

Abstract
We assessed the role of nitric oxide (NO) and the kinin B2 receptor in mediating tissue kallikrein’s actions in intramyocardial inflammation and cardiac remodeling after ischemia/reperfusion (I/R) injury. Adenovirus carrying the human tissue kallikrein gene was delivered locally into rat hearts 4 days prior to 30-minute ischemia followed by 24- hour or 7-day reperfusion with or without administration of icatibant, a kinin B2 receptor antagonist, or N(ω)-nitro-L-arginine methyl ester (L-NAME), a nitric oxide synthase inhibitor. Kallikrein gene delivery improved cardiac contractility and diastolic function, reduced infarct size at 1 day after I/R without affecting mean arterial pressure. Kallikrein treatment reduced macrophage/monocyte and neutrophil accumulation in the infarcted myocardium in association with reduced intercellular adhesion molecule-1 levels. Kallikrein increased cardiac endothelial nitric oxide synthase phosphorylation and NO levels and decreased superoxide formation, TGF-β1 levels and Smad2 phosphorylation. Furthermore, kallikrein reduced I/R-induced JNK, p38MAPK, IκB-α phosphorylation and nuclear NF-κB activation. In addition, kallikrein improved cardiac performance, reduced infarct size and prevented ventricular wall thinning at 7 days after I/R. The effects of kallikrein on cardiac function, inflammation and signaling mediators were all blocked by icatibant and L-NAME. These results indicate that tissue kallikrein through kinin B2 receptor and NO formation improves cardiac function, prevents inflammation and limits left ventricular remodeling after myocardial I/R by suppression of oxidative stress, TGF-β1/Smad2 and JNK/p38MAPK signaling pathways and NF-κB activation.
Keywords: cardiac remodeling, inflammation, ischemia/reperfusion, nitric oxide, tissue kallikrein
Introduction
Left ventricular (LV) remodeling after myocardial ischemia-reperfusion (I/R) is a dynamic and complex biological process that is preceded by apoptosis and inflammation. Myocardial I/R injury induces upregulation of inflammatory cytokines in the infarcted heart. These cytokines and the consecutive inflammatory responses are considered to be essential in the early phase post-infarction for scar formation and tissue repair (Li et al., 2005). The inflammatory cascade is a prerequisite for healing of the infarcted myocardium as it aids in the removal of irreversibly injured cells. However, optimal repair and favorable ventricular remodeling requires timely suppression of inflammation and containment of granulation tissue formation in the infarcted heart. Therefore, inhibition of intramyocardial inflammation and pathological remodeling after myocardial I/R injury is essential for the treatment of ischemic cardiomyopathy and heart failure.
Nitric oxide (NO) modulates many processes that contribute to LV performance. It has been reported that overexpression of endothelial nitric oxide synthase (eNOS) improves cardiac function and limits deleterious left ventricular remodeling after myocardial infarction (MI) (Scherrer-Crosbie et al., 2001). NO has also been shown to inhibit TGF-β1/Smad transactivation in endothelial cells (Saura et al., 2005). As upregulation of TGF-β1 expression after I/R may contribute to the accentuation and expansion of the post-infarction inflammatory response, suppression of the TGF-β1/Smad signaling pathway would lead to a reduction in I/R-induced fibrosis and adverse LV remodeling (Frangogiannis et al., 2005). Therefore, NO-mediated inhibition of TGF-β1 expression could play a crucial role in modulating intramyocardial inflammation and cardiac remodeling. Moreover, mitogen-activated protein kinases (MAPKs) are part of the TGF-β1 signaling pathway (Matsumoto-Ida et al., 2006; Engel et al., 1996) and p38MAPK and c-Jun NH2-terminal kinase (JNK) have been shown to be activated in the heart under ischemic conditions (Kaiser et al., 2005 and 2004). In addition, activation of the transcription factor nuclear factor-κB (NF-κB) after p38MAPK phosphorylation is sufficient to trigger transcriptional activation of inflammatory cytokine expression and release in vivo and in vitro (Matsuyama et al., 2004; Zhao et al., 2001). Taken together, these results indicate that induction of the inflammatory responses by stimulation of TGF-β and MAPK activities and thus NF-κB activation can potently affect cardiac chamber remodeling and contractile dysfunction.
Tissue kallikrein cleaves kininogen substrate to produce kinin peptides. Intact kinins bind to kinin B2 receptor and result in increased nitric oxide (NO), cGMP and cAMP formation which trigger a broad spectrum of biological responses (Regoli et al., 1990). A recent study showed that diabetes-induced intramyocardial inflammation was suppressed by overexpression of tissue kallikrein in transgenic rats (Tschope et al., 2005). In addition, we demonstrated that kallikrein gene delivery reduces inflammatory cell infiltration in association with increased NO levels in the brain and kidney after ischemic stroke or salt-induced renal injury by the kinin B2 receptor (Bledsoe et al., 2006; Xia et al., 2006). Moreover, our previous study showed that kallikrein gene delivery inhibited I/R-induced myocardial apoptosis accompanied by increased NO and cGMP levels (Yin et al., 2005; Yoshida et al., 2000). Based on these findings, we hypothesize that kallikrein, via NO formation and kinin B2 receptor activation, improves cardiac function by inhibiting intramyocardial inflammation and limiting left ventricular remodeling through inhibition of NF-κB activation after myocardial I/R injury.
Materials and Methods
Preparation of replication-deficient adenoviral vectors
Adenoviral vector harboring the human tissue kallikrein cDNA (Ad.CMV-TK) under the control of the cytomegalovirus (CMV) enhancer/promoter or adenoviral vector alone (Ad.Null) were constructed and prepared as previously described (Chen et al., 1997).
Catheter-based gene delivery
Wistar rats (male, 240 to 260 g, Sprague-Dawley Harlan, Indianapolis, IN) were employed in this study. All procedures complied with the standards for care and use of animal subjects as stated in the Guide for the Care and Use of Laboratory Animals (National Academy of Sciences, Bethesda, MD). Four days before coronary artery occlusion, 200 μl adenoviral solution (2 × 1010 pfu/ml in PBS) were delivered using a catheter-based strategy by intracoronary injection of adenovirus as previously described (Yin et al., 2005). Expression and localization of human tissue kallikrein in rat ventricles after gene delivery were identified immunohistochemically.
Myocardial ischemia/reperfusion (I/R)
Myocardial I/R surgery was performed four days after gene delivery as previously described (Yin et al., 2005). Rats were anesthetized, intubated, and mechanically ventilated. Myocardial infarction was established by occlusion of left anterior descending coronary artery for 30 minutes. The study was divided into short-term (24 hours) and long-term (7 days) reperfusion duration. In the short-term study, rats were randomly divided into seven groups. The sham groups, in which the chest was opened but did not undergo I/R surgery consisted of saline (n=6) or Ad.CMV-TK injection (n=6). The rats undergoing I/R surgery were divided into five groups. Control group (n=6) was injected with saline, the second group received Ad.Null (n=7) and the third group received Ad.CMV-TK (n=7). The fourth group received Ad.CMV-TK together with administration of kinin B2 antagonist, icatibant, delivered IP by osmotic mini-pump (Alza) at 1 μmol/kg body weight per day (Ad.CMV-TK/icatibant, n=6). The fifth group was injected with Ad.CMV-TK followed by L-NAME administration (35 mg/kg IV injection twice, at 15 minutes before reperfusion and 12 hours after reperfusion) (Ad.CMV-TK/L-NAME, n=6). In the long-term study, there were five groups including sham (n=5), I/R receiving saline (n=6), Ad.Null (n=7), Ad.CMV-TK (n=8) or Ad.CMV-TK followed by L-NAME delivery (35 mg/kg IV injection 15 minutes before reperfusion followed by IP implantation of osmotic mini-pumps at 2 mg/hr/kg (n=7).
Cardiac function measurements
Upon the completion of reperfusion, the carotid artery was cannulated by advancing PE-50 tubing (Becton Dickingson) to the left ventricle. Heart rate (HR), mean arterial pressure (MAP), left ventricular end-diastolic pressure (LVEDP), left ventricular end-systolic pressure (LVESP), cardiac contractility (dP/dt maximum and dP/dt minimum) and time constant of isovolumic relaxation (Tau) were recorded and analyzed by a polygraph system (BIOPAC).
Myocardial infarct size determination
After cardiac function measurements, the LAD was reoccluded and 1 ml 1% Evan’s blue was injected into the apex to stain the non-infarcted area. The heart was then isolated, perfused with PBS and sectioned rapidly in the middle of left ventricle along the long axis. Heart sections (1 mm) were then incubated in 1.5% 2,3,5-triphenyltetrazolium chloride (TTC) (Sigma) for 5 minutes at 37°C to determine the infarcted area. Each myocardial slice was quantified for area at risk (AAR) and infarcted area (IA) using computer-assisted planimetry (NIH Image 1.57).
Histological analysis
The myocardial specimens at the end of reperfusion were fixed in 4% paraformaldehyde and embedded in paraffin, after which 4-μm-thick sections were stained with hematoxylin-eosin (H & E) and Masson’s trichrome. Inflammatory cells (macrophages/monocytes) in the heart were stained with anti-ED-1 antibody (Chemicon). Quantitative analysis of macrophages/monocytes and neutrophils (assessed by H & E staining) were performed on 10 randomly chosen high-power fields of the infarct region in each section.
Western blot
Western blot analysis was performed using the cytosolic fraction of left venctricle extracts to detect the total and/or phosphorylated forms of eNOS, p38MAPK, JNK, Smad2, phospho-IκB-α (Ser32), IκB-α (Cell Signaling), ICAM-1 and TGF-β1 (Santa Cruz). Membranes were incubated with secondary antibody conjugated to LumiGLO chemiluminescent reagent. Chemiluminescence was detected using an ECL-Plus kit (Perkin Elmer) and visualized by Kodak X-ray film. Phosphorylated and corresponding total protein bands were quantified by optical densitometry.
Nitrate/nitrite and superoxide assays
Nitrate/nitrite (NOx) levels in cardiac extracts, an indicator of NO levels, were measured by a fluorometric assay as previously described (Yoshida et al., 2000). Superoxide levels were measured by a spectrophotometric assay based on rapid reduction of ferricytochrome c to ferrocytochrome c by mixing cytochrome c (810 μM) with cardiac extracts. Non-superoxide-dependent reduction of cytochrome c was corrected for by deducting the activity not inhibited by adding 50 units of superoxide dismutase into the reaction mixture (Xia et al., 2006).
Electrophoretic mobility shift assay (EMSA) for NF-κB activation
Nuclear proteins were isolated from cardiac extracts as previously described (Zhao et al., 2001). NF-κB DNA binding site oligonucleotides AGTTGAGGGGACTTTCCCAGGC) (Integrated DNA Technologies) were labeled using the Biotin 3’ End DNA Labeling kit (Pierce) according to the manufacturer’s protocol. The gel shift assay was performed using the Light Shift Chemiluminescent electrophoretic mobility shift analysis kit (Pierce) following the manufacturer’s instructions. No-specific binding was determined by adding 200-fold molar excess of unlabeled NF-κB DNA. The relative nuclear NF-κB-DNA binding activities were quantified by scanning densitometry.
Statistical analysis
Data were expressed as mean ± SEM and were compared between experimental groups with the use of one-way ANOVA followed by Fisher’s PLSD. Probability values of P<0.05 were considered statistically significant.
Results
Expression of human kallikrein reduces infarct size and improves cardiac function
Expression and localization of immunoreactive human tissue kallikrein were identified in the infarcted area of the left ventricle at 1 and 7 days after myocardial I/R (5 and 11 days after local gene delivery) (Figure 1A). No specific staining was found in the left ventricle of sham rats. Representative TTC staining and quantitative analysis showed that kallikrein gene transfer significantly reduced infarct size in the left ventricle compared with I/R control groups with or without injection of Ad.Null (21.5 ± 6.1% vs. 49.7 ± 9.2 % and 49.1 ± 8.2%, n=6 to 7, P<0.01). Both icatibant and L-NAME abrogated kallikrein’s effects (47.8 ± 6.3 % and 51.5 ± 5.3% vs. 21.5 ± 6.1%, n=6 to 7, P<0.01) (Figure 1B & C). No alterations were observed in left ventricular infarct thickness among all the groups subjected to I/R (data not shown).
Figure 1.
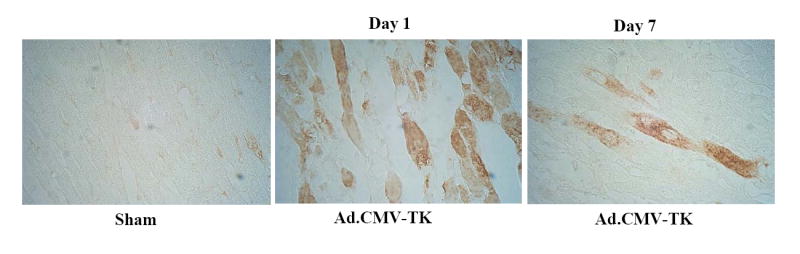

Myocardial infarct size at 1 day after I/R. (A) Representative photomicrographs show immunohistochemical identification of human tissue kallikrein in the infracted area of rats at 1 and 7 days after I/R (5 and 11 days after local gene delivery). Original magnification is 400×. (B) Representative photomicrographs show ventricular infarct size stained with TTC. (C) Quantitative analysis of infarct size from I/R-injured cardiac sections. Data are expressed as mean ± SEM., n=6-7. *P<0.01 vs. other groups.
Table 1 shows the effects of kallikrein on cardiac function after 24-hour reperfusion. HR and MAP were not changed among all the groups. Characteristic impairments in contractility (dP/dtmax) and diastolic function (LVEDP, dP/dtmin, Tau) after I/R were significantly improved after kallikrein gene delivery, and kallikrein’s effects were blocked by icatibant and L-NAME administration. These results indicate that kallikrein, mediated by kinin B2 receptor and NO, improves cardiac function independent of blood pressure reduction at the early stage after myocardial I/R.
Table 1.
Hemodynamic Parameters at 1 Day after Ischemia-Reperfusion
| I/R
|
|||||||
|---|---|---|---|---|---|---|---|
| Sham | Control | Ad.Null | Ad.TK | Ad.TK/Icatibant | Ad.TK/L-NAME | ||
| HR, bpm | 384 ± 24.3 | 377 ± 31.4 | 386 ± 36.9 | 374 ± 26.5 | 388 ± 30.6 | 368 ± 27.3 | |
| MAP, mmHg | 106.3 ± 4.9 | 84.4 ± 4.3 | 83.0 ± 6.9 | 97.2 ± 6.5* | 85.4 ± 6.6 | 82.8 ± 5.3 | |
| LVEDP, mmHg | 2.6 ± 0.3 | 11.6 ± 1.1 | 13.4 ± 0.9 | 6.6 ± 0.6* | 12.7 ± 1.9 | 12.6 ± 1.6 | |
| dP/dtmax, mmHg/s | 4226.6 ± 90.1 | 3140.0 ± 76.2 | 3019.8 ± 104.6 | 3620.8 ± 113.5* | 3038.0 ± 73.8 | 3018.8 ± 158.7 | |
| dP/dtmin, mmHg/s | 3593.6 ± 87.8 | 2393.4 ± 111.2 | 2212.2 ± 81.4 | 3002.8 ± 113.0* | 2452.4 ± 85.2 | 2356.2 ± 162.4 | |
| Tau, ms | 1.88 ± 0.2 | 2.9 ± 0.3 | 3.2 ± 0.2 | 1.6 ± 0.2* | 3.1 ± 0.4 | 3.34 ± 0.2 | |
HR indicates heart rate; bpm, beats per minute; MAP, mean arterial pressure; LVEDP, left ventricular end-diastolic pressure; dP/dtmax, maximum first derivative of pressure; dP/dtmin minimun first derivative of pressure; Tau, time constant of isovolumic relaxation; ms, milisecond.
P<0.01 vs. other I/R groups
Kallikrein improves ventricular remodeling
We further evaluated the effects of kallikrein on ventricular remodeling at 7 days after reperfusion. Masson’s trichrome staining showed characteristic impaired remodeling of enlarged ventricular infarct size and LV free wall thinning after I/R, and kallikrein prevented the deleterious remodeling (Figure 2A). Quantitative analysis showed that kallikrein significantly reduced infarct size and restored LV wall thinning (31.6 ± 2.7% vs. 45.3 ± 3.3 % for infarct size; 2.4 ± 0.33 mm vs. 1.05 ± 0.0 8mm for wall thickness, n= 6 to 8, P<0.01) (Figure 2B & C). Both effects were blocked by L-NAME administration (43.8 ± 4.0% vs. 31.6 ± 2.7% for infarct size; 1.4 ± 0.21mm vs. 2.4 ± 0.33mm for wall thickness, n= 7 to 8, P<0.01). Measurements of hemodynamic parameters showed impaired contractility (dP/dtmax), and diastolic function (LVEDP, dP/dtmin and Tau) along with reduced heart rate and MAP at 7 days after I/R (Table 2). These parameters were worsened compared to those at 1 day after I/R. Kallikrein prevented deterioration of both systolic and diastolic function in conjunction with ameliorated LV remodeling, and the effects were abrogated by L-NAME. The results indicate that kallikrein, through NO formation, effectively improved cardiac function, reduced infarct size and prevented impaired ventricular remodeling at the late stage after I/R injury.
Figure 2.
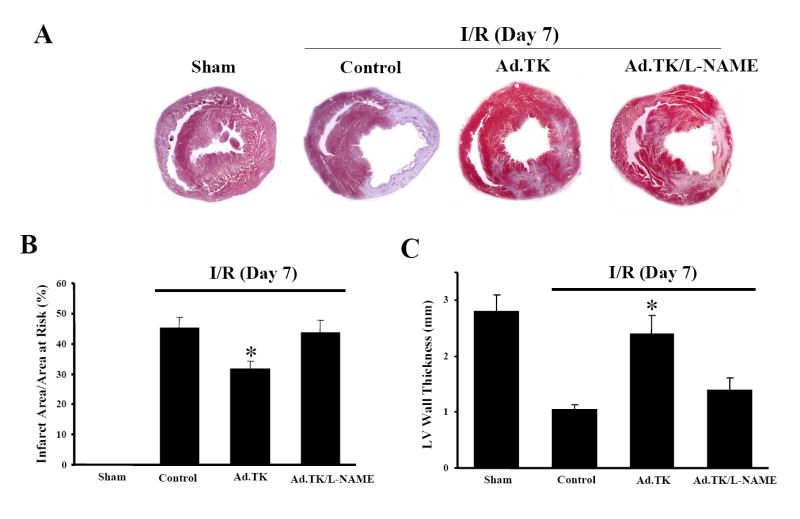
Ventricular remodeling at 7 days after I/R. (A) Representative photomicrographs show ventricular sections stained with Masson’s trichrome. (B) Quantitative analysis of ventricular infarct size. (C) Left ventricular wall thickness. Data are expressed as mean ± SEM., n = 5-8. *P<0.01 vs. other I/R groups. Original magnification is 10×.
Table 2.
Hemodynamic Parameters at 7 Days after Ischemia-Reperfusion
| I/R
|
||||||
|---|---|---|---|---|---|---|
| Sham | Control | Ad.Null | Ad.TK | Ad.TK/L-NAME | ||
| HR, bpm | 390 ± 9.8 | 331 ± 12.7 | 319 ± 8.7 | 363 ± 9.6* | 315 ± 10.6 | |
| MAP (mmHg) | 101.3 ± 4.7 | 70.5 ± 5.6 | 73.5 ± 5.9 | 89.2 ± 6.7* | 68.4 ± 5.8 | |
| LVEDP, mmHg | 3.4 ± 0.6 | 17.8 ± 1.7 | 17.3 ± 1.7 | 11.1 ± 0.7* | 16.8 ± 1.4 | |
| dP/dtmax, mmHg/s | 4130.8 ± 61.3 | 2623.3 ± 106.8 | 2567.4 ± 97.0 | 3019.0 ± 106.6* | 2484.9 ± 104.4 | |
| dP/dtmin, mmHg/s | 3527.8 ± 71.7 | 2043.3 ± 104.4 | 1887.3 ± 104.7 | 2348.9 ± 125.4* | 1933.7 ± 80.9 | |
| Tau, ms | 2.02 ± 0.3 | 8.2 ± 0.8 | 7.3 ± 0.6 | 3.9 ± 0.5* | 7.1 ± 0.8 | |
HR indicates heart rate; bpm, beats per minute; MAP, mean arterial pressure; left ventricular end-diastolic pressure; dP/dtmax, maximum first derivative of pressure; dP/dtmin minimun first derivative of pressure; Tau, time constant of isovolumic relaxation; ms, milisecond.
P<0.01 vs. other I/R groups
Kallikrein attenuates inflammation after I/R
At 24 hours after I/R, ED-1 immunostaining, a marker of macrophages/monocytes, was markedly increased in the infarcted heart with or without injection of Ad.Null, whereas kallikrein gene delivery reduced I/R-induced intramyocardial macrophage/monocyte accumulation (Figure 3A). Quantitative analysis showed that kallikrein gene delivery significantly reduced monocyte/macrophage infiltration compared with the Ad.Null group (38.8 ± 5.6 vs 67.1 ± 9.1 cell/mm2, n=6 to 7, P<0.01) (Figure 3B). Similarly, quantitative analysis from H & E staining showed that kallikrein reduced I/R-induced neutrophil recruitment into the infarcted heart compared with the control group (124.3 ± 16.6 vs. 263.8 ± 38.3 cell/mm2, n=6 to 7, P<0.01) (Figure 3C). Local injection of Ad.Null or Ad.CMV-TK into sham rat hearts without undergoing ischemic surgery did not induce notable ED-1-positive cell infiltrates (data not shown), indicating that the observed inflammatory cell accumulation after myocardial I/R was not attributed to adenovirus injection alone. Furthermore, inhibition of inflammatory cell infiltration by kallikrein was accompanied by reduced ICAM-1 levels (Figure 3D). Kallikrein’s effects on I/R-induced inflammatory responses and ICAM-1 levels were abrogated by icatibant and L-NAME (Figure 3 A-D). In addition, kallikrein reduced I/R-induced monocyte chemoattractant protein-1 mRNA levels and the effect was blocked by icatibant and L-NAME (data not shown). Collectively, our results demonstrate that inflammatory infiltrates after myocardial I/R is reduced by kallikrein gene transfer, and that kallikrein’s anti-inflammatory effect is mediated by the kinin B2 receptor and NO formation.
Figure 3.
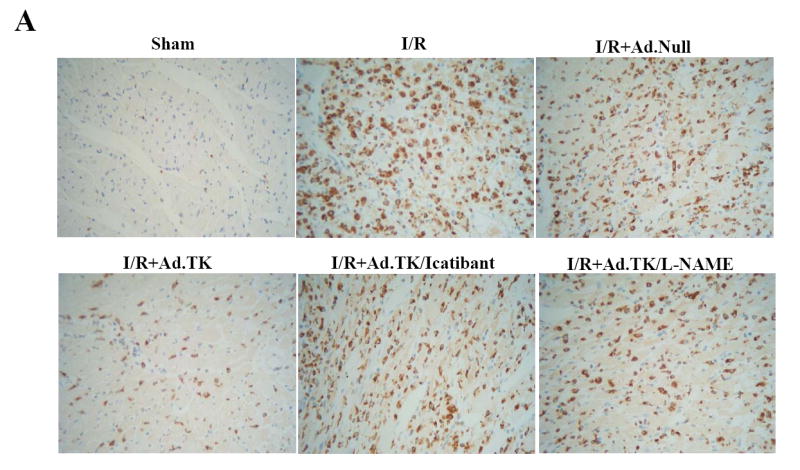

Effects of kallikrein on myocardial inflammation at 1 day after I/R. (A) Representative photomicrographs show ventricular sections stained with ED-1 antibody. Original magnification is 400×. (B) Quantitative analysis of monocyte/macrophage number. (C) Quantitative analysis of neutrophil number. (D) Western blot analysis of ICAM-1 levels. Data are expressed as mean ± SEM., n = 6-7. *P<0.01 vs. other I/R groups.
Effects of kallikrein on NOx levels and superoxide formation
Since endothelial NOS catalyzes NO formation, we evaluated the effect of kallikrein on eNOS activation and NO levels. Western blot analysis showed that I/R reduced eNOS phosphorylation but was restored by kallikrein gene delivery. Total eNOS levels were not altered among all the groups (Figure 4 A). Kallikrein-induced eNOS activation was accompanied by increased cardiac NOx levels in the infarcted heart compared to rats after I/R with or without Ad.Null injection (0.49 ± 0.08 vs. 0.18 ± 0.02 or 0.24 ± 0.03 μmol/mg, n=6 to 7, P<0.01) (Figure 4B). On the other hand, superoxide formation was significantly inhibited by kallikrein (378.2 ± 46.4 vs. 723.1 ± 32.4 or 674.3 ± 79.3 nmol/min/mg, n=6 to 7, P<0.01) (Figure 4C). These effects were all blocked by L-NAME and by icatibant. The results indicate that kallikrein through kinin B2 receptor activation increases NO levels by promoting eNOS activation, and inhibits I/R-induced superoxide formation in the infarcted heart.
Figure 4.

Effects of kallikrein on cardiac eNOS phosphorylation, NOx levels and superoxide formation at 1 day after I/R. (A) Western blot analysis of phosphorylated and total eNOS. (B) Cardiac NOx levels. (C) Superoxide formation. Data are expressed as mean ± SEM., n=6-7. *P<0.01 vs. other groups.
Kallikrein attenuates TGF-β1 levels, Smad2 and MAPK phosphorylation
Western blot analysis showed that cardiac TGF-β1 levels were dramatically increased after 24-hour I/R and the effect was inhibited by kallikrein treatment. Since Smad2 is a critical intracellular mediator that transmits TGF-β signals from cytoplasm to nucleus, we further evaluated Smad2 phosphorylation. Similar to TGF-β1, increased phosphorylation of Smad2 after I/R was reduced by kallikrein. Kallikrein’s effects were blocked by icatibant and L-NAME, indicating that TGF-β1 expression is a downstream signaling event of kallikrein-kinin and NO (Figure 5A). In addition, kallikrein reduced I/R-induced JNK and p38MAPK phosphorylation. Again, kallikrein’s effects on JNK and p38MAPK activation were blocked by icatibant and L-NAME (Figure 5B). No alterations of these signaling mediators were observed in sham rats after kallikrein gene transfer. Taken together, these data indicate that NO mediates the protective effect of kallikrein through the kinin B2 receptor by suppressing of TGF-β/Smad2 and JNK/p38MAPK activation after myocardial I/R injury.
Figure 5.

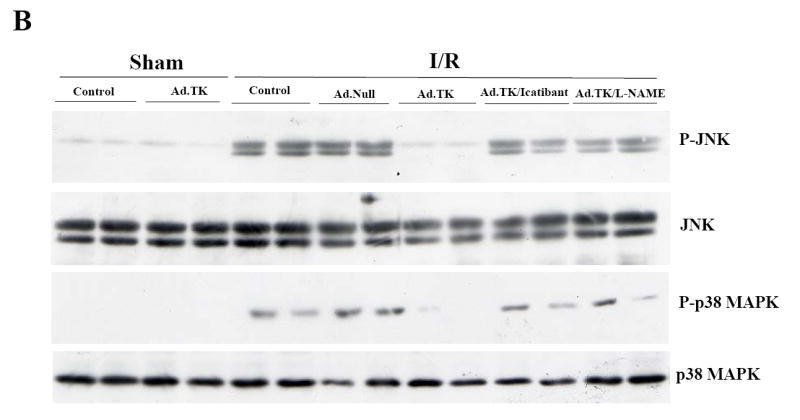
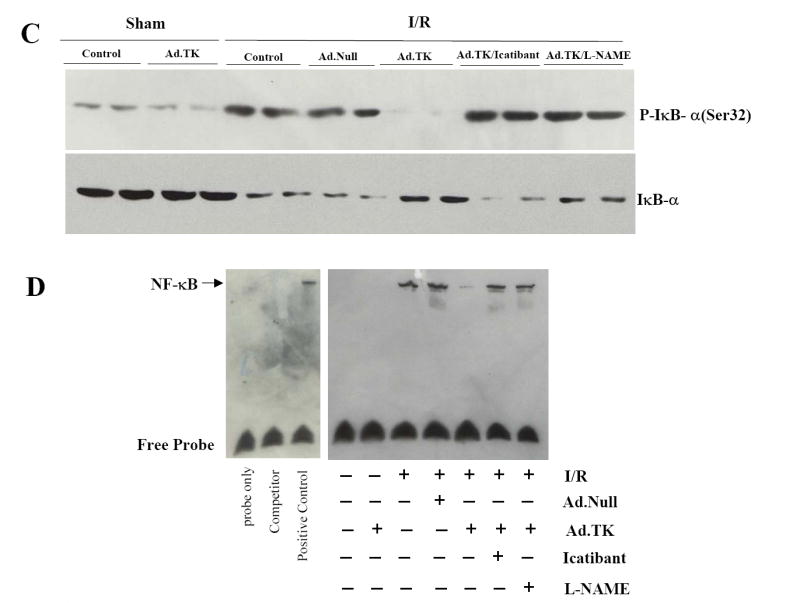
Effects of kallikrein on MAPK phosphorylation and NF-κB translocation at 1 day after I/R. Western blot analysis of (A) cardiac TGF-β levels and Smad2 phosphorylation, (B) JNK and p38MAPK phosphorylation, and (C) IκB-α (Ser 32) phosphorylation. (D) Nuclear NF-κB-DNA EMSA.
Kallikrein inhibits NF-κB translocation
NF-κB is a primary element in regulation of tissue damage in conjunction with oxidative stress. We further investigated the effect of kallikrein on nuclear translocation and activation of NF-κB. Western blot analysis showed that I/R increased IκB-α (Ser32) phosphorylation with or without Ad.Null, and the effect was inhibited by kallikrein (Figure 5C). Conversely, kallikrein increased IκB-α levels after I/R, and icatibant and L-NAME blocked kallikrein’s effect. These results indicate that I/R activated IκB-α phosphorylation, resulting degradation of IκB-α in the cytosol, thus leading to the release and nuclear translocation of NF-κB. EMSA showed that I/R induced a marked increase of NF-κB DNA-binding activity, and kallikrein abrogated the effect (Figure 5D). Again, kallikrein’s inhibitory effect on NF-κB nuclear translocation and activation was reversed by icatibant and L-NAME, indicating that NO mediated the effect of kallikrein-kinin in inhibiting NF-κB activation.
Discussion
This is the first study to demonstrate that NO mediates the effect of tissue kallikrein in improving cardiac function and limiting post-infarction remodeling through inhibition of inflammation. It is intriguing to observe that kallikrein, through the kinin B2 receptor, markedly attenuated I/R-induced inflammatory responses. Contrary to our results, kinins and their receptors are well-known inducers of pro-inflammatory actions (Calixto et al., 2000). One potential explanation for this discrepancy is that kinin B1 and B2 receptors have differentiated functions in I/R tissue injury. Using a model of intestinal I/R, Souza et al. reported that neutrophil migration in inflamed tissues after I/R injury were significantly reduced in kinin B1 receptor-deficient mice, and pretreatment of B2 receptor antagonist reversed this anti-inflammatory effect (Souza et al., 2004). These results indicate that kinin B2 receptor may prevent exacerbated tissue injury in the absence or with very low levels of the B1 receptor. In support of this notion, our recent reports showed that kallikrein gene delivery reduces inflammatory cell infiltrates in the ischemic brain and salt-induced injured kidney (Bledsoe et al., 2006; Xia et al., 2006). Our present study provides new evidence of a kinin B2 receptor-mediated anti-inflammatory effect of kallikrein in myocardial I/R injury. Reduction of excess inflammatory infiltrates by kallikrein could contribute to the repair and remodeling at the late stage after myocardial I/R injury.
NO, known to be a potent vasodilator and antioxidant, has been shown to abolish mitochondrial oxidant damage in adult rat cardiomyocytes (Xu et al., 2005). NO is capable of inhibiting neutrophil superoxide anion production via a direct action on the membrane components of the NADPH oxidase and the assembly of NADH/NADPH oxidase subunits (Clancy et al., 1992; Fujii et al., 1997). In eNOS knockout mice, increased neutrophil infiltration in the I/R myocardium was found to be associated with higher ICAM-1 levels, whereas increased NO formation resulted in reduction of inflammatory reactions and infarct size (Yamakuchi et al., 2005). In addition, the protective effect of NO has been attributed to inhibition of NF-κB activation after injury (Iwata et al.,2001). Kinin is capable of generating NO by increased phosphorylation of eNOS (Chatterjee et al., 2003). Similarly, tissue kallikrein gene transfer has been shown to promote muscular neovascularization by upregulation of eNOS and activation of Akt (Emanueli et al., 2004), as well as to protect against cardiac remodeling through reduction of oxidative stress after MI (Smith et al., 2005). Our present finding showed that icatibant and L-NAME abolished kallikrein’s effects in promoting eNOS activation and NOx (an indicator of NO) levels, and suppressing superoxide production in the infarcted heart. These results indicate that kallikrein/kinin is capable of improving cardiac function through increased NO formation.
The expression of TGF-β1, a multi-functional cytokine, is upregulated after I/R and thus may result in accentuation and expansion of the post-infarction inflammatory reaction, leading to extensive fibrosis and adverse LV remodeling (Frangogiannis et al., 2005). NO has been shown to play a role in modulating TGF-β1 expression as eNOS knockout mice exhibit enhanced TGF-β1 and collagen I expression (Saura et al., 2005). Anti-MCP-1 gene therapy prevented post-MI failure through inhibition of TGF-β1, and gene therapy against TGF-β1 significantly attenuated LV dilation and fibrous formation (Hayashidani et al., 2003; Okada et al., 2005). This is consistent with our current finding that kallikrein reduced I/R-induced TGF-β1 levels and Smad2 phosphorylation. Smad2 activation is the initial and critical step to activate TGF-β1 signaling. Smad2 and Smad3, once phosphorylated, form a complex with Smad4 and translocate into the nucleus to regulate gene expression (Abdollah et al., 1997). Our results indicate that NO mediates the effect of kallikrein in suppression of TGF-β/Smad 2 activation, thereby improving ventricular remodeling.
MAPKs are activated by different cellular stress signals, such as oxidative stress and inflammatory cytokines, which play a vital role in cardiomyocyte apoptosis, inflammation and myocardial injury. The activation of MAPK has been linked to the increased activation of the transcription factor NF-κB. NF-κB is considered to be an oxidative stress-responsive transcription factor regulating the expression of molecules involved in inflammatory and immune responses. Furthermore, lipopolysaccharide (LPS)-mediated activation of JNK, p38MAPK and NF-κB resulted in uncontrolled production of proinflammatory cytokines (Lim et al., 2002). Our present study showed that kallikrein, via NO production, reduced p38MAPK and JNK activation and inhibited NF-κB nuclear translocation and activation. This study implicates that p38MAPK, JNK and NF-κB are responsible for the post-infarction inflammatory actions in the early phase of I/R.
In summary, our data show that kallikrein, through kinin B2 receptor activation, improves cardiac function after myocardial I/R injury by suppression of intramyocardial inflammation and improving deteriorated post-infarct ventricular remodeling. Kallikrein’s effects were mediated by NO formation, leading to inhibition of TGF-β1/Smad2, JNK, p38MAPK and NF-κB activation. This study raises the intriguing possibility of the potential therapeutic value of kallikrein in the treatment of ischemic heart disease.
Acknowledgments
This work was supported by National Institutes of Health grants HL29397, DK066350 and C06 RR015455 from the Extramural Research Facilities Program of the National Center for Research Resources.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdollah S, Macias-Silva M, Tsukazaki T, Hayashi H, Attisano L, Wrana JL. TbetaRI phosphorylation of Smad2 on Ser465 and Ser467 is required for Smad2-Smad4 complex formation and signaling. J Biol Chem. 1997;272:27678–27685. doi: 10.1074/jbc.272.44.27678. [DOI] [PubMed] [Google Scholar]
- Bledsoe G, Shen B, Yao YY, Zhang JJ, Chao L, Chao J. Reversal of renal fibrosis, inflammation and glomerular hypertrophy by kallikrein gene delivery. Human Gene Therapy. 2006;17:545–555. doi: 10.1089/hum.2006.17.545. [DOI] [PubMed] [Google Scholar]
- Calixto JB, Carbrini DA, Ferreira J, Campos MM. Kinins in pain and inflammation. Pain. 2000;97:8140–8145. doi: 10.1016/S0304-3959(00)00335-3. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Cao D, Peterson TE, Simari RD, Shah V. Inhibition of GTP-dependent vesicle trafficking impairs internalization of plasmalemmal eNOS and cellular nitric oxide production. J Cell Sci. 2003;116(pt 17):3645–3655. doi: 10.1242/jcs.00664. [DOI] [PubMed] [Google Scholar]
- Chen LM, Chao L, Chao J. Adenovirus-mediated delivery of human kallistatin gene reduces blood pressure of spontaneously hypertensive rats. Hum Gene Ther. 1997;8:341–347. doi: 10.1089/hum.1997.8.3-341. [DOI] [PubMed] [Google Scholar]
- Clancy RM, Leszczynska-Pizizk J, Abramson SB. Nitric oxide, an endothelial cell relaxation factor, inhibits neutrophil superoxide anion production via a direct action on the NADPH oxidase. J Clin Invest. 1992;90:1116–11121. doi: 10.1172/JCI115929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanueli C, Salis MB, Linthout SV, Meloni M, Desortes E, Silvestre JS. Akt/protein kinase B and endothelial nitric oxide synthase mediate muscular neovascularization induced by tissue kallikrein gene transfer. Circulation. 2004;110:1638–1644. doi: 10.1161/01.CIR.0000142051.36244.83. [DOI] [PubMed] [Google Scholar]
- Engel ME, McDonnell MA, Law BK, Moses HL. Interdependent SMAD and JNK signaling in transforming growth factor-beta-mediated transcription. J Biol Chem. 1999;274:37413–37420. doi: 10.1074/jbc.274.52.37413. [DOI] [PubMed] [Google Scholar]
- Fujii H, Ichimori K, Hoshiai K, Nakazawa H. Nitric oxide inactivates NADPH oxidase in pig neutrophils by inhibiting its assembling process. J Biol Chem. 1997;272:32773–32778. doi: 10.1074/jbc.272.52.32773. [DOI] [PubMed] [Google Scholar]
- Hayashidani S, Tsutsui H, Shiomi T, Ikeuchi M, Matsusaka H, Suematsu N. Anti-monocyte chemoattractant protein-1 gene therapy attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation. 2003;108:2134–2140. doi: 10.1161/01.CIR.0000092890.29552.22. [DOI] [PubMed] [Google Scholar]
- Iwata A, Sai S, Nitta Y, Chen M, de Fries-Hallstrand R, Dalesandro J. Liposome-mediated gene transfection of endothelial nitric oxide synthase reduces endothelial activation and leukocyte infiltration in transplanted hearts. Circulation. 2001;103:2753–2759. doi: 10.1161/01.cir.103.22.2753. [DOI] [PubMed] [Google Scholar]
- Kaiser RA, Bueno O, Lips DJ, Doevendans PA, Jones F, Kimball TF. Targeted inhibition of p38 mitogen-activated protein kinase antagonizes cardiac injury and cell death following ischemia-reperfusion in vivo. J Biol Chem. 2004;279:15524–15530. doi: 10.1074/jbc.M313717200. [DOI] [PubMed] [Google Scholar]
- Kaiser RA, Liang Q, Bueno O, Huang Y, Lackey T, Klevitsky R. Genetic inhibition or activation of JNK1/2 protects the myocardium from ischemia-reperfusion-induced cell death in vivo. J Biol Chem. 2005;280:32602–32608. doi: 10.1074/jbc.M500684200. [DOI] [PubMed] [Google Scholar]
- Li M, Georgakopoulos D, Lu G, Hester L, Kass DA, Hasday J. p38 MAP kinase mediates inflammatory cytokine induction in cardiomyocytes and extracellular matrix remodeling in heart. Circulation. 2005;111:2494–2502. doi: 10.1161/01.CIR.0000165117.71483.0C. [DOI] [PubMed] [Google Scholar]
- Lim W, Ma W, Gee K, Aucoin S, Nandan D, Diz-Mitoma F. Distinct role of p38 and c-Jun N-terminal kinases in IL-10-dependent and IL-10-independent regulation of the costimulatory molecule B7.2 in lipopolysaccharide-stimulated human monocytic cells. J Immunol. 2002;168:1759–1769. doi: 10.4049/jimmunol.168.4.1759. [DOI] [PubMed] [Google Scholar]
- Matsumoto-Ida M, Takimoto Y, Aoyama T, Akao M, Takeda T, Kita T. Activation of TGF-beta1-TAK1-p38 MAPK pathway in spared cardiomyocytes is involved in left ventricular remodeling after myocardial infarction in rats. Am J Physiol Heart Circ Physiol. 2006;290:H709–H715. doi: 10.1152/ajpheart.00186.2005. [DOI] [PubMed] [Google Scholar]
- Matsuyama W, Wang L, Farrar WL, Faure M, Yoshimura T. Activation of discoidin domain receptor 1 isoform b with collagen up-regulates chemokine production in human macrophages: role of p38 mitogen-activated protein kinase and NF-kappa B. J Immunol. 2004;172:2332–2340. doi: 10.4049/jimmunol.172.4.2332. [DOI] [PubMed] [Google Scholar]
- Okada H, Takemura G, Kosai KI, Li Y, Takahashi T, Esaki M. Postinfarction gene therapy against transforming growth factor-beta signal modulates infarct tissue dynamics and attenuates left ventricular remodeling and heart failure. Circulation. 2005;111:2430–2437. doi: 10.1161/01.CIR.0000165066.71481.8E. [DOI] [PubMed] [Google Scholar]
- Regoli D, Rhaleb NE, Dion S, Drapeau G. New selective bradykinin receptor antagonists and bradykinin B2 receptor characterization. Trends Pharmacol Sci. 1990;11:156–161. doi: 10.1016/0165-6147(90)90067-I. [DOI] [PubMed] [Google Scholar]
- Saura M, Zaragoza C, Herranz B, Griera M, Diez-Marqués L, Rodriguez-Puyol D. Nitric oxide regulates transforming growth factor-beta signaling in endothelial cells. Circ Res. 2005;97:1115–11123. doi: 10.1161/01.RES.0000191538.76771.66. [DOI] [PubMed] [Google Scholar]
- Scherrer-Crosbie M, Ullrich R, Bloch KD, Nakajima H, Nasseri B, Aretz HT. Endothelial nitric oxide synthase limits left ventricular remodeling after myocardial infarction in mice. Circulation. 2001;104:1286–1291. doi: 10.1161/hc3601.094298. [DOI] [PubMed] [Google Scholar]
- Smith RS, Jr, Agata J, Xia CF, Chao L, Chao J. Human endothelial nitric oxide synthase gene delivery protects against cardiac remodeling and reduces oxidative stress after myocardial infarction. Life Sci. 2005;76:2457–2471. doi: 10.1016/j.lfs.2004.11.028. [DOI] [PubMed] [Google Scholar]
- Souza DG, Lomez ES, Pinho V, Pesquero JB, Bader M, Pesquero JL. Role of bradykinin B2 and B1 receptors in the local, remote, and systemic inflammatory responses that follow intestinal ischemia and reperfusion injury. J Immunol. 2004;172:2542–2548. doi: 10.4049/jimmunol.172.4.2542. [DOI] [PubMed] [Google Scholar]
- Tschope C, Walther T, Escher F, Spillmann F, Du J, Altmann C. Transgenic activation of the kallikrein-kinin system inhibits intramyocardial inflammation, endothelial dysfunction and oxidative stress in experimental diabetic cardiomyopathy. FASEB J. 2005;19:2057–2059. doi: 10.1096/fj.05-4095fje. [DOI] [PubMed] [Google Scholar]
- Xia CF, Yin H, Yao YY, Borlongan CV, Chao L, Chao J. Kallikrein protects against ischemic stroke by inhibiting apoptosis and inflammation and promoting angiogenesis and neurogenesis. Hum Gene Ther. 2006;17:206–219. doi: 10.1089/hum.2006.17.206. [DOI] [PubMed] [Google Scholar]
- Xu Z, Park SS, Mueller PA, Bagnell RC, Patterson C, Boysen PG. Adenosine produces nitric oxide and prevents mitochondrial oxidant damage in rat cardiomyocytes. Cardiovasc Res. 2005;65:803–812. doi: 10.1016/j.cardiores.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Yamakuchi M, Greer JJ, Cameron SJ, Matsushita K, Morrell CN, Tallbot-Fox K. HMG-CoA reductase inhibitors inhibit endothelial exocytosis and decrease myocardial infarct size. Circ Res. 2005;96:1185–1192. doi: 10.1161/01.RES.0000170229.49776.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Chao L, Chao J. Kallikrein/kinin protects against myocardial apoptosis after ischemia/reperfusion via Akt-glycogen synthase kinase-3 and Akt-Bad.14-3-3 signaling pathways. J Biol Chem. 2005;280:8022–8030. doi: 10.1074/jbc.M407179200. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Zhang JJ, Chao L, Chao J. Kallikrein gene delivery attenuates myocardial infarction and apoptosis after myocardial ischemia and reperfusion. Hypertension. 2000;35(1 Pt 1):25–31. doi: 10.1161/01.hyp.35.1.25. [DOI] [PubMed] [Google Scholar]
- Zhao TC, Taher MM, Valerie KC, Kukreja RC. p38 Triggers late preconditioning elicited by anisomycin in heart: involvement of NF-kappaB and iNOS. Circ Res. 2001;89:915–922. doi: 10.1161/hh2201.099452. [DOI] [PubMed] [Google Scholar]