

Summary
Polycomb genes encode critical regulators of both normal stem cells and cancer stem cells. A gene signature that includes polycomb genes and additional genes co-regulated with polycomb genes was recently identified. The expression of this signature has been reported to identify tumors with the cancer stem cell phenotypes of aggressive growth, metastasis and therapy resistance. Most members of this 11-gene signature encode proteins with well-defined roles in human cancer. However the function of the signature member USP22 remains unknown. We report that USP22 is a previously uncharacterized subunit of the human SAGA transcriptional cofactor complex. Within SAGA, USP22 deubiquitylates histone H2B. Furthermore, USP22 is recruited to specific genes by activators such as the MYC oncoprotein, where it is required for transcription. In support of a functional role within the polycomb/cancer stem cell signature, USP22 is required for appropriate progression through the cell cycle.
Keywords: SAGA, MYC, USP22, polycomb, BMI-1, transcription, ubiquitin hydrolase, histone, chromatin, UBP8
Introduction
The development of high throughput mRNA expression profiling raised the possibility that a small number of transcripts might be identified whose expression could accurately predict the therapeutic response of individual cancer patients (Raetz and Moos, 2004). The recent identification of an 11-gene polycomb/cancer stem cell signature, has brought this type of assessment closer to practical reality (Glinsky, 2005; Glinsky et al., 2005; Widschwendter et al., 2007). This signature contains polycomb genes such as BMI-1 and RNF2/Ring1b, and also a number of genes that are co-regulated with BMI-1, potentially by the elevation of BMI-1 itself. In fact, the polycomb protein BMI-1 regulates transcription of 8 of the 11 genes in the signature (Glinsky, 2005, 2006; Glinsky et al., 2005). In initial tests, this signature was able to distinguish patients whose tumors would eventually metastasize from those whose tumors would remain localized. Remarkably, the predictive power of this signature remains high even when assessing early stage cancer patients and when examining tumors derived from a wide variety of epithelial and non-epithelial tissues. Expression of the polycomb profile is critical for maintaining the self-renewal properties of stem cells (Lessard and Sauvageau, 2003; Valk-Lingbeek et al., 2004) and the identification of this signature has added support to the cancer stem cell hypothesis (Blagosklonny, 2006; Wicha, 2006; Widschwendter et al., 2007). This hypothesis argues that tumors contain therapy-resistant cells with stem cell properties and that these cells are responsible for tumor recurrence after treatment, and for metastases (Wicha, 2006).
Interestingly, proteins encoded by the 11 gene polycomb/stem cell signature are not simply bystander markers of tumor progression. Instead, most play well-documented, causal roles in human cancer (Glinsky, 2006). For example, both BUB1 and HEC1 function in kinetochore assembly and are important for integrity of the mitotic checkpoint (Hori et al., 2003; McCleland et al., 2003). The aberrant expression of these genes is predicted to induce the aneuploid genotype observed in aggressive tumors. Similarly, the signature gene cyclin B1 (CCNB1) is another critical regulator of mitosis, as is another member of this signature, the cyclin B1/cdc2 substrate Ki67 (MacCallum and Hall, 1999). A number of the genes within this signature encode critical components of signaling pathways that are altered in cancer. These signature members include GBX2, the FGF receptor 2 and Ankyrin 3 (Glinsky, 2006). Finally, this signature includes the polycomb protein RNF2/Ring1b, a ubiquitin ligase that forms a complex with BMI-1 to ubiquitylate nucleosomal histones (Ben-Saadon et al., 2006; Li et al., 2006; Voncken et al., 2003). Thus, the polycomb/cancer stem cell signature genes encode proteins whose biochemical functions play an active role in modulating tumor growth.
Unlike the other genes in this cancer stem cell signature, no direct mechanistic link to human cancer has been ascribed to USP22. Sequence analysis reveals that USP22 belongs to a large family of proteins with ubiquitin hydrolase activity, and recombinant USP22 is able to cleave a synthetic ubiquitin molecule in vitro. In order to assess whether USP22 might also participate in pathways important for cancer progression, we initiated studies aimed at defining the in vivo function of this protein. We report here that USP22 is a dedicated subunit of the human SAGA complex (hSAGA), a multiprotein transcriptional cofactor complex broadly required for the function of sequence-specific transcription activators in eukaryotes (Lee and Workman, 2007). Proteomics screens in yeast had previously identified Ubp8p, a protein with significant homology to USP22, as a constitutive subunit of the yeast SAGA complex (Gavin et al., 2002; Ho et al., 2002). Subsequent empirical studies by a number of groups have shown that Ubp8p is required for SAGA-dependent transcription at some yeast genes (Daniel et al., 2004; Henry et al., 2003; Lee et al., 2005; Mutiu et al., 2007; Sanders et al., 2002). Mechanistically, hSAGA is recruited by sequence-specific activators to individual genes (Bhaumik and Green, 2001; Larschan and Winston, 2001), where at least part of its function relies on its ability to acetylate nucleosomal histones (Grant et al., 1997). Confirming a functional role for USP22 within hSAGA, we show that endogenous USP22 contributes deubiquitylating activity to hSAGA. Furthermore, this activity is directed at the core histone H2B. Thus, like the polycomb/cancer stem cell signature members RNF2/Ring1b and BMI-1 (de Napoles et al., 2004; Fang et al., 2004; Li et al., 2006), USP22 may regulate transcription via alterations in levels of histone ubiquitylation. Among the activators that have been reported to recruit hSAGA in human cells is the MYC oncoprotein (Bouchard et al., 2001; Frank et al., 2001; McMahon et al., 2000). We demonstrate here that USP22 is required for the activation of target gene transcription by MYC. Most importantly, depletion of USP22 compromises MYC functions, including transformation. Consistent with a critical role in cell cycle progression, depletion of USP22 results in a specific G1 phase cell cycle arrest.
These data establish three essential points. First, they provide the first advance in our understanding of the function of the cancer stem cell signature member USP22 by demonstrating that it functions within the hSAGA complex to regulate activator-driven transcription. Second, they establish that hSAGA, like its yeast counterpart, possesses a second, previously unknown enzymatic activity that is critical for transcription. Finally, these data establish a direct biochemical link between human cancer and hSAGA function.
Results
USP22 is a functionally uncharacterized member of the recently identified polycomb/cancer stem cell signature (Glinsky, 2006). USP22 contains a carboxy terminal ubiquitin hydrolase domain that defines the C-19 class of peptidases (Figure 1A), and recombinant USP22 is able to remove a ubiquitin moiety from a synthetic substrate in vitro (Lee et al., 2006). In addition, this and other ubiquitin hydrolases contain an amino terminal zinc finger motif that may mediate the association of these enzymes with other proteins (Ingvarsdottir et al., 2005). In order to gain insight into the biochemical pathway in which USP22 participates, the protein complex associated with USP22 was investigated. The rationale for this was that the identification of proteins physically associated with USP22 in human cells would provide critical information regarding the function of this uncharacterized member of the polycomb/cancer stem cell signature. For this purpose, a FLAG epitope-tagged expression vector was generated and stably introduced into the human lung cancer cell line H1299 (Figure 1A). After confirming the expression of tagged USP22 in this line, nuclear extracts from large-scale cultures were produced. Under non-denaturing conditions, the tagged USP22 protein was affinity purified from these extracts. Western blotting of elutates from this purification revealed that the FLAG-USP22 protein was specifically captured, purified and eluted (Figure 1C). Silver staining of precipitates revealed a number of bands specifically associated with USP22 in the nucleus (Figure 1B). Remarkably, MS/MS analysis demonstrated that the majority of USP22 associated proteins are subunits of the human SAGA complex (Figure 1 and Supplemental Table 1). As mentioned above, hSAGA is a multi-subunit transcriptional cofactor that acetylates nucleosomal histones (Lee and Workman, 2007). The subunits of hSAGA identified as USP22 interacting proteins include TRRAP, ADA2, ADA3, TAF9L, TAF10, ataxin 7 and the acetyltransferase hGCN5. Western blot analysis for two known hSAGA subunits, hGCN5 and TRRAP, confirmed the association of these proteins with USP22 (Figure 1C). These data suggest that USP22 is a previously uncharacterized component of the hSAGA transcriptional cofactor complex.
Figure 1. The cancer stem cell marker USP22 is a subunit of the hSAGA cofactor complex.
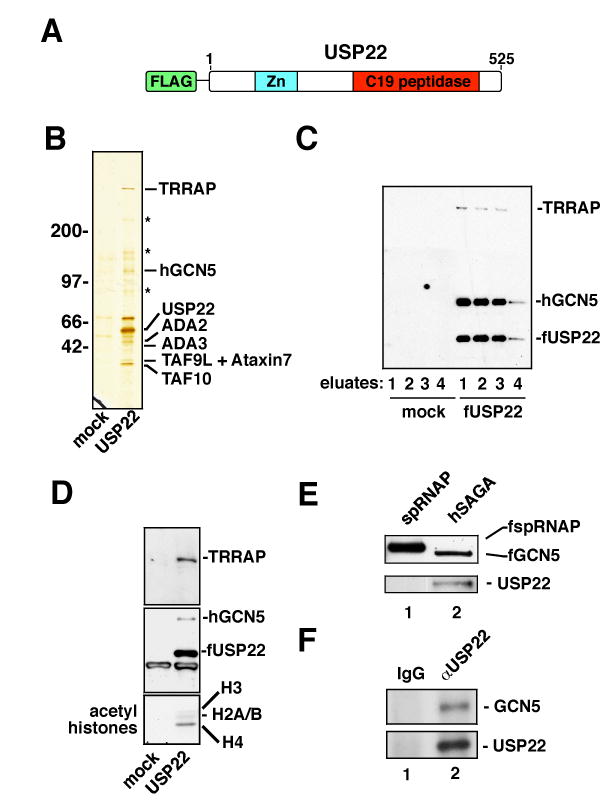
(A) USP22 is a putative ubiquitin hydrolase containing a C-terminal peptidase domain of the C19 family. In addition, USP22 contains an amino terminal zinc finger motif. (B) A stable cell line expressing a FLAG-epitope tagged version of USP22 (fUSP22) was generated in the human non-small cell lung carcinoma line H1299. The FLAG-USP22 protein was affinity purified and co-purified proteins detected by silver staining. A mock purification was performed in parallel using extracts from parental cells. MS/MS identification of USP22 associated proteins was performed on bands excised from duplicate samples run in adjacent lanes and colloidal-stained. The polypeptides listed were identified in the MS/MS analysis and represent known components of the human SAGA complex. Peptide sequences from the recovered proteins are listed in Supplemental Table 1, with the total coverage ranging from 1-5%. Asterisks indicate known contaminants, including keratin and myosin. (C) The purified FLAG-USP22 complex was analyzed by western blotting for USP22 and the hSAGA subunits TRRAP and hGCN5. During affinity purification, four sequential eluates were harvested, as indicated. (D) Purified USP22 complex was assessed for HAT activity using an in vitro reaction containing core histones and radiolabeled acetyl-CoA (lower panel). Western blotting again confirmed the association of USP22 with the hSAGA subunits TRRAP and hGCN5, as well as the presence of FLAG-USP22 itself (upper panels). (E) Western blots confirmed that endogenous USP22 is associated with hSAGA. hSAGA was affinity purified from cells stably expressing FLAG-hGCN5. As a control, a cell line expressing FLAG-spRNAP was utilized. spRNAP and hGCN5 precipitation was confirmed by blotting for the FLAG epitope (top panel). Probing with anti-USP22 revealed the specific association of endogenous USP22 with the hSAGA complex (lower panel). (F) Endogenous hGCN5 and USP22 coprecipitate from nuclear extracts of human cells. Nuclear extracts from 293T cells were subjected to immunoprecipitation with an antibody recognizing endogenous USP22. Precipitates were resolved by SDS/PAGE and blotted for either USP22 (bottom panel) or hGCN5 (top panel). Control precipitates were conducted in parallel using nonimmune IgG, as indicated.
The association of USP22 with hSAGA predicts that the USP22 complex should contain histone acetyltransferase (HAT) activity encoded by the hSAGA subunit hGCN5 (Lee and Workman, 2007). To assess this, FLAG-USP22 was again purified from H1299 cells. The purified material contained hSAGA, as assessed by western blotting for hGCN5 and TRRAP (Figure 1D). When subjected to an in vitro acetylation reaction with purified core histones as a substrate, the USP22 complex demonstrated specific HAT activity. This HAT activity was broadly directed at several of the core histones, consistent with published studies (Kuo et al., 1996; Poux and Marmorstein, 2003; Schiltz et al., 1999). Despite this documented promiscuity of GCN5, the acetyltransferase activity of the purified USP22 complex was directed more strongly towards H4 than expected. This raises the possibility that association with USP22 slightly alters the substrate preference of GCN5.
While ectopic expression of USP22 resulted in its association with hSAGA, it remained possible that this interaction was observed as the result of the high level of USP22 expression in this system. In order to examine whether the hSAGA/USP22 interaction was evident without overexpression of USP22, an antisera against human USP22 was generated. hSAGA was then purified as described previously using a 293T cell line stably expressing an epitope-tagged version of the hGCN5 subunit (Ogryzko et al., 1998). As a control, epitope tagged spRNAP was purified from a stable 293T cell line in parallel. Purified hSAGA contained endogenous USP22, while no USP22 was observed in association with the control spRNAP protein (Figure 1E). Finally, co-immunoprecipitation of endogenous proteins was performed in order to test whether USP22 associates with the hGCN5 complex under conditions where both proteins are expressed at physiological levels. Immunoprecipitation of USP22 from nuclear extracts of human cells revealed the specific coprecipitation of endogenous hGCN5 (Figure 1F). Considered together, these data suggest that even when expressed at endogenous levels, USP22 is a subunit of the hSAGA transcriptional cofactor complex.
USP22 is a histone ubiquitin hydrolase
Recombinant USP22 is capable of hydrolyzing a ubiquitin linkage on a synthetic substrate in vitro (Lee et al., 2006). In addition, proteomic analysis in yeast identified a USP22 ortholog, Ub8p, as a component of the yeast SAGA complex (Gavin et al., 2002; Ho et al., 2002). Further analysis of Ubp8p has demonstrated that one of its functions is to remove ubiquitin from histones during the transcription cycle (Henry et al., 2003). Based on these facts, and the known substrate preference of other hSAGA subunits for histones, we examined whether USP22 could remove ubiquitin from histone H2B. For this analysis, USP22 was ectopically expressed in human cells and then affinity purified as above. In parallel, the canonical hSAGA complex was similarly affinity purified via tagged hGCN5. The presence of USP22 and hGCN5 was confirmed by western blotting for the FLAG epitope carried by the ectopic proteins (Figure 2A, top panel). As a negative control the unrelated acetyltransferase HBO1 was purified in parallel using the same approach. The polypeptide composition of these purified complexes was assessed by silver staining, which revealed a pattern of 10-12 co-migrating proteins shared by the USP22 and hGCN5 complexes (Supplemental Figure 1). These complexes were then assessed in vitro for their ability to remove ubiquitin from histone H2B. The ubiquitylated H2B (uH2B) substrate from porcine thymus was highly purified such that all H2B present in the reactions contains a ubiquitin conjugate (Thorne et al., 1987). As assessed by the appearance of non-ubiquitylated H2B in these reactions, purified USP22 has ubiquitin hydrolase activity that is directed towards uH2B (Figure 2A, lower panel). The hSAGA complex also displayed ubiquitin hydrolase activity on uH2B, consistent with endogenous USP22 being present in this complex, while the HBO1 complex displayed no ubiquitin hydrolase activity. The activity of the holo hSAGA complex appears more robust than that of purified USP22, suggesting that USP22 may be active only when associated with the other hSAGA subunits. This requirement has been observed for yeast Ubp8p (Lee et al., 2005). In order to formally test whether the hSAGA-associated ubiquitin hydrolase activity was dependent on endogenous USP22, hSAGA was purified from cells in which endogenous USP22 had been depleted by shRNA treatment (Figure 2B). USP22 depletion of 70-80% was achieved, as assessed by western blotting and qRT-PCR (Figure 2B, middle panels). When purified from cells expressing reduced levels of USP22, the ubiquitin hydrolase activity of the hSAGA complex was also reduced (Figure 2B, lower panel), consistent with USP22 being the enzyme within hSAGA responsible for this activity. These data are unlikely to result from off-target effects of the USP22 shRNA as identical results were obtained when USP22 was depleted using a second shRNA construct targeting a distinct region of the USP22 transcript (data not shown).
Figure 2. USP22 catalyzes the deubiquitylation of histone H2B in vitro.

(A) FLAG-tagged USP22 or HBO1 were expressed in H1299 cells and affinity purified under non-denaturing conditions. In parallel, hSAGA was purified via FLAG-tagged hGCN5. The purification of USP22, hGCN5 and HBO1 was confirmed by western blotting for the FLAG epitope (upper panel). Purified USP22 complex, hSAGA and HBO1 complex were incubated in vitro with ubiquitylated H2B (uH2B). Reaction mixtures were then resolved by SDS/PAGE and developed with antisera against H2B (lower panel). In addition to deubiquitylation reactions that included the USP22 complex (lane 3), hSAGA (lane 4) and HBO1 (lane 5), a mock reaction was analyzed in parallel (lane 2). Purified non-ubiquitylated H2B was run as a migration standard (lane 1). (B) The contribution of USP22 to the deubiquitylation activity associated with hSAGA was assess by using shRNA to deplete USP22 from the FLAG-hGCN5 expressing cells. Control cells were infected with shRNA targeting luciferase (luc). Western blotting confirmed the purification of hGCN5 and the knockdown of endogenous USP22 within purified hSAGA (upper panels). Efficient USP22 depletion was also documented at the mRNA level using qRT-PCR (middle panel). hSAGA purified from control (luc) and USP22-depleted cells was subjected to an in vitro deubiquitylation assay with uH2B as in (A), (lower panel). Error bars represent standard deviation. (C) Recombinant USP22 was produced by baculovirus infection of insect cells and then purified via the FLAG epitope tag. The histone demethylase LSD1 was affinity purified in parallel, as a negative control. Purified proteins were detected by western blotting for the FLAG epitope (upper panel). The proteins were then subjected to an in vitro deubiquitylation assay as in A and B, using uH2B as a substrate, with H2B and uH2B detected by western blotting (lower panel).
As a final test of whether the H2B ubiquitin hydrolase activity observed in Figures 2A and 2B can be directly ascribed to USP22, recombinant human USP22 was produced and purified from baculovirus-infected insect cells. As negative controls, mock-infected cells and cells infected with an expression vector for the demethylase LSD1 were also examined. Western blotting demonstrated that relatively equal levels of USP22 and LSD1 were produced (Figure 2C). When used in the in vitro uH2B deubiquitylation assay, recombinant USP22 showed specific ubiquitin hydrolase activity towards uH2B. No enzymatic activity was observed in reactions performed using LSD1 or mock-infected cells.
USP22 is required for activator-driven transcription
The SAGA complex is broadly required for activator-driven transcription in eukaryotes (Lee and Workman, 2007). While at least part of this requirement can be ascribed to the ability of SAGA to acetylate nucleosomal histones and to stabilize TBP interactions (Dudley et al., 1999; Grant et al., 1997), we set out to examine whether USP22 activity was also important for the transcriptional coactivator role played by SAGA. In humans, the MYC oncoprotein and the p53 tumor suppressor are among the activators whose function depends on hSAGA recruitment (Barlev et al., 2001; Bouchard et al., 2001; Frank et al., 2001; McMahon et al., 2000; Wang et al., 2001). To assess whether activation of MYC target genes requires USP22, a conditional allele of MYC was utilized. In this allele, MYC is expressed as a fusion protein with a modified ligand-binding domain from the estrogen receptor (Littlewood et al., 1995). This configuration allows MYC to be activated by treatment of cells with the estrogen analog 4-hydroxytamoxifen (4-OHT). This system has been used extensively to selectively activate the transcription of MYC target genes in the absence of other cellular changes (Zhang et al., 2005). Using this system, MYC was activated in primary diploid human fibroblasts and the transcript level of various MYC target genes quantitated by real-time RT-PCR (qRT-PCR). Analysis of the MYC targets JPO1, cyclin D2, ODC, CAD and MTA1 confirmed that MYC activated their transcription as expected (Figure 3A) (Bello-Fernandez et al., 1993; Bouchard et al., 2001; Miltenberger et al., 1995; Prescott et al., 2001; Zhang et al., 2005). Notably, shRNA-mediated depletion of USP22 resulted in a decrease in the ability of MYC to activate the transcription of these targets. As above, a second USP22 shRNA yielded the same result (data not shown).
Figure 3. USP22 is required for activator-driven transcription.

(A) Normal diploid human fibroblasts were engineered to express a conditional allele of MYC that is activated by 4-OHT treatment. These cells were subjected to shRNA-mediated depletion of USP22 (shUSP22) or to control shRNA infection (shLUC). After MYC activation, mRNA levels of the indicated genes were determined by qRT-PCR. USP22 depletion efficiency was also determined by qRT-PCR (left panel). Fold induction for several MYC target genes was measured in the presence or absence of USP22 shRNA. These targets included cyclin D2 (CCND2), ODC, JPO1, CAD and MTA1. As a negative control, vimentin levels were quantified. All samples were normalized to ELF1a mRNA. (B) H1299 cells expressing a tet-regulated p53 allele were treated with USP22 or control shRNA (shLUC). p53 was induced for 8 or 24 hours by tetracycline treatment (1.0μg/ml), as indicated. mRNA levels for USP22 and the p53 target genes PUMA, p21 and PIG3 were quantitated, normalized and displayed as in (A). Error bars represent standard deviation.
In order to distinguish whether USP22 was selectively required for MYC-driven transcription, or instead more broadly required for activator-driven transcription, a conditional p53 system was utilized. Here p53 expression was rescued in the p53 null epithelial carcinoma line H1299 by activation of a tetracycline (tet)-regulated p53 allele. Upon tet treatment, p53 levels rapidly increased, resulting in increased transcription of the p53 target genes p21, PIG3 and PUMA (Figure 3B). As for MYC, USP22 depletion blocked the ability of p53 to activate the transcription of its targets. For p53, the induction of some targets was independent of USP22 activity. For example, PUMA induction by p53 occurred in both the presence and absence of USP22 (Figure 3B). This is consistent with the function of the yeast SAGA complex where some target genes require UBP8 activity and others do not (Henry et al., 2003). The two classes of SAGA targets are presumably defined by differences in their chromatin context.
MYC recruits USP22 to target gene promoters
The current model for SAGA complex function in transcription suggests that DNA sequence-specific activators interact directly with SAGA via TRRAP or other subunits and recruit it to individual target genes (Brown et al., 2001; Klein et al., 2003; Park et al., 2001). Once localized to specific genes, SAGA performs functions such as histone acetylation and TBP stabilization that are important for transcription (Lee and Workman, 2007). As the presence of the ubiquitin hydrolase USP22 within hSAGA is required for the transcription of specific MYC targets, we examined whether MYC binding to these targets results in the recruitment of USP22. To test this hypothesis, chromatin immunoprecipitation (ChIP) analysis was performed at the MYC targets CAD and MTA1, where MYC binding sites have been well characterized (Miltenberger et al., 1995; Zhang et al., 2005). H1299 cells, like the majority of epithelial cancer lines, express high levels of endogenous MYC protein (Figure 4A). MYC levels in these cells were depleted via treatment with shRNA targeting the c-myc transcript. This treatment led to a 90-95% decrease in MYC protein levels (Figure 4A, upper panel). As expected, depletion of MYC resulted in a decrease in the level of CAD and MTA1 transcription (Figure 4A) and in the level of MYC bound to the CAD and MTA1 promoters (Figure 4B and 4C). Using antibodies directed against endogenous USP22, ChIP analysis revealed that USP22 recruitment to the CAD and MTA1 promoter regions depended on the presence of MYC (Figure 4B and 4C). However, while MYC was not present at the 3′ end of these genes, USP22 was localized there in a MYC-dependent manner. These data are consistent with evidence from yeast demonstrating that the SAGA complex is recruited to promoter regions by sequence-specific activators and then migrates into the adjacent coding region (Govind et al., 2007). During this process, the activators remain tethered at the promoter while the SAGA complex travels into the body of the gene, as observed here for the CAD and MTA1 loci. It is worth noting that some transcription of CAD and MTA1 still occurs even when MYC is depleted. In addition, some USP22 is detected at the both the 5′ and 3′ ends of these genes, even in MYC-depleted cells. These two observations are consistent with other activators being responsible for some of the transcription at these loci, in part via the recruitment of hSAGA.
Figure 4. Activator-dependent recruitment of USP22 to target gene loci.

(A) H1299 cells were treated with shRNA directed against c-myc. MYC protein levels were assessed by western blotting (left panel), and qRT-PCR used to define levels of the MYC target genes CAD and MTA1 (right panel). (B) and (C) Within the CAD and MTA1 loci, binding of endogenous MYC and USP22 were evaluated by ChIP. The intron (solid line) and exon (dark boxes) structure of CAD and MTA1 are displayed. For both genes, documented MYC binding sites (labeled 5′) occur near the transcriptional start site (indicated by arrow). ChIP primer sets in the 3′ end of each locus were used to assess MYC and USP22 occupancy. As a control, nonimmune rabbit antibody (IgG) was used. The relative binding of USP22 and MYC to the CAD and MTA1 loci in the presence (white bars) or absence (black bars) of MYC shRNA is displayed. Error bars represent standard deviation.
It remained possible that the transcriptional defect observed for MYC target genes upon depletion of USP22 (Figure 3) might result from a defect in promoter binding by MYC rather than a post-binding defect in hSAGA function. In order to test this possibility, the conditional MYC/ER system was again utilized. In cells in which USP22 had been depleted, MYC was able to bind target gene loci with an affinity indistinguishable for that observed in control cells (Supplemental Figure 2). Thus, the transcriptional defect observed at MYC target loci upon USP22 depletion results from a defect in hSAGA function that is independent of an effect on the activator/DNA interaction.
USP22 is required for transformation by MYC
The enhanced transcription of MYC target genes is a critical component of malignant transformation in mammalian cells (Dang et al., 2006). Given the presence of USP22 within the polycomb/cancer stem cell signature and its role in MYC-driven transcription, we examined whether USP22 function is required for MYC-induced transformation. Telomerase-immortalized human fibroblasts that carry germline mutations in the p16 tumor suppressor locus can grow in soft agar upon introduction of the MYC oncogene (Drayton et al., 2003). Using this assay, the role of USP22 activity in MYC-mediated transformation was assessed. As expected, the parental cells demonstrated vigorous growth in soft-agar (Figure 5A). Upon depletion of USP22, the number of colonies visible by low power microscopy was inhibited by 70%, suggesting that USP22 is required for the biological activity of the MYC oncoprotein.
Figure 5. USP22 is required for MYC-mediated transformation and for appropriate cell cycle progression.

(A) After USP22 depletion, Tert-immortalized, p16 null human fibroblasts transformed by MYC, were analyzed for growth in soft agar. Luciferase shRNA-treated cells served as a control. Images show representative colony size and number at 10 days after USP22 knockdown (left panels). Colony number was also quantitiated by using microscopy to count all visible colonies, under low power in three 6 cm plates (right panel). This methodology resulted in scoring of all colonies containing > 15-20 cells. The soft agar assay was performed on three independent occasions using cells infected with separate batches of USP22 shRNA and the quantitation shown is a representative assay. Statistical analysis was performed on the colony number data from the USP22 knockdown and control groups (Student's t test), resulting in a P value of 0.001. Error bars represent standard deviation. (B) H1299 cells were infected with lentivirus expressing shRNA molecules directed against either USP22 (closed triangles) or luciferase (closed diamonds). After selection for infected cells, cells were plated in 6-well plates. Cell numbers were determined by direct counting of triplicate wells at each of the time points indicated. (C) H1299 cells from (B) were also stained with propidium iodide and cell cycle profile determined by flow cytometry.
USP22 is required for cell cycle progression
MYC is among the most potent activators of cell cycle progression encoded in the mammalian genome (Dang et al., 2006), and much of its role in cancer is based on this ability to drive proliferation. The transformation defect observed upon USP22 depletion might therefore be explained by a specific requirement for USP22 during cell cycle progression. In order to examine this possibility, proliferation assays were conducted in the H1299 cells. These cells were chosen because they lack p53, whose activation can trigger cell cycle arrest. While parental cells proliferated rapidly, shRNA-mediated depletion of USP22 resulted in a substantial defect in proliferation (Figure 5B). Doubling time for parental H1299 cells was approximately 24 hours, while H1299 cells with USP22 depletion doubled only once every 45 hours. This analysis was extended in order to define whether the requirement for USP22 in proliferation was due to a specific cell cycle phase defect. Staining of these cells for DNA content revealed that USP22 depletion resulted in the accumulation of cells in the G1 phase of the cell cycle, with concomitant decreases in the S and G2/M phases (Figure 5C). Both the proliferative defect and the G1 arrest upon USP22 depletion were observed consistently over the course of several experiments. In addition, this phenotype was also observed in normal diploid human fibroblasts (data not shown). A specific requirement for the hSAGA component USP22 in mammalian cell cycle progression is consistent with its role in the polycomb/cancer stem cell signature as an accurate predictor of aggressive tumor cell growth.
Discussion
The data presented here establish that the cancer stem cell marker USP22 is a novel enzymatic subunit of the hSAGA transcriptional cofactor complex. As suspected from its domain structure, USP22 is able to hydrolyze a ubiquitin linkage from histone H2B in vitro and endogenous USP22 contributes ubiquitin hydrolase activity to the hSAGA complex. Functionally, USP22 is required for the transcription of target genes regulated by the MYC oncoprotein and other sequence-specific activators that require hSAGA activity. We further demonstrate that USP22 is recruited by MYC to specific target gene loci and that USP22 is required for MYC-mediated transformation of mammalian cells. Finally, USP22 is also required for cell cycle progression and its depletion results in a G1 phase cell cycle arrest. These data establish that, like the other members of this small, 11-gene signature, USP22 plays a direct functional role in regulating cell cycle progression. This role likely reflects the requirement for USP22 in transcriptional activation of critical cell cycle genes. However, it is noteworthy that our data suggests that the transcription of the cell cycle inhibitor p21 also requires USP22 function. Thus the G1 arrest observed upon USP22 depletion likely reflects a complex phenotype that depends on the cumulative effect of defects in the transcription of numerous genes.
As mentioned above, two members of the USP22/cancer stem cell signature belong to the polycomb group of genes (Glinsky, 2005, 2006; Glinsky et al., 2005). Polycomb function relies in part on the ubiquitylation of histones by the BMI-1 complex (Ben-Saadon et al., 2006; Cao et al., 2005; Lessard and Sauvageau, 2003; Li et al., 2006; van der Lugt et al., 1996). This enzymatic activity, along with histone methylation catalyzed by a distinct polycomb complex (Cao et al., 2002; Kuzmichev et al., 2002), is responsible for the global transcriptional repression attributed to the polycomb group (Shilatifard, 2006). These observations suggest a model in which the simultaneous induction of polycomb ubiquitin ligases such as RNF2 and the ubiquitin hydrolase USP22 is critical during cancer progression because USP22 induction allows certain essential cell cycle genes to be transcriptionally activated in the face of the global transcriptional repression catalyzed by the BMI-1 complex. Consistent with this model, USP22 is required for MYC function and BMI-1 was originally discovered as an oncogene capable of cooperating with MYC during transformation (Haupt et al., 1991; van Lohuizen et al., 1991). It will ultimately be informative to define the distinct sets of genes regulated by BMI-1 and USP22.
The finding that depletion of USP22 results in specific defect in hSAGA function and G1 transition suggests that there is little or no apparent redundancy within the large USP family. Members of this family therefore exhibit highly restricted substrate specificity. USP44, for example, is essential during mitosis because it deubiquitylates cdc20, an inhibitor of the anaphase promoting complex (Stegmeier et al., 2007). Despite their shared requirement in cell cycle progression, neither USP22 nor USP44 can compensate for the loss of the other family member.
As mentioned, USP22 is overexpressed in aggressive tumors with other genes that are linked to the polycomb group. The requirement for USP22 in MYC function is consistent with a number of studies over the past two decades that have linked MYC and polycomb group proteins. In addition to MYC and BMI-1 cooperating to transform mammalian cells (Haupt et al., 1991; van Lohuizen et al., 1991), the Sedivy group has recently demonstrated that MYC regulates transcription of the BMI-1 gene itself, a link that ultimately results in repression of the p16(INK4a) tumor suppressor gene (Guney et al., 2006). The repression of p16(INK4a) may explain the requirement for both BMI-1 and MYC in stem cell self renewal (Honeycutt and Roop, 2004; Lessard and Sauvageau, 2003). Additional evidence for a link between MYC and polycomb function has come from studies in Drosophila. Here it has been shown on a genome-wide scale that MYC regulated genes show a remarkable overlap with those regulated by polycomb (Goodliffe et al., 2005). These studies support a model in which transcriptional activation by MYC requires that it somehow overcome the repressive effects of polycomb proteins at many of their shared target loci. The data presented here suggest that the recruitment of USP22 to MYC targets as part of the hSAGA complex may provide this critical function.
An outstanding question raised by these studies is the identity of the gene(s) whose USP22-dependent transcription is important for cell cycle progression. While cyclin D2, the HOX genes, the BMI-1 gene itself and p16(INKA) are attractive candidates, genetic studies will be required to define the essential transcriptional target(s) of USP22. Furthermore, it remains unexplained how USP22 overexpression within the polycomb/cancer stem cell is selectively achieved in the subset of human cancer that is destined to resist therapy and ultimately to metastasize.
The link established here between the metastasis marker USP22, hSAGA-dependent transcription and cell cycle progression suggests the possibility that loci regulated by USP22 may encode attractive therapeutic targets. In fact, the selective overexpression of USP22 in aggressive cancer suggests that small molecule inhibitors of the enzymatic activity of USP22 could have therapeutic value.
USP22 is one of a small set of marker genes capable of predicting metastatic potential and therapeutic outcome in human cancer (Glinsky, 2005, 2006; Glinsky et al., 2005). Remarkably, the altered expression of an 11 gene MYC pathway signature provides a predictor of cancer prognosis with accuracy similar to that provided by the USP22/BMI-1/polycomb signature (Glinsky, 2006). The studies reported here shed light on the biochemical function of the cancer stem cell marker USP22. They also expand our understanding of transcriptional regulation by demonstrating that the hSAGA histone acetyltransferase complex encodes a second enzymatic activity in the form of USP22-mediated deubiquitylation. The co-expression of both a histone ubiquitin ligase and histone ubiquitin hydolase within the 11-gene cancer stem cell signature suggests several intriguing models worthy of additional study. In addition, future studies will be focused on determining whether the requirement for USP22 in SAGA-dependent transcription depends on controlling other histone modifications or recruitment of critical components of the transcription apparatus. Of particular interest will be determining whether crosstalk exists between USP22 function and the other activities of the hSAGA complex.
Experimental Procedures
Protein purification and identification by mass spectroscopy
A stable cell line expressing a FLAG-epitope tagged version of USP22 (fUSP22) was generated in the human non-small cell lung carcinoma line H1299. In order to identify USP22 associated proteins, the FLAG-USP22 protein was affinity purified from nuclear extracts. As a negative control, mock purification was performed in parallel using nuclear extracts from parental H1299 cells. FLAG-USP22 was eluted with peptide. USP22 associated proteins were identified by liquid chromatography-tandem mass spectroscopy at the Wistar Institute Proteomics Facility. hSAGA, HBO1 and spRNAP complexes were purified using the same FLAG affinity approach.
Recombinant FLAG-tagged USP22 and LSD1 were purified from insect cells infected with baculovirus, as described previously (Huang et al., 2007). Equal concentrations of the two proteins were documented by western blotting for the shared FLAG epitope. Proteins were then utilized in deubiquitylation assays with purified uH2B as a substrate, as described below.
Cell lines, viral infection, proliferation and transformation assays
Human diploid fibroblast strain 2091, the lung cancer cell line H1299 and 293T cells were obtained from ATCC. Cell lines were maintained in 10% FBS-DMEM with penicillin and streptomycin. MYC/ER expressing cells have been described previously (Zhang et al., 2005). Where indicated, MYC/ER was activated by adding 200nM 4-OHT (Sigma). Assays for proliferation and transformation have been described previously (Zhang et al., 2005). Briefly, soft agar assays were performed using p16(INK4A) null human fibroblasts that have been immortalized by Tert and transformed by the introduction of MYC. These cells were infected with lentivrial shRNA vectors. Cells were then plated in soft agar and colony formation measured at 10 days post-plating using low powered microscopy to score all colonies containing greater that 15-20 cells. Cell cycle analysis was performed using propidium iodide staining, as described (Sykes et al., 2006).
H1299-TO-p53 cells were generated by cloning the p53 cDNA into pENTR™/D-TOPO (Invitrogen). pENTR™/D-TOPO-p53 was then recombined into the pLenti4/TO/V5-DEST vector (Invitrogen). H1299 cells stably expressing the Tet repressor were then infected with pLenti4/TO/p53 recombinant lentiviruses. All procedures were conducted according to the manufacturer's protocol (Invitrogen).
mRNA Analysis, shRNA treatment and western blotting
mRNA was harvested from cells using RNeasy (Qiagen), and converted to cDNA using SuperScript (Invitrogen). Quantitative real-time PCR was performed using SYBR Green (Applied Biosystems) as described (Zhang et al., 2005). Primer sequences are available upon request. In all cases, mRNA levels between samples were normalized to actin levels. Lentiviral shRNA plasmids corresponded to c-MYC, USP22 and control luciferase shRNA were obtained from the TRC collection (Sigma). Western blots were performed as described (Zhang et al., 2005), using antibodies to MYC (Santa Cruz), USP22, FLAG (Sigma) and Actin (Sigma). USP22 antibody was generated in rabbits and then affinity purified using the peptide FYHKQFLEYE.
Histone acetylation and deubiquitylation assays
Histone acetylation reactions were performed as described previously (McMahon et al., 2000). For deubiquitylation assays, equal volumes of hSAGA, HBO1 or USP22 complex, purified as described above, were incubated with purified ubiquitylated H2B (uH2B) isolated from porcine thymus in DUB assay buffer (100mM Tris, pH 8.0, 1mM EDTA, 1mM DTT, 5% glycerol). Following incubation at 37°C for two hours, reactions were stopped by the addition of SDS-PAGE sample buffer. Samples were then run on 4-20% SDS-PAGE gels (Invitrogen) and analyzed by Western blot with an anti-H2B antibody (Upstate).
Chromatin immunoprecipitation
ChIP analysis was conducted as described previously (Zhang et al., 2005). Briefly, cells were plated and treated with 4-OHT for 2.5 hours. Cells were then fixed in 1% formaldehyde, lysed and chromatin sheared to an average size of 500-1000 bp by sonication (Branson Model 250, USA). After immunoprecipitation using the antibodies indicated, precipitated DNA fragments were quantified using qPCR. For ChIP assays using H1299 cells, shRNA viral infection (targeting USP22 or luciferase) was performed 4 days prior to crosslinking and lysis.
Supplementary Material
Acknowledgments
We thank Tom Beer, Kaye Speicher of the Wistar Institute Proteomics Facility. In addition, we thank Alex Mazo, Ramin Shiekhattar and Amanda Norvell for helpful discussions. Immortalized human fibroblasts were generously provided by Dr. Gordon Peters. This work was supported by the grants from the NIH: CA090465 (SBM), CA098172 (SBM), and GM55360 (SLB) and NSF: MCB-9604208 (SLB). An NIH training grant supported AW (T32 GM008216). In addition, this work was partially supported by funds from the Commonwealth Universal Research Enhancement Program, Pennsylvania Department of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barlev NA, Liu L, Chehab NH, Mansfield K, Harris KG, Halazonetis TD, Berger SL. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol Cell. 2001;8:1243–1254. doi: 10.1016/s1097-2765(01)00414-2. [DOI] [PubMed] [Google Scholar]
- Bello-Fernandez C, Packham G, Cleveland JL. The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc Natl Acad Sci U S A. 1993;90:7804–7808. doi: 10.1073/pnas.90.16.7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Saadon R, Zaaroor D, Ziv T, Ciechanover A. The polycomb protein Ring1B generates self atypical mixed ubiquitin chains required for its in vitro histone H2A ligase activity. Mol Cell. 2006;24:701–711. doi: 10.1016/j.molcel.2006.10.022. [DOI] [PubMed] [Google Scholar]
- Bhaumik SR, Green MR. SAGA is an essential in vivo target of the yeast acidic activator Gal4p. Genes Dev. 2001;15:1935–1945. doi: 10.1101/gad.911401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagosklonny MV. Target for cancer therapy: proliferating cells or stem cells. Leukemia. 2006;20:385–391. doi: 10.1038/sj.leu.2404075. [DOI] [PubMed] [Google Scholar]
- Bouchard C, Dittrich O, Kiermaier A, Dohmann K, Menkel A, Eilers M, Luscher B. Regulation of cyclin D2 gene expression by the Myc/Max/Mad network: Myc-dependent TRRAP recruitment and histone acetylation at the cyclin D2 promoter. Genes Dev. 2001;15:2042–2047. doi: 10.1101/gad.907901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CE, Howe L, Sousa K, Alley SC, Carrozza MJ, Tan S, Workman JL. Recruitment of HAT complexes by direct activator interactions with the ATM-related Tra1 subunit. Science. 2001;292:2333–2337. doi: 10.1126/science.1060214. [DOI] [PubMed] [Google Scholar]
- Cao R, Tsukada Y, Zhang Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol Cell. 2005;20:845–854. doi: 10.1016/j.molcel.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- Dang CV, O'Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Seminars in cancer biology. 2006;16:253–264. doi: 10.1016/j.semcancer.2006.07.014. [DOI] [PubMed] [Google Scholar]
- Daniel JA, Torok MS, Sun ZW, Schieltz D, Allis CD, Yates JR, 3rd, Grant PA. Deubiquitination of histone H2B by a yeast acetyltransferase complex regulates transcription. J Biol Chem. 2004;279:1867–1871. doi: 10.1074/jbc.C300494200. [DOI] [PubMed] [Google Scholar]
- de Napoles M, Mermoud JE, Wakao R, Tang YA, Endoh M, Appanah R, Nesterova TB, Silva J, Otte AP, Vidal M, et al. Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev Cell. 2004;7:663–676. doi: 10.1016/j.devcel.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Drayton S, Rowe J, Jones R, Vatcheva R, Cuthbert-Heavens D, Marshall J, Fried M, Peters G. Tumor suppressor p16INK4a determines sensitivity of human cells to transformation by cooperating cellular oncogenes. Cancer Cell. 2003;4:301–310. doi: 10.1016/s1535-6108(03)00242-3. [DOI] [PubMed] [Google Scholar]
- Dudley AM, Rougeulle C, Winston F. The Spt components of SAGA facilitate TBP binding to a promoter at a post-activator-binding step in vivo. Genes Dev. 1999;13:2940–2945. doi: 10.1101/gad.13.22.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Chen T, Chadwick B, Li E, Zhang Y. Ring1b-mediated H2A ubiquitination associates with inactive X chromosomes and is involved in initiation of X inactivation. J Biol Chem. 2004;279:52812–52815. doi: 10.1074/jbc.C400493200. [DOI] [PubMed] [Google Scholar]
- Frank SR, Schroeder M, Fernandez P, Taubert S, Amati B. Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes Dev. 2001;15:2069–2082. doi: 10.1101/gad.906601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- Glinsky GV. Death-from-cancer signatures and stem cell contribution to metastatic cancer. Cell Cycle. 2005;4:1171–1175. doi: 10.4161/cc.4.9.2001. [DOI] [PubMed] [Google Scholar]
- Glinsky GV. Genomic models of metastatic cancer: functional analysis of death-from-cancer signature genes reveals aneuploid, anoikis-resistant, metastasis-enabling phenotype with altered cell cycle control and activated Polycomb Group (PcG) protein chromatin silencing pathway. Cell Cycle. 2006;5:1208–1216. doi: 10.4161/cc.5.11.2796. [DOI] [PubMed] [Google Scholar]
- Glinsky GV, Berezovska O, Glinskii AB. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Invest. 2005;115:1503–1521. doi: 10.1172/JCI23412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodliffe JM, Wieschaus E, Cole MD. Polycomb mediates Myc autorepression and its transcriptional control of many loci in Drosophila. Genes Dev. 2005;19:2941–2946. doi: 10.1101/gad.1352305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govind CK, Zhang F, Qiu H, Hofmeyer K, Hinnebusch AG. Gcn5 promotes acetylation, eviction, and methylation of nucleosomes in transcribed coding regions. Mol Cell. 2007;25:31–42. doi: 10.1016/j.molcel.2006.11.020. [DOI] [PubMed] [Google Scholar]
- Grant PA, Duggan L, Cote J, Roberts SM, Brownell JE, Candau R, Ohba R, Owen-Hughes T, Allis CD, Winston F, et al. Yeast Gcn5 functions in two multisubunit complexes to acetylate nucleosomal histones: characterization of an Ada complex and the SAGA (Spt/Ada) complex. Genes Dev. 1997;11:1640–1650. doi: 10.1101/gad.11.13.1640. [DOI] [PubMed] [Google Scholar]
- Guney I, Wu S, Sedivy JM. Reduced c-Myc signaling triggers telomere-independent senescence by regulating Bmi-1 and p16(INK4a) Proc Natl Acad Sci U S A. 2006;103:3645–3650. doi: 10.1073/pnas.0600069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt Y, Alexander WS, Barri G, Klinken SP, Adams JM. Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in E mu-myc transgenic mice. Cell. 1991;65:753–763. doi: 10.1016/0092-8674(91)90383-a. [DOI] [PubMed] [Google Scholar]
- Henry KW, Wyce A, Lo WS, Duggan LJ, Emre NC, Kao CF, Pillus L, Shilatifard A, Osley MA, Berger SL. Transcriptional activation via sequential histone H2B ubiquitylation and deubiquitylation, mediated by SAGA-associated Ubp8. Genes Dev. 2003;17:2648–2663. doi: 10.1101/gad.1144003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K, Boutilier K, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- Honeycutt KA, Roop DR. c-Myc and epidermal stem cell fate determination. The Journal of dermatology. 2004;31:368–375. doi: 10.1111/j.1346-8138.2004.tb00687.x. [DOI] [PubMed] [Google Scholar]
- Hori T, Haraguchi T, Hiraoka Y, Kimura H, Fukagawa T. Dynamic behavior of Nuf2-Hec1 complex that localizes to the centrosome and centromere and is essential for mitotic progression in vertebrate cells. J Cell Sci. 2003;116:3347–3362. doi: 10.1242/jcs.00645. [DOI] [PubMed] [Google Scholar]
- Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M, Opravil S, Shiekhattar R, Bedford MT, Jenuwein T, Berger SL. p53 is regulated by the lysine demethylase LSD1. Nature. 2007;449:105–108. doi: 10.1038/nature06092. [DOI] [PubMed] [Google Scholar]
- Ingvarsdottir K, Krogan NJ, Emre NC, Wyce A, Thompson NJ, Emili A, Hughes TR, Greenblatt JF, Berger SL. H2B ubiquitin protease Ubp8 and Sgf11 constitute a discrete functional module within the Saccharomyces cerevisiae SAGA complex. Mol Cell Biol. 2005;25:1162–1172. doi: 10.1128/MCB.25.3.1162-1172.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein J, Nolden M, Sanders SL, Kirchner J, Weil PA, Melcher K. Use of a genetically introduced cross-linker to identify interaction sites of acidic activators within native transcription factor IID and SAGA. J Biol Chem. 2003;278:6779–6786. doi: 10.1074/jbc.M212514200. [DOI] [PubMed] [Google Scholar]
- Kuo MH, Brownell JE, Sobel RE, Ranalli TA, Cook RG, Edmondson DG, Roth SY, Allis CD. Transcription-linked acetylation by Gcn5p of histones H3 and H4 at specific lysines. Nature. 1996;383:269–272. doi: 10.1038/383269a0. [DOI] [PubMed] [Google Scholar]
- Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16:2893–2905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larschan E, Winston F. The S. cerevisiae SAGA complex functions in vivo as a coactivator for transcriptional activation by Gal4. Genes Dev. 2001;15:1946–1956. doi: 10.1101/gad.911501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Kim MS, Shin JM, Park TJ, Chung HM, Baek KH. The expression patterns of deubiquitinating enzymes, USP22 and Usp22. Gene Expr Patterns. 2006;6:277–284. doi: 10.1016/j.modgep.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Lee KK, Florens L, Swanson SK, Washburn MP, Workman JL. The deubiquitylation activity of Ubp8 is dependent upon Sgf11 and its association with the SAGA complex. Mol Cell Biol. 2005;25:1173–1182. doi: 10.1128/MCB.25.3.1173-1182.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn't fit all. Nature reviews. 2007;8:284–295. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255–260. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- Li Z, Cao R, Wang M, Myers MP, Zhang Y, Xu RM. Structure of a Bmi-1-Ring1B polycomb group ubiquitin ligase complex. J Biol Chem. 2006;281:20643–20649. doi: 10.1074/jbc.M602461200. [DOI] [PubMed] [Google Scholar]
- Littlewood TD, Hancock DC, Danielian PS, Parker MG, Evan GI. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 1995;23:1686–1690. doi: 10.1093/nar/23.10.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacCallum DE, Hall PA. Biochemical characterization of pKi67 with the identification of a mitotic-specific form associated with hyperphosphorylation and altered DNA binding. Exp Cell Res. 1999;252:186–198. doi: 10.1006/excr.1999.4600. [DOI] [PubMed] [Google Scholar]
- McCleland ML, Gardner RD, Kallio MJ, Daum JR, Gorbsky GJ, Burke DJ, Stukenberg PT. The highly conserved Ndc80 complex is required for kinetochore assembly, chromosome congression, and spindle checkpoint activity. Genes Dev. 2003;17:101–114. doi: 10.1101/gad.1040903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon SB, Wood MA, Cole MD. The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol Cell Biol. 2000;20:556–562. doi: 10.1128/mcb.20.2.556-562.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miltenberger RJ, Sukow KA, Farnham PJ. An E-box-mediated increase in cad transcription at the G1/S-phase boundary is suppressed by inhibitory c-Myc mutants. Mol Cell Biol. 1995;15:2527–2535. doi: 10.1128/mcb.15.5.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutiu AI, Hoke SM, Genereaux J, Liang G, Brandl CJ. The role of histone ubiquitylation and deubiquitylation in gene expression as determined by the analysis of an HTB1(K123R) Saccharomyces cerevisiae strain. Mol Genet Genomics. 2007;277:491–506. doi: 10.1007/s00438-007-0212-6. [DOI] [PubMed] [Google Scholar]
- Ogryzko VV, Kotani T, Zhang X, Schiltz RL, Howard T, Quin J, Nakatani Y. Histone-like TAFs within the PCAF histone acetylase complex. Cell. 1998;94:35–44. doi: 10.1016/s0092-8674(00)81219-2. [DOI] [PubMed] [Google Scholar]
- Park J, Kunjibettu S, McMahon SB, Cole MD. The ATM-related domain of TRRAP is required for histone acetyltransferase recruitment and Myc-dependent oncogenesis. Genes Dev. 2001;15:1619–1624. doi: 10.1101/gad.900101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poux AN, Marmorstein R. Molecular basis for Gcn5/PCAF histone acetyltransferase selectivity for histone and nonhistone substrates. Biochemistry. 2003;42:14366–14374. doi: 10.1021/bi035632n. [DOI] [PubMed] [Google Scholar]
- Prescott JE, Osthus RC, Lee LA, Lewis BC, Shim H, Barrett JF, Guo Q, Hawkins AL, Griffin CA, Dang CV. A novel c-Myc-responsive gene, JPO1, participates in neoplastic transformation. J Biol Chem. 2001;276:48276–48284. doi: 10.1074/jbc.M107357200. [DOI] [PubMed] [Google Scholar]
- Raetz EA, Moos PJ. Impact of microarray technology in clinical oncology. Cancer investigation. 2004;22:312–320. doi: 10.1081/cnv-120030219. [DOI] [PubMed] [Google Scholar]
- Sanders SL, Jennings J, Canutescu A, Link AJ, Weil PA. Proteomics of the eukaryotic transcription machinery: identification of proteins associated with components of yeast TFIID by multidimensional mass spectrometry. Mol Cell Biol. 2002;22:4723–4738. doi: 10.1128/MCB.22.13.4723-4738.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiltz RL, Mizzen CA, Vassilev A, Cook RG, Allis CD, Nakatani Y. Overlapping but distinct patterns of histone acetylation by the human coactivators p300 and PCAF within nucleosomal substrates. J Biol Chem. 1999;274:1189–1192. doi: 10.1074/jbc.274.3.1189. [DOI] [PubMed] [Google Scholar]
- Shilatifard A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem. 2006;75:243–269. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- Stegmeier F, Rape M, Draviam VM, Nalepa G, Sowa ME, Ang XL, McDonald ER, 3rd, Li MZ, Hannon GJ, Sorger PK, et al. Anaphase initiation is regulated by antagonistic ubiquitination and deubiquitination activities. Nature. 2007;446:876–881. doi: 10.1038/nature05694. [DOI] [PubMed] [Google Scholar]
- Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006;24:841–851. doi: 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne AW, Sautiere P, Briand G, Crane-Robinson C. The structure of ubiquitinated histone H2B. Embo J. 1987;6:1005–1010. doi: 10.1002/j.1460-2075.1987.tb04852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valk-Lingbeek ME, Bruggeman SW, van Lohuizen M. Stem cells and cancer; the polycomb connection. Cell. 2004;118:409–418. doi: 10.1016/j.cell.2004.08.005. [DOI] [PubMed] [Google Scholar]
- van der Lugt NM, Alkema M, Berns A, Deschamps J. The Polycomb-group homolog Bmi-1 is a regulator of murine Hox gene expression. Mechanisms of development. 1996;58:153–164. doi: 10.1016/s0925-4773(96)00570-9. [DOI] [PubMed] [Google Scholar]
- van Lohuizen M, Verbeek S, Scheijen B, Wientjens E, van der Gulden H, Berns A. Identification of cooperating oncogenes in E mu-myc transgenic mice by provirus tagging. Cell. 1991;65:737–752. doi: 10.1016/0092-8674(91)90382-9. [DOI] [PubMed] [Google Scholar]
- Voncken JW, Roelen BA, Roefs M, de Vries S, Verhoeven E, Marino S, Deschamps J, van Lohuizen M. Rnf2 (Ring1b) deficiency causes gastrulation arrest and cell cycle inhibition. Proc Natl Acad Sci U S A. 2003;100:2468–2473. doi: 10.1073/pnas.0434312100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Kobayashi T, Takimoto R, Denes AE, Snyder EL, el-Deiry WS, Brachmann RK. hADA3 is required for p53 activity. Embo J. 2001;20:6404–6413. doi: 10.1093/emboj/20.22.6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicha MS. Cancer stem cells and metastasis: lethal seeds. Clin Cancer Res. 2006;12:5606–5607. doi: 10.1158/1078-0432.CCR-06-1537. [DOI] [PubMed] [Google Scholar]
- Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, Laird PW. Epigenetic stem cell signature in cancer. Nat Genet. 2007;39:157–158. doi: 10.1038/ng1941. [DOI] [PubMed] [Google Scholar]
- Zhang XY, DeSalle LM, Patel JH, Capobianco AJ, Yu D, Thomas-Tikhonenko A, McMahon SB. Metastasis-associated protein 1 (MTA1) is an essential downstream effector of the c-MYC oncoprotein. Proc Natl Acad Sci U S A. 2005;102:13968–13973. doi: 10.1073/pnas.0502330102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.