec- were less active as those from PA-immunized mice.
Long-term antibody levels, memory response, and toxin-neutralizing activity
Because the DNI and Oligo- mutants were significantly more potent than PA in inducing protective antibodies after three immunizations, we further evaluated their potential as anthrax vaccine candidates by examining the longevity of the anti-PA response in mice immunized with these proteins (Fig. 5A). Six months after the first immunization, serum anti-PA IgG levels were 76.0 ± 5.3, 65.6 ± 4.0, and 45.5 ± 3.4 μg/ml in mice immunized with DNI, Oligo-, and PA respectively. Anti-PA IgG levels in the DNI and Oligo- groups were significantly higher than those of the PA-immunized mice (p = 0.00025 and p = 0.0018, respectively). After one year, PA-specific IgG remained at levels of 42.2 ± 0.9, 42.2 ± 1.5, and 33.1 ± 1.5 μg/ml in mice immunized with DNI, Oligo-, and PA, respectively. Anti-PA levels were still significantly higher in DNI- and Oligo--immunized mice than PA-immunized mice, with p values of 0.00015 (DNI vs. PA). and 0.00088 (Oligo- vs. PA). To test the memory response, mice were boosted with a fourth dose of these proteins. One week after the boost, anti-PA IgG levels increased to 126.8 ± 4.7, 121.9 ± 5.7, and 92.8 ± 10.3 μg/ml in mice immunized with DNI, Oligo-, and PA, respectively. The first two values were significantly higher than the last, with p values of 0.008 and 0.024, respectively.
FIG. 5.
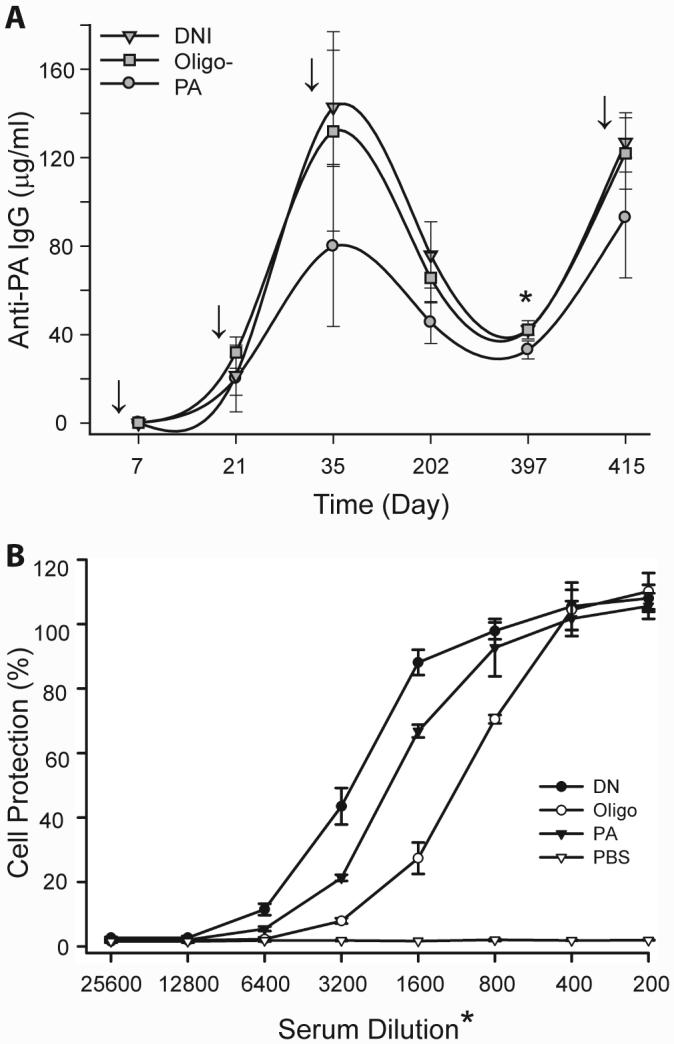
(A) Long-term antibody and memory responses in mice immunized with wild-type PA, Oligo-, or DNI. Mice were immunized four times as indicated by arrows and sera were obtained seven days after each immunization. After the 3rd immunization and over the course of more than one year, DNI- or Oligo--immunized mice consistently maintained significantly higher levels of anti-PA IgG than mice immunized with wild-type PA. (B) Toxin-neutralizing activities of mouse sera obtained more than one year (day 397) after immunization with wild-type PA, Oligo-, or DNI. Sera from DNI-immunized mice were more active that those from PA-immunized mice. Interestingly, Oligo- induced high amounts of anti-PA (A) but decreased levels of toxin-neutralizing activity (B).
We then tested the toxin-neutralizing potency of serum obtained one year after the first immunization. The toxin-neutralizing titers achieving 50% protection were 1:3,200, 1:2,000, and 1:1,100 for sera obtained on day 397 from mice immunized with DNI, PA, and Oligo-, respectively (Fig. 5B). Sera from DNI-immunized mice were consistently more potent that those obtained from PA-immunized mice. However, sera obtained from Oligo--immunized mice were less potent than those from PA-immunized mice. This time-dependent loss of toxin-neutralizing potency of Oligo--induced antisera diminishes the enthusiasm for its use as an alternative antigen in future anthrax vaccine formulations. In contrast, vaccination with DNI induced sustained high levels of toxin-neutralizing antibodies and a robust booster (immunological memory) response. These findings significantly strengthen the notion that the DNI mutant may not only be a much safer but also a more potent vaccine than wild-type PA.
Discussion
Cytotoxic pathway vs. antigen processing pathway
In this study, we demonstrated that certain PA mutants display enhanced immunogenicity. A plausible explanation is that these mutations may have altered the cellular fate of PA to a pathway that is more optimal for the induction of antibodies. In the anthrax intoxication process, PA participates in a well-defined cellular pathway that efficiently translocates the toxic LF and EF enzymes into the cytosol (Fig. 6) [3, 5]. First, the 83-kDa PA molecules bind to a host cell receptor [20]. Second, cell-bound PA is cleaved by furin or a furin-like protease into two components [12], an N-terminal 20-kDa fragment (PA20) that diffuses away and a cell-bound 63-kDa fragment (PA63). Third, cell-bound PA63 molecules self-assemble into ring-shaped (PA63)7 heptamers and form complexes with LF or EF [5]. Fourth, upon receptor-mediated endocytosis and within the acidic endosome, (PA63)7 changes from a ring-shaped core to a membrane-inserting β barrel [15] and forms an ion-conductive pore in the endosomal membrane. LF and EF then shuttle across the endosomal membrane through the (PA63)7 channels into the cytosol, where they enzymatically modify substrates and exert toxic effects. The PA-mediated anthrax intoxication pathway has presumably evolved to optimize the targeted delivery of LF and EF to kill the host while minimizing the induction of host immunity, e.g., by impairing the function of dendritic cells, macrophages, neutrophils, and T- and B-lymphocytes [22-24].
FIG. 6.
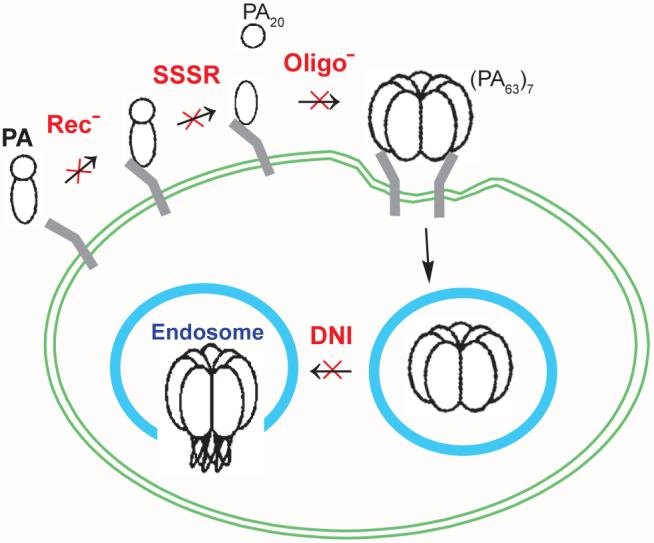
Model of PA cellular trafficking in anthrax toxin action. Upon binding cellular receptors, PA is cleaved into two components. The small fragment PA20 diffuses away, and the cell-bound fraction PA63 self-assembles into heptameric (PA63)7 cores. The heptamers then undergo receptor-mediated endocytosis. Once inside the acidic endosomal compartment, (PA63)7 complexes change conformation and insert into the endosomal membrane. As marked in red, Rec-, SSSR, Oligo-, and DNI mutants are defective in different steps of PA cellular trafficking.
Hence we hypothesize that altering the cellular trafficking of PA may not only eliminate its toxic function but may also channel PA into a pathway that is more advantageous for the induction of a robust anti-PA host immunity. We selected the Rec-, SSSR, Oligo-, and DNI mutants (Figs. 6 and 7), each carrying 2-4 point mutations (Fig. 7) that render them dysfunctional in one of the four distinct steps of the PA intoxication process (Fig. 6). As revealed in this study, all four mutants are highly potent in inducing specific antibody responses, with immunogenicity increasing in the order of PA < Rec- < SSSR < Oligo- < DNI. Whereas the quantitative differences between Rec- or SSSR and PA are smaller and not statistically significant, DNI and Oligo- are significantly more immunogenic than wild-type PA.
FIG. 7.
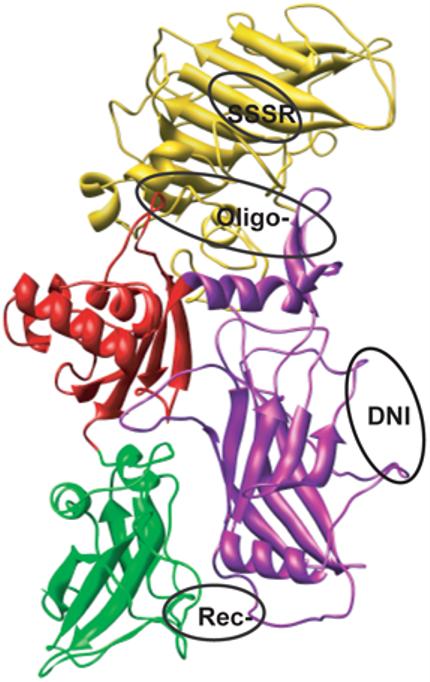
PA structure showing the mutation sites of DNI, SSSR, Oligo-, and Rec- mutants. PA is composed of four functionally independent domains. Domain 1 (gold, aa 1-258) contains the cleavage site for furin (RKKR, residues 164-167) in a surface loop. Domain 2 (purple, aa 249-487) is involved in pore formation. Domain 3 (red, aa 488-595) functions primarily in oligomerization. Domain 4 (green, aa 596-735) functions in receptor binding. The different functional families of mutations are encircled by ovals, representing SSSR, Oligo-, DNI, and Rec- mutants from top to bottom, respectively.
In order to induce a potent antibody response, protein antigens need to be processed and presented by antigen-presenting cells (APCs) to recruit T-cell help. In this process, protein antigens are internalized by APCs, such as dendritic cells and macrophages, and degraded within three increasingly acidic endosomal compartments: early endosomes, late endosomes, and lysosomes. Acid-dependent proteolytic enzymes degrade antigens into peptides of 13-18 amino acid residues. These peptides are then presented to T-cell receptors by MHC class II molecules leading to activated T helper cells [25]. Although the eventual cellular fate of PA is not currently known, it has been demonstrated that membrane insertion of (PA63)7 pores occur already in early endosomes [26]. The insertion of PA pores into the endosomal membrane may disrupt the integrity of the endosome and thus compromise its ability to process antigens. For example, it is possible that PA pore formation changes the pH or ionic environment inside the endosomal lumen, thus compromising the processing machinery. Furthermore, PA may disrupt or otherwise “leak” out of the endosome and enter the cytosol (as is the case for the LF and EF subunits) and thus escape the efficient endosomal antigen processing required for a potent antibody response. Hence, targeted modification of PA’s cellular fate, especially its endosomal fate, may enhance its immunogenicity. This hypothesis would explain the enhanced immunogenicity of PA mutants.
We found that DNI was the most potent mutant. We repeated immunization of mice with DNI and wild-type PA at least four times, and DNI has consistently proven significantly more potent than PA. We proposed that the enhanced immunogenicity of DNI is due to an “endosomal trapping” mechanism [3, 27]. The DNI mutant and PA differ primarily in their endosomal fates. While internalized PA heptamers change their conformation in the acidic endosomes and form transmembrane channels to allow translocation of anthrax toxin enzymes LF and EF into the cytosol, DNI heptamers are unable to change their conformation. Consequently, the endosomes would not be damaged and DNI would remain trapped in the endosomes. Hence, while PA compromises the function of endosomes and may actually escape from endosomal processing, DNI remains trapped within the endosome and thus could undergo efficient antigen processing. We suggest that endosomal trapping is a major contributor to the enhanced immunogenicity of DNI.
We also showed that the Oligo- mutant is significantly more potent than wild-type PA in inducing antibody production. Oligo- molecules cannot self-assemble to form heptamers and, consequently, cannot form pores and insert into endosomal membranes. Consequently, Oligo- may also remain trapped within endosomes and become subject to efficient antigen processing. Therefore, the enhanced immunogenicity of Oligo- may also be due to an endosomal trapping mechanism. The SSSR mutant appeared to be slightly more immunogenic than PA, although the difference was not statistically significant. Since SSSR is resistant to furin-cleavage, it is also not expected to form trans-endosomal membrane heptamer pores. Although the Rec- mutant also appeared to be very slightly more potent than PA (not statistically significant), it was the weakest immunogen of all PA mutants. Rec- carries two point mutations in a PA-receptor binding loop, which virtually prevents it from binding to the two known PA-receptors [28]. If Rec- does not bind to a cell receptor, one would expect that Rec- would remain extracellular, eventually be degraded, and thus fail to induce any significant antibody response. However, it is possible that Rec- binds to other, currently undefined cellular receptors and/or is internalized through other mechanisms. Of note, the loss of function of Rec- has thus far been tested only in CHO and macrophage cells. Therefore, it is possible that Rec- may bind to other cells such as dendritic cells to elicit an immune response.
Are protective epitopes retained in DNI but lost in Oligo- , SSSR, and Rec- mutants?
In addition to the ability to induce a potent and sustained antibody response, another critical parameter for use of a PA mutant as anthrax vaccine antigens is whether the mutant retains the protective, toxin-neutralizing epitopes of native PA. If a mutated residue were to be involved in a protective epitope of PA, this mutant is not likely a good vaccine candidate. Using in vitro toxin-neutralizing assays, we found that immune sera from DNI-immunized mice are significantly more active than sera from mice immunized with wild-type PA, even at high serum dilutions (Figs. 4 and 5B) and over a long time (more than one year; Fig. 5B). The higher toxin-neutralizing activity of DNI over PA antisera corresponds to the higher anti-PA IgG concentrations (Figs. 3 and 5A). In contrast, although the Oligo- mutant also induced significantly higher levels of anti-PA IgG after three immunizations than wild-type PA (Figs. 3 and 5), the toxin-neutralizing activity of Oligo- immune sera was less than that of PA antisera (Figs. 4 and 5B). Similarly, sera from mice immunized with SSSR- and Rec- were significantly less active that sera from PA-immunized mice (Figs. 3 and 4). These results suggest that DNI most likely retained the key protective toxin-neutralizing epitopes of native PA, whereas Oligo-, Rec-, and SSSR may have lost some of the protective epitopes.
To verify this conclusion, we examined the mutation sites with respect to known protective epitopes of PA. Wild-type PA (735 residues, 83 kDa) is folded into four functionally independent domains (Fig. 7) [5, 29]. Domain 1 (residues 1-258) contains the cleavage site (RKKR, residues 164-167) for furin in a surface loop. PA20 corresponds to residues 1-167. PA63, the C-terminal fragment, corresponds to residues 168-735. Domain 2 (residues 259-487) is involved in pore formation. Domain 3 (residues 488-595) is believed to function primarily in self-association of PA63. Domain 4 (residues 596-735) is responsible for binding to cell surface receptors. Rec- carries two mutations (N682A and D683A) in domain 4 and does not bind to the known PA receptors [9, 28, 30]. DNI carries two mutations (K397D and D425K) in domain 2 and cannot form endosomal transmembrane pores to translocate LF or EF [15]. Oligo- carries four mutations (K199E, R468A, R470D, and D512K), one in domain 2 and three in domain 3, and is not able to form heptamers [13, 14]. SSSR carries three mutations (R164S, K165S, and K166S) in domain 1 and is resistant to cleavage by furin [11, 12]. As expected and confirmed in this study (Fig. 2), all four mutants are non-functional in anthrax toxin action.
Several toxin-neutralizing epitopes of PA have been identified. A recent epitope mapping study using phage display techniques revealed that the 312SFFD315 peptide in the 2β2-2β3 loop of PA contains a dominant neutralizing epitope [31, 32]. None of the four mutants carry mutations at this site. Another potential toxin-neutralizing epitope was mapped to the peptide sequence 412SKNLAPI419, corresponding to a loop within domain 2 between 2β9 and 2β10 [33]. This epitope lies between the two mutations of DNI (residues 397 and 425) and is most likely preserved in the DNI mutant. Another study revealed that a toxin-neutralizing mAb 14B7 recognizes a region of PA between residues 671 and 721 [34]. In particular, residue Asn682 was found to be critical for both toxicity and interaction with a neutralizing mAb and residue Asp683 was most critical for cell binding and toxicity but not crucial for interaction with the mAb [30]. Therefore, the use of Rec- (carrying N682A and D683A mutations) as a vaccine candidate is not desirable due to the role of Asn682 as a part of the toxin-neutralizing epitope.
It should be noted, however, that the identification of protective epitopes of native PA is far from complete. Potentially, many other protective epitopes await discovery. The seemingly contraintuitive finding of high antibody levels and low toxin-neutralizing titers of Oligo-, Rec-, and SSSR do not support their use as potential anthrax vaccine antigens. However, this finding could be due to the fact that the mutation sites of these mutants overlap with sites of protective epitopes of PA and, hence, the mutation sites of these mutants may pinpoint unstudied protective PA epitopes.
For further development of new PA-mutant anthrax vaccines, it will need to be shown in direct animal anthrax infection studies that the vaccines protect against infection/intoxication, moderate the clinical course, or increase survival. Such experiments will require the use of fully virulent strains of B. anthracis and are currently very much limited due to associated biosafety concerns.
In summary, PA mutants hold great promise as improved antigens for use in future anthrax vaccine formulations. Strategic mutations impairing specific steps of the anthrax intoxication process render these mutants non-toxic and safe for postexposure vaccination. The mutations may also alter cellular trafficking and result in an overall enhanced humoral response. Enhanced immunogenicity and diminished toxicity offer strong support for the use of PA mutants, particularly DNI, as candidates for a next generation of better and safer anthrax vaccines.
Acknowledgements
We are grateful to Dr. R. John Collier for the wild-type and mutant PA plasmids. Our work was partially supported by Grants R01 AI057926 and R01 AI068826 from the National Institute of Allergy and Infectious Diseases. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health. We thank Paul Guttry of the Brigham and Women’s Hospital Editorial Services for editorial assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
The authors have no financial conflict of interest.
References
- [1].Joellenbeck L, Zwanziger L, Durch J, Strom B, editors. Is It Safe? Does It Work? National Academy Press; Washington, DC: 2002. The Anthrax Vaccine. [PubMed] [Google Scholar]
- [2].Smith H. Discovery of the anthrax toxin: the beginning of in vivo studies on pathogenic bacteria. Trends Microbiol. 2000;8(5):199–200. doi: 10.1016/s0966-842x(00)01755-8. [DOI] [PubMed] [Google Scholar]
- [3].Aulinger BA, Roehrl MH, Mekalanos JJ, Collier RJ, Wang JY. Combining anthrax vaccine and therapy: a dominant-negative inhibitor of anthrax toxin is also a potent and safe immunogen for vaccines. Infect Immun. 2005;73(6):3408–14. doi: 10.1128/IAI.73.6.3408-3414.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rhie GE, Roehrl MH, Mourez M, Collier RJ, Mekalanos JJ, Wang JY. A dually active anthrax vaccine that confers protection against both bacilli and toxins. Proc Natl Acad Sci U S A. 2003;100(19):10925–30. doi: 10.1073/pnas.1834478100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Collier RJ, Young JA. Anthrax toxin. Annu Rev Cell Dev Biol. 2003;19:45–70. doi: 10.1146/annurev.cellbio.19.111301.140655. [DOI] [PubMed] [Google Scholar]
- [6].Park JM, Greten FR, Li ZW, Karin M. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science. 2002;297(5589):2048–51. doi: 10.1126/science.1073163. [DOI] [PubMed] [Google Scholar]
- [7].Agrawal A, Lingappa J, Leppla SH, Agrawal S, Jabbar A, Quinn C, et al. Impairment of dendritic cells and adaptive immunity by anthrax lethal toxin. Nature. 2003;424(6946):329–34. doi: 10.1038/nature01794. [DOI] [PubMed] [Google Scholar]
- [8].Leppla SH. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc Natl Acad Sci U S A. 1982;79(10):3162–6. doi: 10.1073/pnas.79.10.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. Identification of the cellular receptor for anthrax toxin. Nature. 2001;414(6860):225–9. doi: 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- [10].Lacy DB, Wigelsworth DJ, Melnyk RA, Harrison SC, Collier RJ. Structure of heptameric protective antigen bound to an anthrax toxin receptor: a role for receptor in pH-dependent pore formation. Proc Natl Acad Sci U S A. 2004;101(36):13147–51. doi: 10.1073/pnas.0405405101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Klimpel KR, Molloy SS, Thomas G, Leppla SH. Anthrax toxin protective antigen is activated by a cell surface protease with the sequence specificity and catalytic properties of furin. Proc Natl Acad Sci U S A. 1992;89(21):10277–81. doi: 10.1073/pnas.89.21.10277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Beauregard KE, Collier RJ, Swanson JA. Proteolytic activation of receptor-bound anthrax protective antigen on macrophages promotes its internalization. Cell Microbiol. 2000;2(3):251–8. doi: 10.1046/j.1462-5822.2000.00052.x. [DOI] [PubMed] [Google Scholar]
- [13].Mogridge J, Mourez M, Collier RJ. Involvement of domain 3 in oligomerization by the protective antigen moiety of anthrax toxin. J Bacteriol. 2001;183(6):2111–6. doi: 10.1128/JB.183.6.2111-2116.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mogridge J, Cunningham K, Lacy DB, Mourez M, Collier RJ. The lethal and edema factors of anthrax toxin bind only to oligomeric forms of the protective antigen. Proc Natl Acad Sci U S A. 2002;99(10):7045–8. doi: 10.1073/pnas.052160199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sellman BR, Mourez M, Collier RJ. Dominant-negative mutants of a toxin subunit: an approach to therapy of anthrax. Science. 2001;292(5517):695–7. doi: 10.1126/science.109563. [DOI] [PubMed] [Google Scholar]
- [16].Santelli E, Bankston LA, Leppla SH, Liddington RC. Crystal structure of a complex between anthrax toxin and its host cell receptor. Nature. 2004;430(7002):905–8. doi: 10.1038/nature02763. [DOI] [PubMed] [Google Scholar]
- [17].Scobie HM, Wigelsworth DJ, Marlett JM, Thomas D, Rainey GJ, Lacy DB, et al. Anthrax toxin receptor 2-dependent lethal toxin killing in vivo. PLoS Pathog. 2006;2(10):e111. doi: 10.1371/journal.ppat.0020111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wei W, Lu Q, Chaudry GJ, Leppla SH, Cohen SN. The LDL receptor-related protein LRP6 mediates internalization and lethality of anthrax toxin. Cell. 2006;124(6):1141–54. doi: 10.1016/j.cell.2005.12.045. [DOI] [PubMed] [Google Scholar]
- [19].Wolfe JT, Krantz BA, Rainey GJ, Young JA, Collier RJ. Whole-cell voltage clamp measurements of anthrax toxin pore current. J Biol Chem. 2005;280(47):39417–22. doi: 10.1074/jbc.M509049200. [DOI] [PubMed] [Google Scholar]
- [20].Rainey GJ, Wigelsworth DJ, Ryan PL, Scobie HM, Collier RJ, Young JA. Receptor-specific requirements for anthrax toxin delivery into cells. Proc Natl Acad Sci U S A. 2005;102(37):13278–83. doi: 10.1073/pnas.0505865102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mourez M, Yan M, Lacy DB, Dillon L, Bentsen L, Marpoe A, et al. Mapping dominant-negative mutations of anthrax protective antigen by scanning mutagenesis. Proc Natl Acad Sci U S A. 2003;100(24):13803–8. doi: 10.1073/pnas.2436299100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Paccani SR, Tonello F, Ghittoni R, Natale M, Muraro L, D’Elios MM, et al. Anthrax toxins suppress T lymphocyte activation by disrupting antigen receptor signaling. J Exp Med. 2005;201(3):325–31. doi: 10.1084/jem.20041557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Comer JE, Chopra AK, Peterson JW, Konig R. Direct inhibition of T-lymphocyte activation by anthrax toxins in vivo. Infect Immun. 2005;73(12):8275–81. doi: 10.1128/IAI.73.12.8275-8281.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fang H, Xu L, Chen TY, Cyr JM, Frucht DM. Anthrax lethal toxin has direct and potent inhibitory effects on B cell proliferation and immunoglobulin production. J Immunol. 2006;176(10):6155–61. doi: 10.4049/jimmunol.176.10.6155. [DOI] [PubMed] [Google Scholar]
- [25].Bryant P, Ploegh H. Class II MHC peptide loading by the professionals. Curr Opin Immunol. 2004;16(1):96–102. doi: 10.1016/j.coi.2003.11.011. [DOI] [PubMed] [Google Scholar]
- [26].Abrami L, Lindsay M, Parton RG, Leppla SH, van der Goot FG. Membrane insertion of anthrax protective antigen and cytoplasmic delivery of lethal factor occur at different stages of the endocytic pathway. J Cell Biol. 2004;166(5):645–51. doi: 10.1083/jcb.200312072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wang JY, Roehrl MH. Anthrax vaccine design: strategies to achieve comprehensive protection against spore, bacillus, and toxin. Med Immunol. 2005;4(1):4. doi: 10.1186/1476-9433-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chen KH, Liu S, Bankston LA, Liddington RC, Leppla SH. Selection of anthrax toxin protective antigen variants that discriminate between the cellular receptors TEM8 and CMG2 and achieve targeting of tumor cells. J Biol Chem. 2007 doi: 10.1074/jbc.M611142200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Petosa C, Collier RJ, Klimpel KR, Leppla SH, Liddington RC. Crystal structure of the anthrax toxin protective antigen. Nature. 1997;385(6619):833–8. doi: 10.1038/385833a0. [DOI] [PubMed] [Google Scholar]
- [30].Rosovitz MJ, Schuck P, Varughese M, Chopra AP, Mehra V, Singh Y, et al. Alanine-scanning mutations in domain 4 of anthrax toxin protective antigen reveal residues important for binding to the cellular receptor and to a neutralizing monoclonal antibody. J Biol Chem. 2003;278(33):30936–44. doi: 10.1074/jbc.M301154200. [DOI] [PubMed] [Google Scholar]
- [31].Zhang J, Xu J, Li G, Dong D, Song X, Guo Q, et al. The 2beta2-2beta3 loop of anthrax protective antigen contains a dominant neutralizing epitope. Biochem Biophys Res Commun. 2006;341(4):1164–71. doi: 10.1016/j.bbrc.2006.01.080. [DOI] [PubMed] [Google Scholar]
- [32].Gubbins MJ, Berry JD, Corbett CR, Mogridge J, Yuan XY, Schmidt L, et al. Production and characterization of neutralizing monoclonal antibodies that recognize an epitope in domain 2 of Bacillus anthracis protective antigen. FEMS Immunol Med Microbiol. 2006;47(3):436–43. doi: 10.1111/j.1574-695X.2006.00114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Brossier F, Levy M, Landier A, Lafaye P, Mock M. Functional analysis of Bacillus anthracis protective antigen by using neutralizing monoclonal antibodies. Infect Immun. 2004;72(11):6313–7. doi: 10.1128/IAI.72.11.6313-6317.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Little SF, Novak JM, Lowe JR, Leppla SH, Singh Y, Klimpel KR, et al. Characterization of lethal factor binding and cell receptor binding domains of protective antigen of Bacillus anthracis using monoclonal antibodies. Microbiology. 1996;142(Pt 3):707–15. doi: 10.1099/13500872-142-3-707. [DOI] [PubMed] [Google Scholar]