

Abstract
Autophagy is an evolutionally conserved “self-eating” process. Although the genes essential for autophagy (termed Atg) have been identified in yeast, the molecular mechanism of how these Atg proteins control autophagosome formation in mammalian cells remains to be elucidated. Here, we demonstrate that Bif-1 (also known as Endophilin B1) interacts with Beclin 1 through UVRAG and acts as a positive mediator of the class III PI3-kinase (PI3KC3). In response to nutrition deprivation, Bif-1 localizes to autophagosomes where it colocalizes with Atg5, as well as LC3. Furthermore, loss of Bif-1 suppresses autophagosome formation. While the SH3 domain of Bif-1 is sufficient for binding to UVRAG, both the BAR and SH3 domains are required for Bif-1 to activate PI3KC3 and induce autophagosome formation. We also found that Bif-1 ablation prolongs cell survival under nutrient starvation. Moreover, knockout of Bif-1 significantly enhances the development of spontaneous tumors in mice. These findings suggest that Bif-1 joins the UVRAG-Beclin 1 complex as a potential activator of autophagy and tumor suppressor.
Autophagy is a tightly orchestrated intracellular process for bulk degradation of cytoplasmic proteins or organelles that appears to be essential for many physiological processes such as cellular homeostasis, development, differentiation, tissue remodeling, cell survival and death, innate immunity, and pathogenesis in various organisms1-4. The process of autophagic degradation is initiated when a portion of the cytosolic components are sequestered in cup-shaped membrane structures called isolation membranes1, 2, 5, 6. The isolation membranes are elongated and eventually sealed to become double-membrane vesicles called autophagosomes, which are then fused with lysosomes resulting in degradation of the enclosed components. Eighteen autophagy-related (Atg) genes have been characterized in S. cerevisiae and can be categorized into four functional groups: (1) the Atg1 protein kinase complex regulating the induction of autophagy, (2) the class III PI3-kinase (PI3KC3) lipid kinase complex controlling vesicle nucleation, (3) the Atg12-Atg5 and Atg8-phosphatidylethanolamine conjugation pathways for vesicle expansion and completion, and (4) the Atg protein retrieval system2, 7. Beclin 1, the mammalian homologue of yeast Atg6, is a key component of the PI3KC3 complex, which plays an essential role in autophagosome formation8-11. Although the phosphatidylinositol 3-phosphate (PtdIns-3-P) generated by PI3KC3 has been proposed to control membrane dynamics during autophagosome formation3, the molecular mechanism underlying this process remains unknown.
Endophilins are cytosolic proteins which contain an N-terminal N-BAR (Bin-Amphiphysin-Rvs) domain and a C-terminal SH3 (Src-homology 3) domain. The N-BAR domain has been shown to bind to membranes and drive membrane curvature12-14. The Endophilins can be categorized into two groups, the Endophilin A family and the Endophilin B family15. The Endophilin A family is well-characterized; proteins in this class are involved in endosome formation at the fission step15. In contrast, the physiological function of the Endophilin B family of proteins is not fully understood. Bif-1, also known as SH3GLB1 or Endophilin B1, was originally discovered as a Bax-binding protein16, 17. Although Bif-1 has liposome tubulation activity in vitro, it appears as though this protein does not localize to transferrin-positive endosomes18. Recent studies have shown that Bif-1 associates with membranes of intracellular organelles, such as the Golgi apparatus18, 19 and mitochondria20, 21, and has been implicated in vesicle formation and membrane dynamics. Here, we show that Bif-1 forms a complex with Beclin 1 through UVRAG and regulates the formation of autophagosomes.
Results
Loss of Bif-1 suppresses caspase-independent cell death
We have previously reported that Bif-1 localizes to mitochondria and regulates the activation of Bax and Bak during apoptosis induced by intrinsic death stimuli21. To examine Bif-1 localization in mouse embryonic fibroblast (MEF) cells during serum deprivation, we added a pancaspase inhibitor, z-VAD-fmk, to prevent apoptosis-induced cell detachment. Surprisingly, we found that z-VAD-fmk could not prevent, but rather enhanced serum deprivation-induced cell death of wild-type MEFs, indicating that a caspase-independent pathway regulates cell death when apoptosis is blocked (see Supplementary Information, Fig. S1a). Indeed, it has been shown that inhibition of apoptosis (type I programmed cell death (PCD)) by treatment with a caspase inhibitor triggers type II PCD, also known as autophagic cell death, in L929 fibroblasts22. Similar cell death has been observed in MEF cells lacking the proapoptotic Bax and Bak proteins when treated with etoposide23. Interestingly, addition of z-VAD-fmk had no effect on the cell death of Bif-1 -/- MEFs (see Supplementary Information, Fig. S1a), suggesting that Bif-1 may contribute to type II PCD.
To examine whether Bif-1 is involved in autophagy-dependent cell death, spontaneously immortalized Bif-1 +/+ and -/- MEFs21 were cultured in Earl's Balanced Salt Solution (EBSS), an amino acid and growth factor-free medium. As shown in Fig. 1a, Bif-1 -/- cells exhibited a resistance to trypan blue staining after EBSS culture, suggesting that loss of Bif-1 protects cells from death during nutrient starvation. This was confirmed by clonogenic survival (see Supplementary Information, Fig. S1b), MTT (see Supplementary Information, Fig. S1c) and LDH release (data not shown) assays. Similar results were obtained by using HeLa cells stably transfected with shRNA against Bif-1 (see Supplementary Information, Fig. S1d). The suppression of cell death in Bif-1 -/- cells was diminished by restoration of Bif-1 expression (Fig. 1b). To further determine whether this cell death is caspase-dependent, we treated the cells with z-VAD-fmk. Addition of z-VAD-fmk had no effect on the death of wild type cells (Fig. 1c), demonstrating that MEFs could undergo cell death independently of caspase activity after nutrient starvation. In contrast, the caspase inhibitor significantly suppressed the cell death of Bif-1 -/- MEFs (Fig. 1c), suggesting that the observed cell death of Bif-1-deficient cells is apoptosis-dependent. The ability of MEFs to undergo caspase-independent cell death under starvation conditions was further confirmed in Bax and Bak double-knockout MEFs (Fig. 1d), which are resistant to apoptosis induced by a wide variety of death stimuli24.
Figure 1.

Loss of Bif-1 suppresses caspase-independent cell death induced by nutrition deprivation. The percentage of dead cells was determined at the indicated timepoints by trypan blue exclusion assay (mean ± s.d.; n = 3). (a) Bif-1 +/+ (WT) and -/- (KO) MEFs were cultured in EBSS for 0, 3, 6 or 12 h. (b) Bif-1 KO MEFs stably transfected with a Bif-1 expression plasmid or control empty vector were incubated in EBSS for 0 or 12 h. Recovery of Bif-1 expression in Bif-1 KO cells was determined by immunoblot analysis (inset). (c) Bif-1 WT and KO MEFs were cultured in EBSS in the presence of 50 μM z-VAD-fmk or control DMSO for 0, 3, 6 or 12 h. (d) Bax+/+Bak+/+ (WT) and Bax-/-Bak-/- (DKO) MEFs were cultured in EBSS in the presence of 50 μM z-VAD-fmk or control DMSO for 0, 6 or 12 h.
Autophagy is required for caspase-independent cell death
To determine whether autophagy is required for cell death induced by nutrition starvation, we inhibited autophagy either by silencing Atg genes or by treating cells with PI3KC3 inhibitors. We found that starvation-induced cell death was repressed by knockdown of Beclin 1 (Fig. 2a, b). Notably, the reduction of cell death by Beclin 1 shRNA was much greater in Bax and Bak double-knockout MEFs as compared to wild type cells after 12 h of culture in EBSS (Fig. 2b), suggesting that, under starvation conditions, autophagy is the primary cell death mechanism when apoptosis is blocked. Similarly, pharmacological inhibition of autophagy by treatment with 3-methyladenine (3-MA) or wortmannin (WM) significantly reduced EBSS-induced cell death in wild type MEFs (Fig. 2c). In contrast, inhibition of autophagy had no effect on the death of Bif-1 -/- MEF cells (Fig. 2a, c). Consistent with previous studies25, caspase-3 activation was promoted by inhibition of autophagy (Fig. 2d and see Supplementary Information, Fig. S2a), in spite of the observed reduction in cell death, suggesting that initiation of autophagy suppresses apoptosis and simultaneously activates a caspase-independent cell death pathway. To confirm this finding, we next examined cell death and caspase-3 activation in the well-characterized, autophagy-deficient, Atg5 knockout MEFs 26. Consistently, impairment of autophagy, by loss of Atg5, suppressed EBSS-induced cell death despite enhanced caspase-3 activity (Fig. 2e, f).
Figure 2.
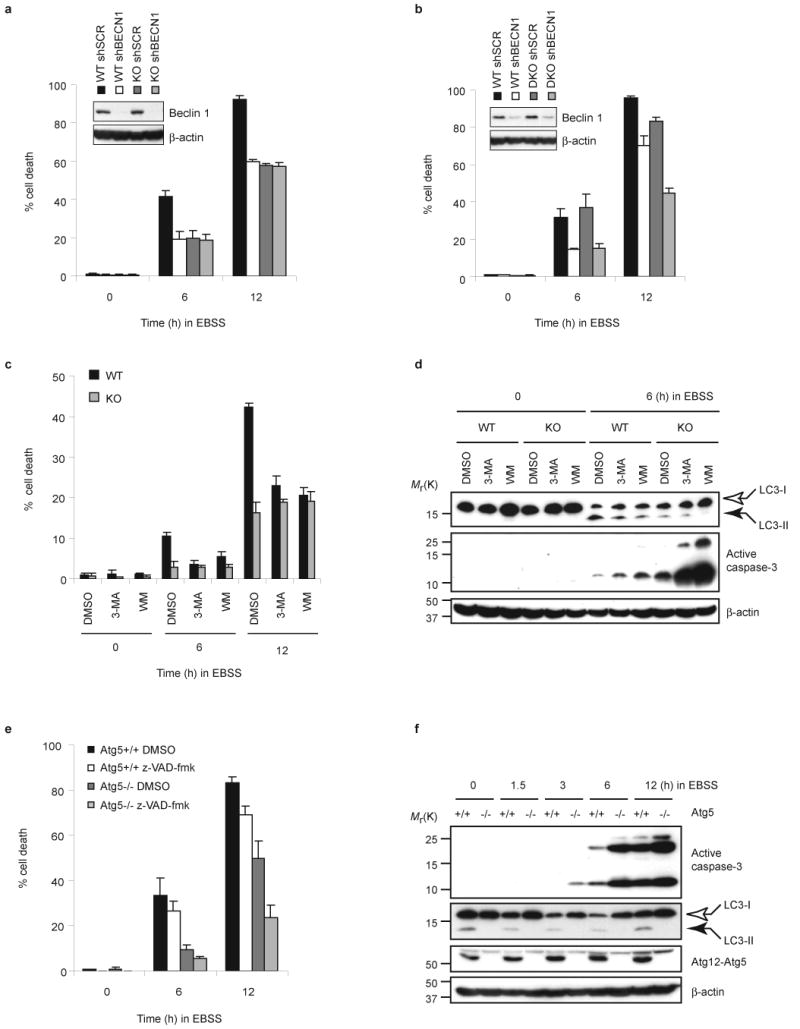
Inhibition of autophagy enhances caspase-3 activation, but suppresses caspase-independent cell death. (a, b) Bif-1 WT and KO MEFs or Bax/Bak WT and DKO MEFs were infected with shBECN1 or control shSCR lentiviruses. Twenty-four hours after infection, the medium was replaced with fresh culture medium and the cells were allowed to recover for an additional 48 h. The cells were then cultured in EBSS for 0, 6 or 12 h, and the percentage of dead cells was determined by trypan blue exclusion assay (mean ± s.d.; n = 3). Knockdown of Beclin 1 expression was confirmed by immunoblot analysis (insets). (c, d) Bif-1 WT and KO MEFs were pre-treated with 10 mM 3-methyladenine (3-MA), 0.2 μM wortmannin (WM) or control DMSO for 30 min followed by culture in EBSS for the indicated times. The percentage of dead cells was determined by trypan blue exclusion assay (mean ± s.d.; n = 3). Immunoblot analyses were performed to detect LC3 modification and caspase-3 activation. (e) Atg5 +/+ and -/- MEFs were cultured in EBSS in the presence of 50 μM of z-VAD-fmk or control DMSO for 0, 6 or 12 h. The percentage of dead cells was determined by trypan blue exclusion assay (mean ± s.d.; n = 3). (f) Total cell lysates (TCL) were prepared from Atg5 +/+ and -/- MEFs cultured in EBSS for the indicated times and subjected to immunoblot analyses. The full scans of the immunoblots shown in a, b, d and f are presented in the Supplementary Information, Fig. S5.
Bif-1 is involved in autophagy
To investigate the intracellular events that occur during nutrition deprivation, we next analyzed the cells by electron microscopy. Incubation in EBSS induced a large number of autophagic vacuoles in the cytoplasm of wild type cells (Fig. 3a). However, the number of autophagosomes was significantly lower in cells lacking Bif-1 (Fig. 3a), suggesting that Bif-1 participates in the formation or processing of autophagic vacuoles. Moreover, lack of Bif-1 resulted in an inhibition of GFP-LC3 fusion protein punctate foci formation (Fig. 3b), which is a well-characterized marker used to visualize autophagosomes and represents the accumulation of a membrane-bound form of LC3 onto autophagic vacuoles27. Inhibition of autophagy by treatment with wortmannin reduced the number of GFP-LC3 positive foci (see Supplementary Information, Fig. S2b). Similar results were obtained using HeLa cells that were stably transfected with Bif-1 shRNA (shBif-1) versus control cells (Fig. 3e). The expression of shBif-1 resistant Bif-1, but not a Bif-1ΔBAR mutant, recovered GFP-LC3 punctate foci formation in Bif-1 knockdown cells (Fig. 3f), suggesting that the BAR domain is required for Bif-1 to induce autophagosome formation. During autophagy, LC3 is processed from the cytosolic form, LC3-I, to the membrane-bound form, LC3-II27. If Bif-1 is involved in the formation of autophagic vacuoles, we reasoned that LC3 modification should be inhibited by deletion of Bif-1. As expected, the processing of LC3-I to LC3-II was suppressed in Bif-1 -/- MEFs after nutrition starvation (Fig. 3c). Importantly, this delayed LC3 modification in Bif-1 -/- cells was recovered by restoration of Bif-1 expression (Fig. 3d). Notably, the basal level of LC3-II in normal cultures was lower in Bif-1 -/- MEFs as compared to wild type cells (Fig. 3c). Interestingly, the decrease in α-tubulin expression was suppressed in Bif-1 -/- cells during EBSS culture (Fig. 3c), suggesting that autophagic degradation of cellular proteins was also inhibited by loss of Bif-1.
Figure 3.
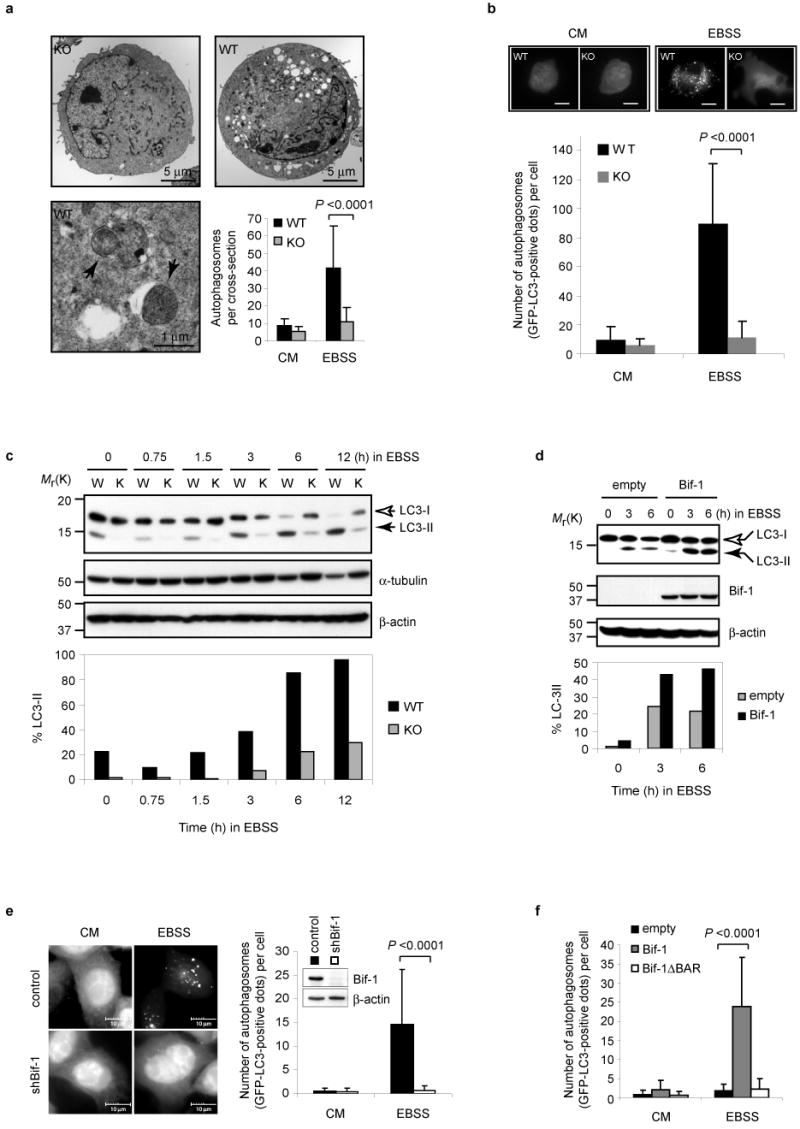
Loss of Bif-1 inhibits autophagy. (a) Bif-1 WT and KO MEFs were cultured in complete medium (CM) or EBSS for 3 h and analyzed by transmission electron microscopy. Arrows indicate autophagosomes. The number of autophagosomes per cross-sectioned cell was counted (mean ± s.d.; n = 42). (b) Bif-1 WT and KO MEFs were transfected with GFP-LC3, cultured in EBSS or CM for 3 h, then analyzed by fluorescent microscopy. Scale bars indicate 10 μm. The number of GFP-LC3 dots per GFP-positive cell was counted (mean ± s.d.; n = 35). (c) Bif-1 WT (W) and KO (K) MEFs were cultured in EBSS for the indicated times and subjected to immunoblot analysis with anti-LC3, α-tubulin and β-actin antibodies. (d) Bif-1 KO MEFs stably transfected with a Bif-1 expression plasmid or control empty vector were cultured in EBSS for 0, 3 or 6 h and subjected to immunoblot analysis. The immunoblotting results of LC3 in c and d were quantified as the percentage of LC3-II out of total LC3 (LC3-I + LC3-II) and are represented in bar graphs. (e) Stable shBif-1 expressing clones or control HeLa cells transfected with GFP-LC3 were cultured in CM or EBSS for 2 h and analyzed by fluorescent microscopy. The number of GFP-LC3 dots per GFP-positive cell was quantified (mean ± s.d.; n = 66). (f) Bif-1 knockdown HeLa cells were transfected with GFP-LC3 in combination with shBif-1 resistant Bif-1-HcRed, Bif-1ΔBAR-HcRed or empty HcRed vector for 20 h. The cells were then cultured in EBSS or CM for 2 h and the number of GFP-LC3 dots per HcRed-positive cell was determined using a fluorescent microscope (mean ± s.d.; n = 35). Statistical significance in a, b, e and f was determined by Student's t test. The full scans of the immunoblots shown in c, d and e are presented in the Supplementary Information, Fig. S5.
Bif-1 localizes to autophagosomal membrane
To examine the role of Bif-1 in the regulation of autophagy, we performed immunofluorescence analyses to determine the intracellular distribution of Bif-1-HcRed in combination with GFP-Atg5 or GFP-LC3. Consistent with previous reports27, 28, both GFP-Atg5 and GFP-LC3 formed punctate dots in the cytoplasm following nutrition deprivation (Fig. 4a). Under nutrition-rich conditions Bif-1 was localized to the cytosol, particularly in perinuclear regions (Fig. 4a). However, upon nutrient withdrawal, Bif-1 accumulated in punctate structures along with Atg5 and LC3 (Fig. 4a and see Supplementary Information, Fig. S3), suggesting that Bif-1 translocated to autophagosomal membranes. Indeed, the autophagosomal localization of Bif-1 was confirmed by immunogold electron microscopy (Fig. 4b). Atg5 has been shown to be localized on the isolation membrane throughout the elongation step of autophagosome formation and to dissociate from autophagosomal membranes once autophagosome formation is complete28. Notably, nearly all GFP-Atg5 dots were Bif-1-HcRed positive (Fig. 4a), indicating that Bif-1 is involved in the early stages of autophagosome formation.
Figure 4.
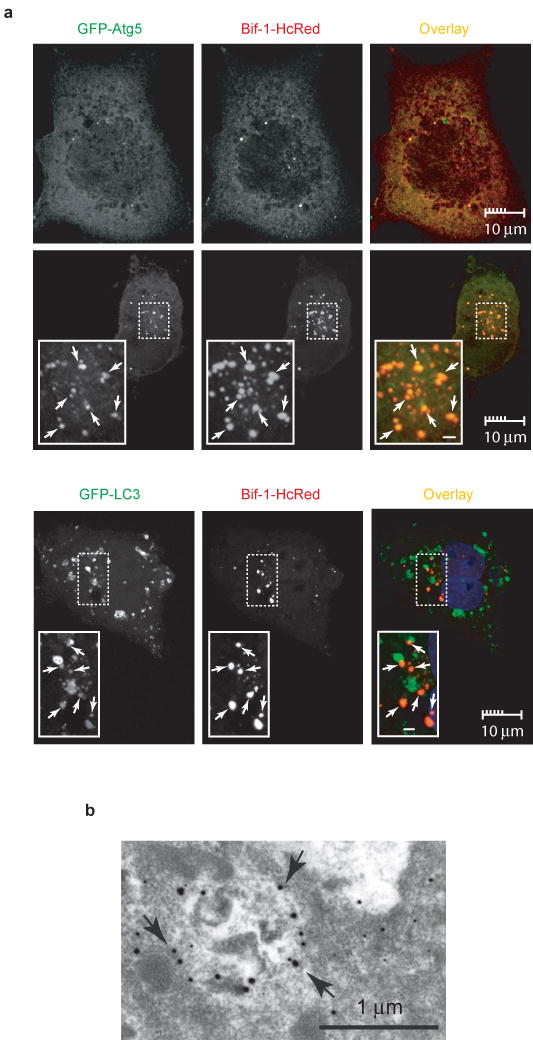
Bif-1 localizes to autophagosomes. (a) COS7 cells were transfected with Bif-1-HcRed together with GFP-Atg5 or GFP-LC3. Twenty hours after transfection, the cells were cultured in EBSS or CM for 2 h and subjected to confocal microscopic analysis. Magnified images are shown as insets. Representative Bif-1-Atg5 or Bif-1-LC-3 double positive foci are indicated by arrows. Scale bars in the insets are 1 μm. (b) COS7 cells transfected with Bif-1-GFP were cultured in EBSS for 2 h and the localization of Bif-1-GFP was examined by immunogold electron microscopy using anti-GFP antibody. Arrows indicate representative gold particles that are detected on autophagosomal membranes.
Bif-1 interacts with Beclin 1 through UVRAG
Since Bif-1 co-localizes with Atg5 and LC3, we next performed co-immunoprecipitation assays to determine whether Bif-1 interacts with Atg proteins during autophagy. As shown in Fig. 5a, endogenous Bif-1 was found to co-precipitate with endogenous Beclin 1, but not LC3. This interaction was confirmed by ectopic expression of epitope tagged Bif-1 and Beclin 1 (see Supplementary Information, Fig. S4a). Interestingly, the Bif-1-Beclin 1 interaction was enhanced by nutrition deprivation (Fig. 5a and data not shown), suggesting that Bif-1 is involved in the autophagic pathway that is regulated by Beclin 1.
Figure 5.
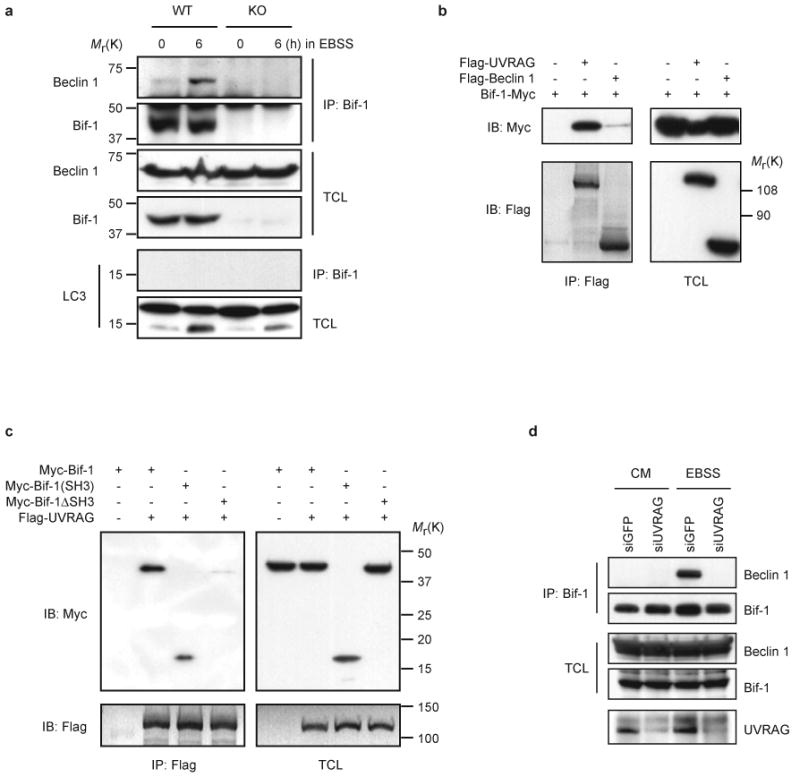
Bif-1 interacts with Beclin 1 through UVRAG. (a) Bif-1 WT and KO MEFs were cultured in EBSS for 0 or 6 h and subjected to immunoprecipitation with anti-Bif-1 monoclonal antibody. The resulting immune complexes and TCL were analyzed by immunoblotting with the indicated antibodies. (b) 293T cells were co-transfected with the indicated plasmids for 24 h and subjected to immunoprecipitation with anti-Flag monoclonal antibody. The resulting immune complexes and TCL were analyzed by immunoblotting with anti-Myc or anti-Flag polyclonal antibodies. (c) 293T cells were co-transfected with the indicated plasmids for 22 h, starved in EBSS for 2 h and subjected to immunoprecipitation with anti-Flag monoclonal antibody. The resulting immune complexes and TCL were analyzed by immunoblotting with anti-Myc or anti-Flag polyclonal antibodies. (d) 293T cells were transfected with siGFP or siUVRAG for 24 h, starved in EBSS for 2 h and subjected to immunoprecipitation with anti-Bif-1 monoclonal antibody. The resulting immune complexes and TCL were analyzed by immunoblotting with anti-Bif-1 and Beclin 1 polyclonal antibodies. The reduction of UVRAG protein expression by siUVRAG was confirmed by immunoprecipitation and immunoblot analysis with anti-UVRAG polyclonal antibody (Abgent). The full scans of immunoblots are presented in the Supplementary Information, Fig. S5.
Recently, it has been shown that UVRAG directly interacts with the Beclin 1-PI3KC3 lipid kinase complex to activate autophagy29. UVRAG contains an amino-terminal proline-rich (PR) sequence followed by a potential calcium-dependent phospholipid binding C2 domain and a central coiled-coil domain (CCD). UVRAG directly binds Beclin 1 through its CCD29. Although the PR and C2 domains are not required for UVRAG to interact with Beclin 1, removal of the N-terminal domains reduces the autophagosome formation activity of UVRAG, suggesting that the PR or C2 domain also contributes to efficient autophagosome formation29. Interestingly, Bif-1 contains a C-terminal SH3 domain, forms a complex with Beclin 1, and is involved in autophagosome formation. Thus, we speculated that UVRAG may function as a bridge molecule for the Bif-1-Beclin 1 interaction. Co-immunoprecipitation experiments revealed that Bif-1 indeed had a relatively high binding affinity for UVRAG compared to Beclin 1 (Fig. 5b). The SH3 domain of Bif-1 was required and sufficient for its interaction with UVRAG (Fig. 5c). Overexpression of the Beclin 1 binding domain of UVRAG, the CCD, disrupted the association of Bif-1 with Beclin 1 (see Supplementary Information, Fig. S4b). Moreover, knockdown of UVRAG by siRNA completely blocked the Bif-1-Beclin 1 interaction at physiological protein levels in 293T cells following nutrient withdrawal (Fig. 5d). These results clearly indicate that Bif-1 interacts with Beclin 1 through UVRAG.
Because the SH3 domain of Bif-1 is sufficient for Bif-1 to bind to UVRAG (Fig. 5c) and the BAR domain of Bif-1 is required for its ability to rescue autophagosome formation in Bif-1 knockdown cells (Fig. 3f), we next examined whether the SH3 domain of Bif-1 has a dominant-negative effect on autophagy. As shown in Fig. 6a, overexpression of the Bif-1 SH3 domain inhibited GFP-LC3 punctate foci formation in a dose-dependent manner in HeLa cells following nutrient deprivation. In marked contrast, overexpression of the SH3 domain of Endophilin A1 had no effect on autophagosome formation (Fig. 6b), which is consistent with the inability of Endophilin A1 to interact with UVRAG (Fig. 6c).
Figure 6.
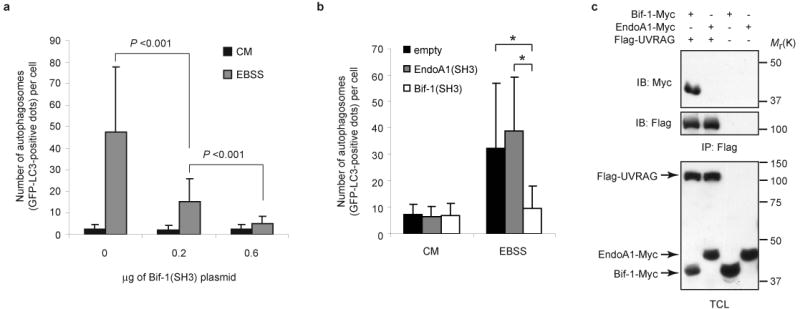
Overexpression of the SH3 domain of Bif-1 but not of Endophilin A1 suppresses autophagosome formation. (a) HeLa cells were transfected with 0.2 μg of GFP-LC3 and 0, 0.2 or 0.6 μg of Bif-1(SH3)-Myc expression plasmids for 24 h. The total amount of plasmid DNA was adjusted at 0.8 μg per transfection with the pcDNA3 vector. The cells were then cultured in EBSS or CM for 3 h and the number of GFP-LC3 dots per GFP-positive cell was determined using a fluorescent microscope (mean ± s.d.; n = 40). (b) HeLa cells were transfected with GFP-LC3 in combination with either Bif-1 (SH3)-HcRed, Endophilin A1 (SH3)-HcRed or empty HcRed vector for 20 h. The cells were then cultured in EBSS or CM for 2 h and the number of GFP-LC3 dots per HcRed-positive cell was counted using a fluorescent microscope (mean ± s.d.; n = 46). Statistical significance in a and b was determined by Student's t test (* p < 0.0001). (c) 293T cells were transfected with the indicated plasmids for 24 h and subjected to immunoprecipitation with anti-Flag monoclonal antibody. The resulting immune complexes and TCL were analyzed by immunoblotting with anti-Myc and anti-Flag polyclonal antibodies. The full scans of the immunoblots shown in c are presented in the Supplementary Information, Fig. S5.
Bif-1 regulates PI3KC3 activity
Given that Bif-1 is a component of the Beclin 1 complex, we next wanted to examine whether Bif-1 affects the lipid kinase activity of PI3KC3 during autophagy. In vitro kinase assays showed that the activation of PI3KC3 in response to nutrient deprivation was suppressed in Bif-1 -/- MEFs as compared to wild type cells (Fig. 7a). This was confirmed by analysis of foci formation of the p40(phox) PX-EGFP fusion protein29, which specifically binds to PtdIns-3-P produced by PI3KC3 (Fig. 7b). Moreover, knockdown of Bif-1 in HeLa cells also significantly reduced the enzymatic activity of PI3KC3 (Fig. 7c). After nutrient deprivation, overexpression of wild type, but not the SH3 deletion mutant of Bif-1 enhanced PI3KC3 activation in 293T cells (Fig. 7d). Taken together, these observations suggest that Bif-1 forms a complex with Beclin 1 through UVRAG to facilitate the activation of the PI3KC3 lipid kinase and autophagosome formation under starvation conditions (Fig. 7e).
Figure 7.
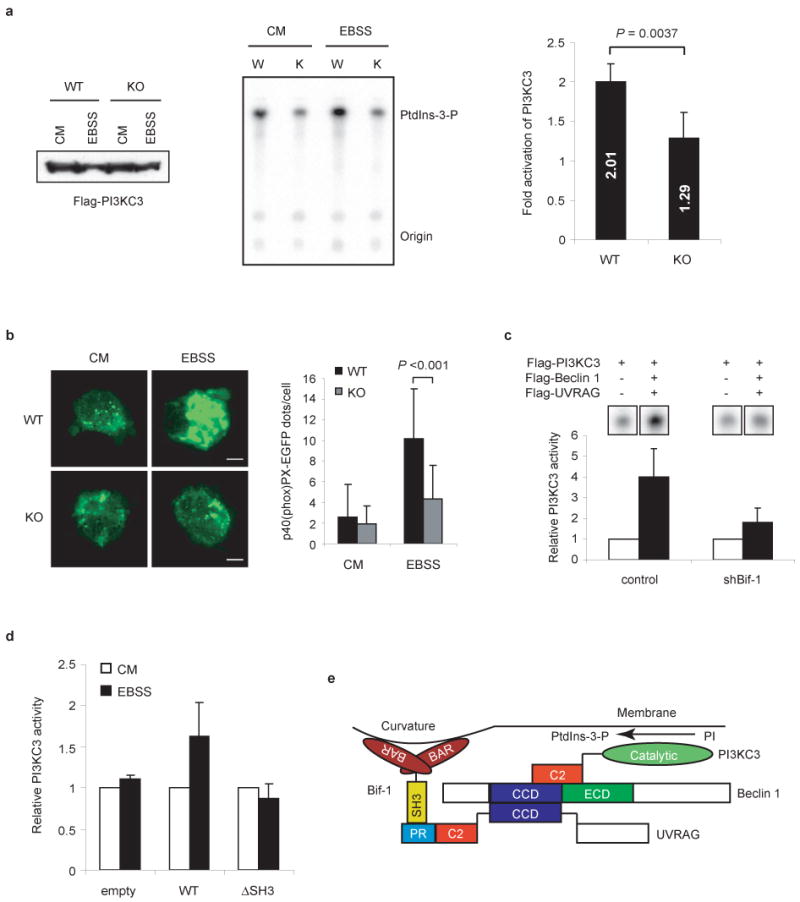
Loss of Bif-1 suppresses PI3KC3 activation during nutrient starvation. (a) Bif-1 WT and KO MEFs were transfected with Flag-PI3KC3, then incubated in CM or EBSS for 2 h and subjected to an in vitro PI3KC3 kinase assay. The expression of Flag-PI3KC3 was confirmed by immunoblot analysis (left panel). A representative autoradiograph is shown (middle panel). The results were quantified by phosphoimaging with a Typhoon scanner and shown as fold activation (right panel). The data are presented as means ± s.d. of five independent experiments. (b) Bif-1 WT and KO MEFs were transfected with p40(phox) PX-EGFP, cultured in CM or EBSS for 2 h and analyzed by fluorescent microscopy. The number of p40(phox) PX-EGFP-positive vesicles per cell was counted (mean ± s.d.; n = 35). Statistical significance in a and b was determined by Student's t test. (c) Stable shBif-1 clones or control HeLa cells were transfected with Flag-PI3KC3 alone or in combination with Flag-tagged Beclin 1 and UVRAG for 48 h and subjected to an in vitro PI3KC3 lipid kinase assay. The results were quantified and are shown as relative activities (mean ± s.d.; n = 3). (d) 293T cells were co-transfected with Flag-PI3KC3 and either wild type Bif-1, Bif-1ΔSH3 or control empty vector for 24 h and subjected to an in vitro PI3KC3 kinase assay (mean ± s.d.; n = 3). (e) Schematic representation of the Bif-1-UVRAG-Beclin 1-PI3KC3 protein complex.
Knockout of Bif-1 promotes spontaneous tumorigenesis in mice
Unlike Beclin 1-deficient mice, which are embryonic lethal10, 30, Bif-1 -/- mice developed normally and were indistinguishable from their wild type littermates. This phenotype is also different from Atg5 or Atg7 knockout mice which cannot overcome severe starvation periods after birth and die at an early neonatal stage26, 31. One possible explanation for the phenotype discrepancy between Bif-1 knockout and Atg knockout mice is that unknown factor(s) exists in specific tissues that functionally compensate for the lack of Bif-1 during embryonic and neonatal development. Anatomical and histological analyses showed that most organs examined in Bif-1 -/- mice appeared to be normal except for the size of the spleen, which was significantly larger than that of age-matched wild type mice (Fig. 8a). However, Bif-1 -/- mice had a significantly higher tumor incidence; 26 of 29 (89.7%) Bif-1 knockout mice developed spontaneous tumors at an average age of 12 months as compared to only 3 of 21 (14.3%) wild type mice (P<0.001, Fisher's exact test; Fig. 8b). The most frequent tumor observed in Bif-1 -/- mice was lymphoma (24 of 29 Bif-1 -/- mice versus 3 of 21 Bif-1 +/+ mice). Bif-1 -/- mice also developed solid tumors (10 of 29 mice) with high-frequency compared to wild-type mice (1 of 21 mice). These results indicate that Bif-1 is a potential tumor suppressor.
Figure 8.
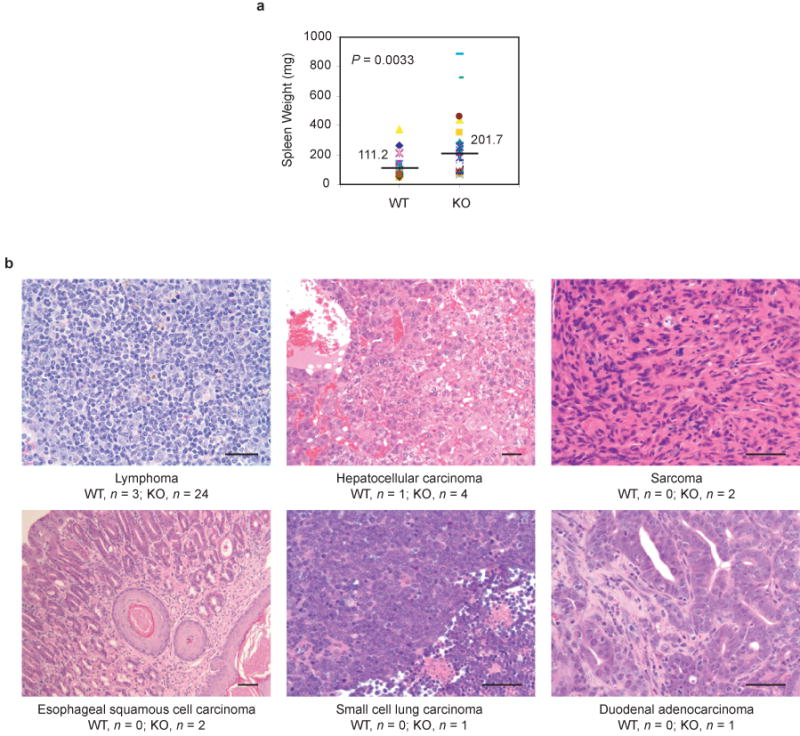
Knockout of Bif-1 enhances spontaneous tumorigenesis in mice. (a) Bif-1 -/- mice exhibit moderate splenomegaly. The weight of spleens extirpated from Bif-1 +/+ (n = 27) and -/- (n = 34) mice (6 to 18 months old) was measured. Statistical significance was determined by Wilcoxon rank test. (b) Hematoxylin and eosin staining of tumor tissues. Tumors were observed in 3 of 21 (14.3%) and 26 of 29 (89.7%) autopsied Bif-1 wild type and knockout mice, respectively. Scale bars indicate 50 μm.
Discussion
In the present study, we have demonstrated that Bif-1 forms a complex with Beclin 1 through UVRAG to enhance PI3KC3 lipid kinase activity and induce autophagosome formation during nutrient starvation. Notably, EBSS-induced PI3KC3 activation was only partially suppressed by loss of Bif-1 (Fig. 7), while the autophagosome formation was greatly blocked in Bif-1-deficient cells (Fig. 3). These results suggest that Bif-1 has other modes of action besides enhancing PI3KC3 activation for the regulation of autophagy. Similarly, whereas Beclin 1 is indispensable for autophagosome formation8, deletion of Beclin 1 only partially suppresses PI3KC3 activity32. Moreover, silencing of Beclin 1 in Bif-1 knockout cells has no significant effect on autophagy-dependent cell death (Fig. 2a), suggesting that Bif-1 and Beclin 1 participate in the same signaling pathway in response to nutrition deprivation. The process of autophagy involves multiple steps, including initiation, cargo packaging, vesicle nucleation, vesicle expansion and completion, retrieval, docking and fusion and lysosomal degradation of the vesicles and their contents 2, 4, 5, 33. Of these steps, vesicle nucleation is one of the least understood aspects of autophagy as the driving force for membrane curvature and deformation is entirely unknown2. It has been shown that Beclin 1 is involved in the nucleation of autophagosomal membranes2 and that Bif-1 binds to lipids and induces membrane curvature18. Therefore, it is possible that Bif-1 interacts with Beclin 1 through UVRAG at the isolation membrane or phagophore to regulate vesicle nucleation by inducing membrane curvature through its N-BAR domain during autophagy (Fig. 7e). Recently, Hsu and colleagues have demonstrated that Bif-1 plays a critical role in the vesicle formation for COPI-mediated retrograde transportation from the trans-Golgi network (TGN) to the endoplasmic reticulum19. Moreover, the majority of Beclin 1 has been shown to be localized in the TGN9. Therefore, it will be interesting to determine whether Bif-1-mediated COPI-vesicle formation contributes to the process of autophagy.
While autophagy is thought to play an essential role in cell survival by recycling nutrients under starvation conditions 3, 7, 34, accumulating evidence suggests that autophagy is also involved in programmed cell death 4, 7. We have observed that suppression of autophagy promotes nutrient deprivation-induced caspase-3 activation (Fig. 2), which is consistent with the notion that autophagy is an adaptive response connected to cell survival. Interestingly, blockade of apoptosis by caspase inhibition does not improve cell survival whereas inactivation of autophagy suppresses cell death under starvation conditions. These results suggest that autophagy coordinates programmed cell death and favors a caspase-independent mechanism over apoptosis. Unlike apoptosis, which is executed by activated caspases, autophagic cell death occurs in a caspase-independent manner by the degradation of cytoplasmic proteins and organelles. It is not difficult to imagine that autophagic degradation of cytoplasmic components promotes the collapse of cellular functions and leads to an irreversible cell death, while at the same time preventing caspase activation and apoptosis by the removal of damaged organelles, such as mitochondria, and pro-apoptotic proteins4.
Evidence from recent studies suggest that autophagy is involved in tumorigenesis 3, 4, 35. It has been shown that the rate of autophagy is reduced during chemical carcinogenesis in rats36-38. In addition, oncogenes, such as Akt and Bcl-2, suppress autophagy, whereas tumor suppressors, such as DAPK, p53 and PTEN, stimulate autophagy39-42. Haploid deletion of Beclin 1 is frequently detected in human breast and ovarian cancers 8, 43. In comparison to wild type mice, Beclin 1 +/- mice have a higher frequency of tumor occurrence including lymphomas, liver carcinomas and lung cancers 10, 30. Similar to Beclin 1, UVRAG promotes autophagy and suppresses tumorigenesis in nude mice 29. It has also been shown that UVRAG is monoallelically mutated at a high frequency in human colon cancer cells44. Consistently, suppression of Bif-1 expression in HeLa cells promotes tumor growth in nude mice 21 and knockout of Bif-1 significantly enhances the development of spontaneous tumors in mice (Fig. 8). Furthermore, homozygous deletion of the Bif-1 gene locus has been identified in mantle cell lymphomas45 and decreased expression of Bif-1 has been reported in gastric carcinomas46. Although further studies are required to elucidate the precise mechanism of Bif-1 in Beclin 1-mediated initiation of autophagy, this study has identified Bif-1 as an activator of autophagy and a tumor suppressor and provides new evidence in support of the concept that induction of autophagy could contribute to the prevention and treatment of cancer.
Methods
RNA interference
The sequences for GFP siRNA (siGFP) and UVRAG siRNA (siUVRAG) were described previously21, 29. All siRNAs were purchased from Dharmacon (Lafayette, CO) and transfected into cells at 10 μg of siRNA per 1 × 106 cells using the nucleofector system (Amaxa, Gaithersburg, MD). The sequences for scrambled control shRNA (shSCR) and Beclin1 shRNA (shBECN1) are 5′-CAACAAGATGAAGAGCACCAA-3′ and 5′-GCGGGAGTATAGTGAGTTTAA-3′, respectively. All lentiviral shRNA constructs were purchased from Open Biosystems (Huntsville, AL) and co-transfected into 293FT cells along with packaging plasmids. The resulting supernatant containing shRNA expressing lentivirus was used to transduce MEFs according to the manufacture's protocol (Invitrogen, Carlsbad, CA).
Autophagy assays
Cells were cultured on 0.1% gelatin-coated dishes or coverslips, washed twice with PBS, once with EBSS and then incubated in EBSS for the indicated times. For the electron microscopic analyses, cells were fixed with 2.5% glutaraldehyde in 0.1M phosphate buffer at pH7.2 for 16 h at 4 °C, postfixed with 1% osmium tetroxide in 0.1M phosphate buffer at pH7.2 for 1 h at 4 °C, embedded, sectioned at a thickness of 80 nm, stained with 6% uranyl acetate and Reynolds lead citrate and analyzed with a Philips CM10 transmission electron microscope. For the analysis of GFP-LC3 punctate foci, cells were transfected with GFP-LC3 using FuGENE HD (Roche, Indianapolis, IN) for 20 h. After incubation in EBSS, the cells were fixed in 3.7% formaldehyde and the number of GFP dots (≥ 300 nm diameter) per GFP-positive cell was counted using an automated Zeiss Axio-Imager Z.1 microscope. To analyze the modification of LC3-I to LC3-II, total cell lysates were prepared in radio-immunoprecipitation assay buffer (150 mM NaCl, 10 mM Tris-HCl, pH 7.4, 0.1% SDS, 1% Triton X-100, 1% Deoxycholate, 5 mM EDTA, pH 8.0) containing protease inhibitors and subjected to SDS-PAGE/immunoblot analysis with anti-rat LC3 polyclonal antibody27.
Co-immunoprecipitation and confocal microscopy
Total cell lysates were prepared in 2% CHAPS lysis buffer (20 mM Tris-HCl, pH 7.5, 137 mM NaCl, 2mM EDTA, 10% glycerol, 2% CHAPS) containing protease inhibitors and subjected to immunoprecipitation with anti-Bif-1 (Imgenex, San Diego, CA), anti-Flag (Sigma, St. Louis, MO) or anti-Myc-tag monoclonal antibodies. The resulting immunocomplexes were washed three times with lysis buffer and subjected to Western blot analysis with anti-Beclin 1 (Abgent, San Diego, CA), anti-Bif-1 (GeneTex, San Antonio, TX), anti-LC3, anti-Myc (Sigma) or anti-Flag (Sigma) polyclonal antibodies. To determine Bif-1 localization during autophagy, COS7 cells were grown on gelatinized coverslips and transfected with Bif-1-HcRed and GFP-Atg5 or GFP-LC3. Twenty hours after transfection, the cells were washed twice with PBS, once with EBSS, incubated in EBSS for 2 h, fixed in 3.7% formaldehyde/PBS and mounted with DAPI containing medium (Vector Laboratories, Burlingame, CA). Fluorescence images were analyzed by a Leica DMI6000 laser scanning confocal microscope.
Immunoelectron microscopy
COS7 cells were transfected with Bif-1-GFP for 20 h and cultured in EBSS for 2 h. The cells were fixed in 4% paraformaldehyde in 0.1 M phosphate buffer (PB) at pH7.2 for 90 min, incubated in 0.1% sodium borohydride in 0.1 M PB at pH7.2 for 30 min and permeabilized with 0.05% Triton X-100 in PBS for 30 min. After 30 min incubation in blocking buffer (5% bovine serum albumen fraction V, 0.1% cold water fish gelatin, 5% normal goat serum, PBS, pH7.4), the cells were stained with anti-GFP rabbit polyclonal antibody (Invitrogen) for 1 h, followed by goat anti-rabbit IgG fab' secondary antibody (Electron Microscopy Science, Hatfield, PA). The cells were fixed in 2.5% glutaraldehyde overnight at 4 °C and post-fixed in 0.5% osmium tetroxide for 30 min. The gold particles were enlarged for 25 min with a silver enhancement kit (Electron Microscopy Science). The cells were then dehydrated, infiltrated with plastic, embedded in plastic, sectioned at a thickness of 80 nm, stained with 6% uranyl acetate and Reynolds lead citrate and analyzed with a Philips CM10 transmission electron microscope.
PI3KC3 activity assays
Cells were transfected with Flag-tagged PI3KC3 or p40(phox) PX-EGFP for 24 h and incubated in EBSS or normal culture medium (CM) for an additional 2 h. For the in vitro PI3KC3 kinase assays, Flag-PI3KC3 was immunoprecipitated with anti-Flag affinity beads (Sigma), washed twice with 1% NP-40 in PBS, twice with washing buffer (100 mM Tris-HCl at pH 7.5, 500 mM LiCl), twice with TNE buffer (10 mM Tris-HCl at pH 7.5, 100 mM NaCl, 1 mM EDTA) and pre-incubated for 10 min at room temperature in 60 μl of reaction buffer (10 mM MnCl2 in TNE) containing 2 μg of sonicated phosphatidylinositol. The reaction was initiated with addition of 5 μl of ATP mix (1 μl of 10 mM unlabelled ATP, 1 μl of γ-32P-ATP, 3 μl H2O) and incubated for 15 min at room temperature. The reaction was terminated by adding 100 μl of 1 N HCl, extracted with CHCl3:MeOH (1:1), washed twice with MeOH:1 N HCl (1:1), dried with SpeedVac and resuspended in 20 μl of CHCl3:MeOH (1:1). The resultant assay product was separated on dried silica, aluminum thin liquid chromatography plates with running buffer (CHCl3:MeOH:4 M NH4OH (9:7:2) and visualized using a Typhoon9400 Variable Mode Imager (Amersham Biosciences, Piscataway, NJ). For p40(phox) PX-EGFP analysis, the cells were trypsinized, cytospun onto slides, fixed in 3.7% formaldehyde/PBS and mounted with DAPI containing medium (Vector Laboratories). The number of p40(phox) PX-EGFP-positive vesicles per cell was determined using an automated Zeiss Axio-Imager Z.1 microscope.
Histological examination
Bif-1 knockout mice were generated by targeted homologous recombination as previously described21. All mice used for examination of spontaneous tumor development were of 129SvJ × C57BL/6 crosses. For histological examination, tissues extirpated from 6 to 18 months old mice were fixed in 10% formaldehyde, embedded, sectioned, stained with hematoxylin and eosin and examined by two independent senior pathologists (DC, HC).
Supplementary Material
Note: Supplementary Information is available on the Nature Cell Biology website.
Acknowledgments
We thank E. Haller for assistance with electron microscopic analyses; G. Gao for statistical help; C. Meyerkord and R. Youle for critical reading of the manuscript; D.C.S. Huang, B. Levine, N. Mizushima, G. Nolan, G. Reuther and T. Yoshimori for reagents. This work was supported by grants from the National Institutes of Health (NIH) and American Cancer Society (ACS) to H.-G.W. and fellowships from the Uehara Memorial Foundation and Japan Society for the Promotion of Science (JSPS) to Y.T.
Footnotes
COMPETING INTERESTS STATEMENT: The authors declare that they have no competing financial interests.
References
- 1.Yoshimori T. Autophagy: a regulated bulk degradation process inside cells. Biochem Biophys Res Commun. 2004;313:453–8. doi: 10.1016/j.bbrc.2003.07.023. [DOI] [PubMed] [Google Scholar]
- 2.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–77. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 3.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–5. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- 5.Eskelinen EL. Maturation of autophagic vacuoles in Mammalian cells. Autophagy. 2005;1:1–10. doi: 10.4161/auto.1.1.1270. [DOI] [PubMed] [Google Scholar]
- 6.Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct. 2002;27:421–9. doi: 10.1247/csf.27.421. [DOI] [PubMed] [Google Scholar]
- 7.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115:2679–88. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liang XH, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 9.Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2001;2:330–5. doi: 10.1093/embo-reports/kve061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077–82. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furuya N, Yu J, Byfield M, Pattingre S, Levine B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy. 2005;1:46–52. doi: 10.4161/auto.1.1.1542. [DOI] [PubMed] [Google Scholar]
- 12.Peter BJ, et al. BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science. 2004;303:495–9. doi: 10.1126/science.1092586. [DOI] [PubMed] [Google Scholar]
- 13.Masuda M, et al. Endophilin BAR domain drives membrane curvature by two newly identified structure-based mechanisms. Embo J. 2006;25:2889–97. doi: 10.1038/sj.emboj.7601176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gallop JL, et al. Mechanism of endophilin N-BAR domain-mediated membrane curvature. Embo J. 2006;25:2898–910. doi: 10.1038/sj.emboj.7601174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huttner WB, Schmidt A. Lipids, lipid modification and lipid-protein interaction in membrane budding and fission--insights from the roles of endophilin A1 and synaptophysin in synaptic vesicle endocytosis. Curr Opin Neurobiol. 2000;10:543–51. doi: 10.1016/s0959-4388(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 16.Cuddeback SM, et al. Molecular cloning and characterization of Bif-1: A novel SH3 domain- containing protein that associates with Bax. J Biol Chem. 2001;276:20559–20565. doi: 10.1074/jbc.M101527200. [DOI] [PubMed] [Google Scholar]
- 17.Pierrat B, et al. SH3GLB, a new endophilin-related protein family featuring an SH3 domain. Genomics. 2001;71:222–34. doi: 10.1006/geno.2000.6378. [DOI] [PubMed] [Google Scholar]
- 18.Farsad K, et al. Generation of high curvature membranes mediated by direct endophilin bilayer interactions. J Cell Biol. 2001;155:193–200. doi: 10.1083/jcb.200107075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang JS, et al. Key components of the fission machinery are interchangeable. Nat Cell Biol. 2006;8:1376–82. doi: 10.1038/ncb1503. [DOI] [PubMed] [Google Scholar]
- 20.Karbowski M, Jeong SY, Youle RJ. Endophilin B1 is required for the maintenance of mitochondrial morphology. J Cell Biol. 2004;166:1027–39. doi: 10.1083/jcb.200407046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takahashi Y, et al. Loss of Bif-1 suppresses Bax/Bak conformational change and mitochondrial apoptosis. Mol Cell Biol. 2005;25:9369–82. doi: 10.1128/MCB.25.21.9369-9382.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu L, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–2. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 23.Shimizu S, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–8. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 24.Willis SN, et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boya P, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–40. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuma A, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–6. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 27.Kabeya Y, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mizushima N, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657–68. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang C, et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–99. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- 30.Qu X, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–20. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Komatsu M, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–34. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kihara A, Noda T, Ishihara N, Ohsumi Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol. 2001;152:519–30. doi: 10.1083/jcb.152.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol. 2001;2:211–6. doi: 10.1038/35056522. [DOI] [PubMed] [Google Scholar]
- 34.Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–48. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- 35.Mathew R, White E. Why sick cells produce tumors: the protective role of autophagy. Autophagy. 2007;3:502–5. doi: 10.4161/auto.4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwarze PE, Seglen PO. Reduced autophagic activity, improved protein balance and enhanced in vitro survival of hepatocytes isolated from carcinogentreated rats. Exp Cell Res. 1985;157:15–28. doi: 10.1016/0014-4827(85)90148-x. [DOI] [PubMed] [Google Scholar]
- 37.Kisen GO, et al. Reduced autophagic activity in primary rat hepatocellular carcinoma and ascites hepatoma cells. Carcinogenesis. 1993;14:2501–5. doi: 10.1093/carcin/14.12.2501. [DOI] [PubMed] [Google Scholar]
- 38.Toth S, Nagy K, Palfia Z, Rez G. Cellular autophagic capacity changes during azaserine-induced tumour progression in the rat pancreas. Up-regulation in all premalignant stages and down-regulation with loss of cycloheximide sensitivity of segregation along with malignant transformation. Cell Tissue Res. 2002;309:409–16. doi: 10.1007/s00441-001-0506-7. [DOI] [PubMed] [Google Scholar]
- 39.Arico S, et al. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem. 2001;276:35243–6. doi: 10.1074/jbc.C100319200. [DOI] [PubMed] [Google Scholar]
- 40.Pattingre S, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 41.Inbal B, Bialik S, Sabanay I, Shani G, Kimchi A. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J Cell Biol. 2002;157:455–68. doi: 10.1083/jcb.200109094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005;102:8204–9. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aita VM, et al. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics. 1999;59:59–65. doi: 10.1006/geno.1999.5851. [DOI] [PubMed] [Google Scholar]
- 44.Ionov Y, Nowak N, Perucho M, Markowitz S, Cowell JK. Manipulation of nonsense mediated decay identifies gene mutations in colon cancer Cells with microsatellite instability. Oncogene. 2004;23:639–45. doi: 10.1038/sj.onc.1207178. [DOI] [PubMed] [Google Scholar]
- 45.Balakrishnan A, et al. Quantitative microsatellite analysis to delineate the commonly deleted region 1p22.3 in mantle cell lymphomas. Genes Chromosomes Cancer. 2006;45:883–92. doi: 10.1002/gcc.20352. [DOI] [PubMed] [Google Scholar]
- 46.Lee JW, et al. Decreased expression of tumour suppressor Bax-interacting factor-1 (Bif-1), a Bax activator, in gastric carcinomas. Pathology. 2006;38:312–5. doi: 10.1080/00313020600820880. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: Supplementary Information is available on the Nature Cell Biology website.