

Abstract
Proteolytic remodeling of the extracellular matrix is an important component of disease progression in many chronic disease states and is the initiating event in the formation of the tumor microenvironment in cancer. It is the balance of extracellular matrix degrading enzymes, the matrix metalloproteinases (MMPs) and their endogenous inhibitors that determine the extent of tissue remodeling. Unchecked MMP activity can result in significant tissue damage, facilitate disease progression and is associated with host responses to pathologic injury such as angiogenesis and inflammation. The tissue inhibitors of metalloproteinases (TIMPs) have been shown to regulate MMP activity. However, recent findings demonstrate that the tissue inhibitor of metalloproteinases-2 (TIMP-2) inhibits the mitogenic response of human microvascular endothelial cells to growth factors, such as VEGF-A and FGF-2 in vitro and angiogenesis in vivo. The mechanism of this effect is independent of metalloproteinase inhibition. Our lab is the first to demonstrate a cell-surface signaling receptor for a member of the TIMP family and suggest that TIMP-2 functions to regulate cellular responses to growth factors. These new findings are discussed in terms of a model of TIMP-2 regulation of cellular functions in the tumor microenvironment.
Keywords: Tissue inhibitor of metalloproteinase, TIMP-2, Anti-angiogenic, Anti-tumorigenic, Cellular differentiation, Cancer therapy
1 Extracellular matrix remodeling and chronic disease
Many chronic disease states are characterized by an imbalance between tissue destruction and endogenous mechanisms of tissue repair, potentially resulting in a vicious cycle of continued and expanding cellular injury coupled with incomplete repair or resolution. These pathologic conditions frequently disrupt the function and structural organization of both the parenchymal (cellular elements) and connective tissue stroma, such that the injury cannot be repaired by simple regeneration of the parenchymal elements alone. The resulting tissue damage is further complicated by host responses elicited during the initial pathologic insult. In cancer progression, the initial proteolytic remodeling of the extracellular matrix signals the progression of tumor and host responses that results in the formation of a pathologic milieu often referred to as the tumor microenvironment.
This tumor microenvironment is composed of a variety of cell types that includes not only tumor cells and stromal fibroblasts but also cells derived from a variety of host responses. One such host response is the proliferation of new blood vessels, termed angiogenesis, and is frequently associated with chronic diseases such as psoriasis, rheumatoid arthritis and cancer. The initiation of new blood vessel formation is itself due to a shift in the local balance of pro-angiogenic factors and endogenous inhibitors of angiogenesis, i.e. the angiogenic switch [1, 2]. The angiogenic response and associated host responses that constitute the tumor microenvironment may exacerbate the underlying local pathology, such as in medullary (inflammatory) carcinoma of the breast or in disease progression, i.e. cancer metastasis.
Pro-angiogenic factors, such as angiogenic growth factors VEGF-A and FGF-2, induce the expression of matrix-degrading proteinases whose activity results in remodeling of the extracellular matrix to facilitate invasion of new blood vessels and formation of the tumor microenvironment [1, 3]. Inhibition of proteinase activity can result in diminution of the angiogenic response, which in some disease states can result in resolution of the underlying pathology and/or arrest disease progression. This finding suggests that protease inhibitors could be a novel therapeutic approach in the treatment of chronic diseases such as cancer. However, the translation of this strategy to the treatment of human cancer has been disappointing [4]. The reasons for this failure remain unclear, but suggest that our understanding of the molecular and cellular events involved in tissue remodeling and host responses such as angiogenesis, are at best incomplete. Further understanding of the mechanisms of tissue homeostasis and repair should lead to novel therapeutic strategies for the treatment of both chronic inflammatory and malignant diseases.
2 TIMPs: MMP inhibitors and activators
Members of the matrix metalloproteinase (MMP) family have been shown to mediate both tissue development (organogenesis) and remodeling. Collectively, the 24 members of the mammalian MMP family can degrade all components of the extracellular matrix, and many of these protease activities have been specifically associated with pathologic tissue destruction in chronic diseases such as cancer and arthritis. The role of metalloproteinases in cancer, inflammation and other diseases have been reviewed elsewhere [3–7].
The Tissue Inhibitors of MetalloProteinases, or TIMPs, have been identified in species ranging from drosophila, zebra fish and C. elegans to humans, suggesting that these proteins are ancient eukaryotic proteins [8–10]. Furthermore, recent studies have shown developmental defects in TIMP-deficient organisms, in both non-mammalian and mammalian systems, suggesting the importance of these proteins during embryonic development, as well as possible functional redundancy of some TIMPs in mammalian development [11–14].
The mammalian TIMP family has four members, which share significant homology and structural identity at the protein level. The features of the TIMP family members are described in Table 1 and have been reviewed in detail elsewhere. [8–10]. TIMP-2 is unique as a member of the TIMP family in that in addition to inhibiting MMPs TIMP-2 selectively interacts with MT1-MMP to facilitate the cell-surface activation of pro-MMP-2 [15]. Thus, TIMP-2 functions both as an inhibitor of MMPs, and is required for the cellular mechanism of pro-MMP-2 activation.
Table 1.
Properties of mammalian TIMPs
| Property | TIMP-1 | TIMP-2 | TIMP-3 | TIMP-4 | References |
|---|---|---|---|---|---|
| Mass (mature protein) | 28.5 | 21 | 22/27 | 22 | [7–10] |
| Core protein size (aa residues) | 195 | 194 | 188 | 184 | [7–10] |
| Protein expression | Inducible | Constitutive | Inducible | Inducible | [7–10] |
| N-glycosylation | Yes | No | Yes | No | [8–10, 40] |
| Diffusible/soluble | Diffusible | Diffusible | ECM bound | Diffusible | [8–10, 40] |
| Chromosome location | Xp11.23–11.4 | 17q23–25 | 22q12.1–13.2 | 3p25 | [8–10] |
| Nested genes | Nested in synapsin 1 | Contains nested DDC8 | Nested in synapsin 3 | Nested in synapsin 2 | [16–18] |
| mRNA (kbp) | 0.9 | 1.0/3.5 | 5.0 | 1.4 | [8–10] |
| Weak inhibitor of MMP | MT-MMPs | None identified | None identified | None identified | [7–10] |
| ADAM Inhibition | 10 | None reported | 12,17,10, TS-4, TS-5 | None reported | [8–10] |
| Growth/apoptosis | Inhibits apoptosis | Inhibits growth | Promotes apoptosis | Inhibits growth & promotes tumor growth | [24–26, 35–38, 43] |
| Cell surface binding | Nanomolar | Low-nanomolar | Nanomolar | Not reported | [24, 26, 35–38, 43, 47, 49, 53] |
| Receptor | CD63 | α3β1 | VEGFR-2, TACE | Not identified | [31 35, 38, 53, 59] |
| Signaling | FAK, PI3K, p27Kip1 | PTP, Shp-1, p27Kip1, p21Cip1, Rap1 | Antagonist, Inhibit TNF release | Not reported | [31, 34, 35, 38, 53, 55, 56, 59] |
Summarized in this table are a number of properties of the mammalian TIMP family. Notable distinguishing features for TIMP family members include the ECM binding of TIMP-3 and that TIMP-2 is the only member whose gene is not nested within a synapsin family gene.
TIMP-2 also has a distinct gene structure compared with the other three members of the TIMP family. An interesting relationship exists between the TIMPs and the synapsin gene family in that three members of the TIMP family are nested within the synapsin genes [16–18]. The synapsin 1 gene nests TIMP-1, synapsin 2 nests TIMP-4 and synapsin 3 nests TIMP-3. TIMP-2 is the only member of the TIMP family that is not nested within a gene of the synapsin family. The synapsin-TIMP gene nesting relationship began phylogenetically as far back as Drosophila [18]. A recent report describes a nested gene within the very large (~60 kb) first intron of the TIMP-2 gene [19], known as DDC8 [20]. Furthermore, these authors demonstrate that the brain of the TIMP-2 knock out mouse described by Wang et al. contains TIMP-2 mRNA encoding exons 2–5 downstream of DDC8, suggesting alternative splicing between these two genes [20, 21].
3 Identification of non-MMP-dependent TIMP functions
Sequencing of the cDNA clone for TIMP-1 revealed identity of this metalloproteinase inhibitor with erythroid-potentiating activity (EPA) [22]. EPA was identified as a T lymphoblastic factor present in serum that supports the growth of erythroid precursors in vitro by a mechanism involving direct cell surface binding [23]. EPA potentiates erythropoietin (EPO)-stimulated colony formation by early or late erythroid stem cells (BFU-E or CFU-E). Subsequently, TIMP-2 was also shown to have EPA, suggesting that this biological activity may be attributed to a common structural element, which remains to be identified [24]. However, some investigators felt that the dual EPA and MMP inhibitor functions for these proteins were “incongruous” [25]. The argument for this conclusion was principally based on the observation that TIMPs are ubiquitously expressed and that the in vivo plasma concentration of TIMP-1 at approximately 17 nM was well above the concentration of 80 pM required for the maximal physiologic effect of EPA. The physiologic significance of EPA and its relationship to other TIMP functions remain unresolved. Although TIMPs have been principally viewed as functioning exclusively as MMP inhibitors, there remains a significant body of data, starting with the original cloning of TIMP-1, which suggests TIMPs may have other biological functions.
Hayakawa and colleagues were the first to report that TIMP-1 present in serum acted as a growth factor to support proliferation in vitro of a variety of cell types that included both normal mesenchymal and epithelial cells, as well as several tumor cell lines [26]. In these experiments depletion of TIMP-1 from the bovine serum used in cell culture was necessary to observe the growth effects of TIMP-1. Interestingly, TIMP-2 did not stimulate cell growth in these experiments, suggesting that these effects are TIMP-1 specific, although the requirement for MMP inhibitory activity was not examined. We and others have clearly demonstrated that TIMP-1 can inhibit apoptosis in a variety of cell types from Burkitt lymphoma cells [27–30] to breast cancer cells[31–34]. However, the mechanism of this effect seems to be cell type specific.
TIMP-3 has been shown to promote apoptosis in several in vitro systems [8, 9]. It remains unclear if this effect is mediated independent of MMP inhibition by TIMP-3. Recent findings in TIMP-3-null mice suggest that TIMP-3 can either promote or inhibit apoptosis depending on the model system examined [12, 13]. In vivo data now show that TIMP-3 deficient mice have an increase in TNF-α converting enzyme (TACE) activity that in a liver regeneration model results in chronic hepatic inflammation and failure of the liver to regenerate [35]. These results suggest that the effects of TIMP-3 on cell fate are mediated by inhibition of metalloproteinase activity, in this case TACE, also known as ADAM17, and not a member of the MMP family. Interestingly, TIMP-3 also functions as a direct antagonist of the VEGFR2, resulting in inhibition of angiogenesis, a function that is clearly independent of MMP inhibition [36]. TIMP-4 reportedly enhances or inhibits the in vivo growth of tumor xenografts, however, the mechanism of these effects has not been described [37, 38]. It has recently been demonstrated that although TIMP-4 does inhibit endothelial cell migration in vitro it does not inhibit FGF-2-induced angiogenesis in the chick chorioallantoic assay [39].
TIMP-3 is unique amongst the TIMP family in that it has been shown to specifically interact with sulfated glycosaminoglycans and as a result is sequestered in the extracellular matrix [40], the other TIMP family members remain soluble and diffusible. Although the biological significance of the matrix association of TIMP-3 has not been determined, it does suggest that its pericellular distribution and availability to interact with cell surface proteins may be more restricted than other members of the TIMP family. Furthermore, many in vitro studies of TIMP-3 cellular functions have been conducted by addition of soluble, exogenous recombinant TIMP-3 and have not addressed the role of matrix binding which is a unique feature of this inhibitor.
The central issue regarding all of these potential biological activities of the TIMPs is: “Are they unique biological activities of these proteins or are they dependent on inhibition of metalloproteinase activity?”
4 TIMP-2: separation of MMP-inhibitory activity from growth regulatory activity
By virtue of their ability to inhibit metalloproteinase activity members of the TIMP family should function as inhibitors of angiogenesis. This general principal is supported by the demonstration that TIMP-1, TIMP-2 and TIMP-3 all demonstrate anti-angiogenic activity in vitro and in vivo [41, 42]. However, this concept is confounded by the recent demonstration that TIMP-4 did not inhibit FGF-2 induced angiogenesis in vivo [39]. Some evidence suggests that the anti-angiogenic effects of TIMPs may be functionally distinct [43]. The synthetic MMP inhibitor (BB94) effectively blocked angiogenesis in a murine hemangioma model in vivo [44], suggesting that inhibition of MMP activity, either by TIMPs or synthetic MMP inhibitors, was sufficient to block angiogenesis in vivo. Combined with additional evidence that synthetic MMP inhibitors could block tumor cell invasion, tumor growth and reduce metastasis formation, these data provided strong support for the development of synthetic MMP inhibitors for the treatment of human cancer [44]. Unfortunately, the enormous industrial effort involved in the development and preclinical testing of synthetic MMP inhibitors has not produced significant results in clinical trials with cancer patients [4], although the reasons for this failure are not completely understood.
In 1990, Moses and colleagues isolated and characterized a novel anti-angiogenic agent from bovine cartilage, the cartilage-derived inhibitor (CDI) of angiogenesis [45]. CDI co-purified with MMP inhibitor activity, and was shown to inhibit angiogenesis in vivo. In addition CDI inhibited endothelial cell proliferation in response to FGF-2 stimulation in vitro and blocked endothelial cell migration. The N-terminal amino acid sequence of CDI suggested that it was TIMP-related, showing close identity with TIMP-2 [45].
Subsequently, both TIMP-1 and TIMP-2 were shown to inhibit polyamine-stimulated angiogenesis in the chick chorioallantoic membrane assay, but the effects on endothelial cell proliferation and/or migration were not examined [46]. It is interesting to note that in these experiments, both TIMP-1 and TIMP-2 inhibited stimulated angiogenesis (polyamine-dependent) but did not alter vascular development in non-stimulated 3 day-old chick chorioallantoic membrane assays. This suggests that the process defined as vasculogenesis (de novo development of an organized vascular system) is functionally distinct from angiogenesis (development of new vessels form existing vasculature), with respect to both requirements for MMP activity and sensitivity to TIMP inhibition. Together, the experiments of Moses and Hayakawa suggested that although both TIMP-1 and TIMP-2 inhibit angiogenesis, the mechanism of these effects might be different.
To address this issue Murphy et al. examined the ability of both TIMP-1 and TIMP-2 to inhibit endothelial cell proliferation and migration [47]. These experiments demonstrated that TIMP-2, but not TIMP-1 or the synthetic MMP inhibitor, BB-94, inhibited the FGF-2-stimulated proliferation of human endothelial cells. This inhibitory effect was not observed using a pro-MMP-2/TIMP-2 complex suggesting that only free TIMP-2 was capable of inhibiting endothelial cell growth in response to FGF-2 stimulation. Furthermore, TIMP-2 slightly inhibited endothelial cell migration in response to FGF-2 stimulation and appeared to promote endothelial cell adhesion. Comparison of these effects of TIMP-2 with the lack of effect of either TIMP-1 or BB-94 led us to conclude that the effects of TIMP-2 on endothelial cell proliferation were unique biological activities of this member of the TIMP family, independent of MMP inhibitory activity and possibly mediated by cell surface receptor mechanism.
Following these observations, investigations began to focus on interaction of TIMP-2 with the cell surface. However, with the demonstration of a cell surface mechanism for activation of pro-MMP-2 by MT1-MMP that is mediated by interaction of TIMP-2 with MT1-MMP [8–10], it became evident that MT-MMPs represent a cell surface-binding site for TIMP-2. This finding suggests that one possible mechanism through which TIMP-2 could influence cell growth was by binding to MT1-MMP that would act as a signaling receptor. However, no evidence has been forthcoming to demonstrate that MT1-MMP can act as a signaling receptor to suppress endothelial cell proliferation.
In 1999, Wingfield et al reported a novel TIMP-2 mutant devoid of MMP inhibitory activity [48]. For the first time this mutant allowed investigators to assess the requirement for MMP inhibitory activity in TIMP-2-mediated suppression of cell growth. This mutant also allowed us to ultimately identify potential TIMP-2 binding sites on the cell surface that did not contain an MMP active site, vide infra. Preparation of this mutant was accomplished through appending a single amino acid, alanine, to the amino-terminus of TIMP-2 to produce Ala+TIMP-2 [48]. Ala+TIMP-2 does not inhibit MMP-2 or MT1-MMP activity or mediate MT1-MMP activation of pro-MMP-2 [48, 49]. Furthermore, the inability of Ala+TIMP-2 to inhibit MMP activity was reversible by treatment with aminopeptidase activity that removes the amino-terminal Ala residue [48]. This suggests that other than the single Ala residue at the amino-terminus, the remainder of the TIMP-2 is correctly folded and capable of inhibiting MMP activity once the Ala residue blocking the amino terminus is enzymatically removed.
Subsequently, this Ala+TIMP-2 mutant was used to explore the interaction of TIMP-2 with the cell surface. Both TIMP-2 and Ala+TIMP-2 bound to the surface of human A549 lung cancer cells with very high affinity (Kd=147 pM) and this binding was not competed by the synthetic MMP inhibitor BB94 or TIMP-1 [49]. Furthermore, the binding of Ala+TIMP-2 showed only partial competition by MT1-MMP blocking antibodies, and immunofluorescence co-localization studies demonstrated that TIMP-2 and Ala+TIMP-2 binding was, at least in part, independent of MT1-MMP. These findings have been confirmed by investigators who have shown two distinct cell surface binding sites for TIMP-2 [50], cell-surface binding of TIMP-2 is independent of the level of MT1-MMP expression [51], and not all cell surface-bound TIMP-2 can be competed by synthetic MMP inhibitors [50].
More recently, Moses and colleagues definitively demonstrated uncoupling of the MMP-inhibitory activity and anti-angiogenic activity of TIMP-2 [52]. This was accomplished using Pichia pastoris expression system to engineer and produce both the N-terminal and C-terminal domains of TIMP-2. These authors found that although both domains of TIMP-2 inhibited angiogenesis in the embryonic CAM assay, the c-terminal domain and wild type TIMP-2 were more effective inhibitors of angiogenesis in the mouse corneal pocket assay (in which angiogenesis is driven by addition of exogenous pro-angiogenic mitogens) than the N-terminal TIMP-2 domain. Furthermore, the ability of the N-terminal domain was dependent on MMP-inhibitory activity, as blocking the amino-terminus of the TIMP-2 amino-terminal fragment by appending glutamic acid (E) and alanine residues (A) reversed the MMP-inhibitory activity and in vivo anti-angiogenic activity of this TIMP-2 domain [52]. The activity of TIMP-2 that inhibits endothelial cell proliferation was localized to the carboxy-terminal domain of TIMP-2, specifically to the carboxy-terminal disulfide loop, referred to as loop 6 [52]. It is interesting to note that this region is encoded by exon 5 of the TIMP-2 gene, and as noted above the TIMP-2 deficient mice may produce alternative splice variants of the DDC8 gene that could express proteins containing a TIMP-2 loop 6 structure [20]. This may in part explain the benign phenotype of this knock out mouse strain.
The binding of TIMP-2 to the endothelial cell surface and the ability to inhibit endothelial cell proliferation were shown to be independent of MMP inhibition as demonstrated by Ala+TIMP-2 [53]. The binding of TIMP-2 to the human microvascular endothelial cell surface was saturable and reversible with a dissociation constant on the order of 900 pM. The binding of TIMP-2 to the endothelial cell surface did not antagonize growth factor (FGF-2 or VEGF-A) binding, was not competed by growth factor receptor blocking antibodies (anti-VEGFR-2 or anti-FGFR-1) or MT1-MMP blocking antibodies. Competition binding studies demonstrated that TIMP-2 binding to the surface of human microvascular endothelial cells could be competed by anti-β1 and anti-α3 integrin blocking antibodies. The interaction of TIMP-2 with α3β1 cell surface integrin was confirmed by immunoprecipitation experiments in which anti-TIMP-2 antibodies resulted in selective co-immunoprecipitation of α3β1 integrin. The ability of TIMP-2 to inhibit growth in response to tyrosine kinase growth factor stimulation was dependent on cell surface expression of β1 integrin subunits, as demonstrated using β1-null fibroblasts.
Subsequent studies have demonstrated that TIMP-2 or Ala+TIMP-2 binding via α3β1 results in G1 growth arrest and enhanced de novo expression of the cyclin-dependent kinase inhibitor p27Kip1 [54]. Further studies revealed that TIMP-2 and Ala+TIMP-2 enhanced the expression of the reversion-enhancing-cysteine-rich protein with Kazal motifs, also known as RECK [55]. TIMP-2 induction of RECK expression was shown to be mediated by inhibition of Src kinase activity resulting in an altered pattern of paxillin phosphorylation at residues 31 and 118 [56]. This altered phosphorylation results in inactivation of the small G protein Rac1 and a reciprocal activation of the small G-protein Rap1, resulting in loss of a migratory phenotype. Collectively, these findings suggest that TIMP-2 inhibits angiogenesis by inducing endothelial cell differentiation to a quiescent state. These findings are summarized in Fig. 1.
Fig. 1.
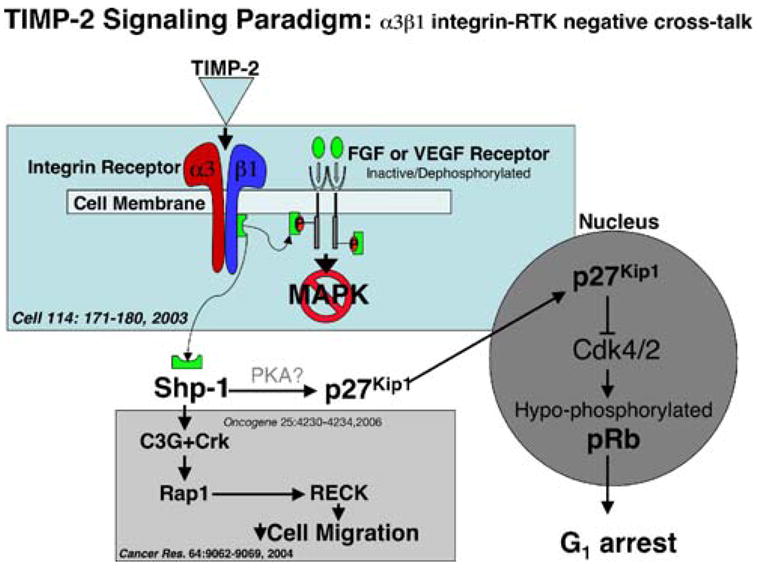
Multiple pathways of TIMP-2/α3β1 signaling. TIMP-2 binding to α3β1 initiates receptor tyrosine kinase inactivation via the action of the protein tyrosine phosphatase activity. Cell cycle arrest is mediated by de novo synthesis of p27Kip1, that down-regulate the cyclin-dependent kinases 4 and 2, resulting in hypophosphorylation of pRb and cell cycle arrest in G1. TIMP-2 also mediates activation of the small G protein Rap1 via a mechanism involving altered association of paxillin scaffolding proteins, guanidine exchange factors and ultimately results in enhanced expression of RECK. This suggests that in addition to arresting cellular proliferation, TIMP-2 also seems to promote expression of cellular differentiation markers
5 TIMP tissue distribution: clues to new functions?
Few studies have examined the expression and localization of TIMPs in adult tissues. However, we do know from many in vitro studies that in many cells the transcriptional activation of TIMP expression is differentially regulated. In most cell types expression of TIMP-2 is constitutive, whereas TIMP-1 and TIMP-3 expression can be induced by a variety of growth factors and cytokines.
A recent study utilizing quantitative PCR demonstrated essentially ubiquitous and abundant expression of all four mammalian TIMPs in most mouse tissues [57]. TIMP-2 was constitutively expressed at high levels in all tissues of the adult mouse, with the expression of the other three TIMPs demonstrating more selective patterns of tissue distribution. These patterns for tissue expression of TIMPs are identical to previous studies and the constitutive high-level expression of at least one TIMP family member in each organ of the adult mouse suggests that TIMPs “provide a crucial checkpoint for tissue degradation” [57]. Although this study did not localize cellular expression of the TIMPs, previous studies of TIMP-2 expression by in situ hybridization suggest selective expression in the stromal compartment, with complete absence of TIMP-2 transcripts in epithelial cells [58]. It should be pointed out that although TIMP concentrations may be significant in some “normal” tissues, the expression of active MMP species in “normal tissues” is usually very low or nonexistent [57]. This raises the question: “What is the functional role of TIMPs in normal tissues lacking MMP activity or evidence of active extracellular matrix remodeling?” We propose that these observations are consistent with TIMP-2 functioning in the absence of MMPs to maintain cellular differentiation and tissue homeostasis.
Consistent with this hypothesis is the recent work of Jaworski and colleagues who have demonstrated that TIMP-2 inhibits growth and promotes neurite differentiation in vitro via an α3β1 integrin dependent mechanism [59]. Interestingly, in this system cell cycle arrest also occurs in G1 but appears to be mediated by enhanced expression of the cyclin-dependent kinase inhibitor p21Cip, not p27Kip1 as we observed in the endothelial cell system. These authors extended this work to demonstrate that TIMP-2 expression correlates with the appearance of microfilament positive neurons and that live cell labeling experiments show TIMP-2 association only with α3 integrin positive cells [60]. These observations led the authors to suggest that up-regulation of TIMP-2 expression by proliferative stimuli implicates TIMP-2 expression in the transition from neuronal proliferation to promotion of terminal neuronal differentiation. This concept is further supported by their subsequent demonstration that TIMP-2 KO mice have abnormal motor deficits, and shows for the first time a significant phenotype for these TIMP-2 deficient mice [61].
6 Model for TIMP-2 in modulating the tumor microenvironment
It is now evident that the matrix metalloproteinase inhibitor TIMP-2 has multiple functions that include inhibition of MMP activity, mediating the cell surface activation of pro-MMP-2 by MT-1-MMP, as well as the metalloproteinase inhibitory independent function of promoting cellular differentiation that is mediated by binding to its cell surface receptor α3β1. The finding that TIMP-2 promotes cellular differentiation in vivo via a mechanism that is independent of MMPs implies a new function for this member of the TIMP family.
These observations suggest the following model for the bifunctional role of TIMP-2 in the tumor microenvironment presented in the context of the angiogenic response (Fig. 2). In quiescent normal tissues the levels of TIMP-2 are sufficient to allow free, uncomplexed TIMP-2 to accumulate in the pericellular milieu. The source of this TIMP-2 may be stromal fibroblasts, perivascular smooth muscle or endothelial cells, all of which have been shown to synthesize and secrete TIMP-2 in vitro. In these quiescent adult tissues the growth suppressing activity of TIMP-2 is functional through binding to available α3β1 integrin receptors on fibroblasts and endothelial cells. In this scenario TIMP-2 functions to suppress fibroblast and endothelial cell responses to transient or minor fluctuations in angiogenic growth factors, i.e. VEGF-A and/or FGF-2.
Fig. 2.
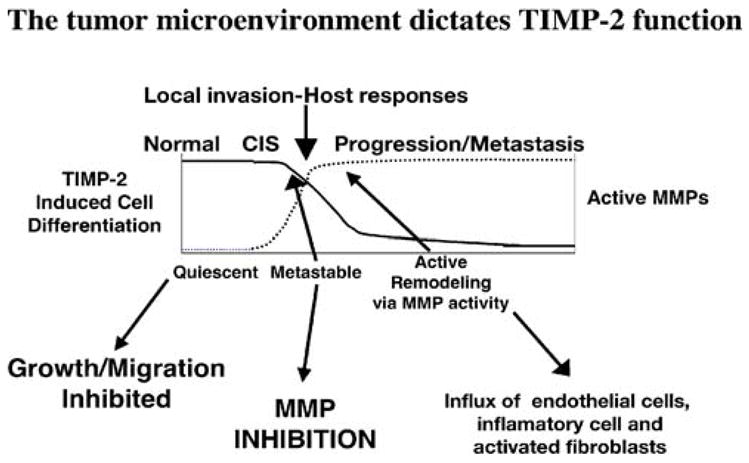
TIMP-2 controls cell behavior directly through α3β1 integrin receptors and indirectly by modulating the activity of MMPs. In physiological quiescent states high levels of free TIMP-2 promotes cellular quiescence and maintenance of the differentiated state. This occurs independently of MMP inhibitory action. As local concentrations of angiogenic factors increase, endothelial cells respond by increasing MMP production and limiting TIMP-2 expression. Increasing concentrations of activated MMPs acts as a sink to reduce free TIMP-2 concentrations, limiting interaction of TIMP-2 with α3β1, thus reducing the growth inhibitory effects. As active MMP concentrations continue to increase, TIMP-2 concentrations may be insufficient to completely inhibit MMP activity. At low concentrations TIMP-2 is insufficient to inhibit MMP activity, and actually enhances MMP-2 activation (via MT1-MMP-dependent mechanism) resulting in remodeling of extracellular matrix, facilitating angio-invasion
However, during tumor progression there is an increase in secretion and activation of MMPs produced by either the tumor cells themselves or tumor-associated fibroblasts. The secretion of these proteases initiates the formation of the tumor microenvironment. The local increase in MMP activity is initially counteracted by the MMP inhibitory activity of endogenous TIMP-2, at the expense of TIMP-2 cell differentiating activity. We speculate that this may facilitate activation of the tumor associated fibroblasts, which may also contribute to the evolving tumor microenvironment. As the tumor progresses, more MMPs are produced overwhelming the local TIMP-2 concentration and contributing to extensive remodeling of the ECM. Furthermore, continued tumor growth leads to tissue hypoxia and MMP-mediated release of matrix sequestered angiogenic factors. These events promote the tumor angiogenic response, which also requires MMP activity. As recently demonstrated, endothelial cells responding to angiogenic factors, such as VEGF-A, decrease the synthesis and secretion of TIMP-2 [62]. This further promotes the local decline in TIMP-2 concentrations and potentiates the proteolytic remodeling of the extracellular matrix. Thus, in this model TIMP-2 initially functions as a rheostat by limiting cellular responsiveness to stimuli leading to cellular proliferation, cellular activation and extracellular matrix remodeling. However, as the tumor microenvironment evolves TIMP-2 function shifts to inhibition of MMP activity, and further depletion of TIMP-2 levels may even facilitate TIMP-2 function in the MT-1-MMP activation of pro-MMP-2.
The primary question that arises is if TIMP-2 functions to maintain tissue homeostasis, why do TIMP-2 deficient mice reproduce normally and show no overt vascular defects, only a motor deficit phenotype?[61] With respect to vascular development in mammals we do know that this process is regulated in a fashion distinct from angiogenesis in the adult. Vascular development occurs through the process of vasculogenesis, which is functionally distinct from angiogenesis [63]. We propose that either: (1) TIMP-2 plays no direct role in vasculogenesis; or (2) that other members of the TIMP family compensate for the loss of TIMP-2 MMP inhibitory activity during embryonic vascular development. Although no developmental defects are observed in TIMP-2 deficient mice [21, 64], a recent report demonstrates that in zebra fish the single TIMP expressed during development is most homologous to TIMP-2 and that ablation of TIMP-2 expression results in abnormal zebra fish development [11]. This finding suggests that in mammalian systems other TIMPs may compensate for the loss of TIMP-2 function. Angiogenesis on the other hand is a process limited to adult tissues responding to a pathologic stimulus. Therefore, the question should be: “Do TIMP-2 deficient animals have a normal or abnormal angiogenic responses to tissue injury?” To our knowledge such experiments have not yet been reported.
The next question that arises is can TIMP-2 be used as a therapeutic in the treatment of cancer? If the concentration of TIMP-2 or even better Ala+TIMP-2, which would not be sequestered by MMP active sites, could the cellular activation of host responses contributing to the tumor microenvironment, such as fibroblast activation and tumor angiogenesis be suppressed by promoting cellular differentiation. That such a therapeutic approach is possible is supported by the recent report of a TIMP-2 transgenic murine model using the MMTV-Wnt-1 mammary carcinogenesis model [65]. Although exact tissue concentrations of TIMP-2 were not determined, the authors demonstrated that enhanced TIMP-2 expression in the mammary glands of the MMTV-Wnt-1 double transgenic mice resulted in increased tumor latency, ~26% reduction of tumor formation, 18% decrease in tumor cell proliferation and a 12% increase in tumor cell apoptotic rate. Although tumor-associated angiogenesis was reduced in the double transgenics, the authors’ primary focus was demonstrating a role for MMPs in the MMTV-Wnt-1 tumor model. Therefore, possible direct effects of TIMP-2 on cellular elements of the tumor microenvironment were not examined.
7 Summary
Although the concept of MMP-independent functions for TIMPs is not new, the demonstration that TIMP-2 promotes cellular differentiation of endothelial cells and neurons is mediated by cell surface receptors definitively demonstrates that TIMPs have functions other than inhibition of MMPs. Additionally, it has recently been reported that CD63 may function as a cell surface receptor for TIMP-1 and mediate the anti-apoptotic activity in breast cancer [31]. This finding has enormous implications for the role of TIMP-1 in modulating the cellular responses that are involved in formation and maintenance of the tumor microenvironment. Not only can TIMP-1 promote carcinogenesis by directly promoting tumor cell growth and tumorigenicity [31–33, 66], TIMP-1 has also been shown to modulate the immune function [28, 30] as well as angiogenesis [67–71].
From their initial identification, TIMPs have been associated with regulation of cell growth and differentiation (i.e. erythroid-potentiating activity, EPA). However, we are now beginning to develop the experimental evidence to define the mechanisms of such activities. The question remains: Do all TIMPs also have cell surface receptors that may mediate regulation of cell growth or differentiation? TIMP-3 has been shown to bind to VEGFR-2 and function as an antagonist of VEGF-A stimulation [36]. However, the significance of this mechanism in preventing cancer progression is unclear. Cell surface binding for TIMP-4 has been suggested, but identification of specific cell surface receptors has not been forthcoming. Characterization of these receptors and understanding the signaling mechanisms involved will identify new therapeutic targets not just for cancer but also for a variety of other chronic disease states.
8 Conclusions
Recent evidence suggests that TIMPs, particularly TIMP-1 and TIMP-2, may have unique biological properties, independent of their ability to inhibit MMPs. This is supported by the identification of cell surface receptors for these two members of the TIMP family. TIMP-2 impacts the tumor microenvironment by initially promoting cellular differentiation of endothelial cells and fibroblasts, and possibly cells in the epithelial compartment. However, subsequent progression and massive production and activation of MMPs by a variety of cell types that comprise the tumor microenvironment, including tumor cells, endothelial cells, immune cells and tumor-associated fibroblasts, shifts the function of TIMP-2 to metalloproteinase inhibitor. Finally, continued progression and further depletion of TIMP-2 levels could again change TIMP-2 function from MMP inhibitor to activator by facilitating MT-1-MMP activation of pro-MMP-2.
9 Key unanswered questions
Do all four members of the TIMP family have cell surface receptors and if so what unique biological activities do they support?
Can over expression of TIMP-2 within the tumor microenvironment overcome the MMP-mediated activation of the tumor microenvironment and promote cellular differentiation of host cells?
Can over expression of TIMP-2 promote differentiation of the malignant carcinoma cells leading to reversal of the epithelial to mesenchymal transition?
Can addition of TIMP-2 over expression enhance the tumoricidal activity of conventional cytotoxic agents?
References
- 1.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J, Hanahan D. Switch to the angiogenic phenotype during tumorigenesis. Princess Takamatsu Symposia. 1991;22:339–347. [PubMed] [Google Scholar]
- 3.Heissig B, Hattori K, Friedrich M, Rafii S, Werb Z. Angiogenesis: vascular remodeling of the extracellular matrix involves metalloproteinases. Current Opinion in Hematology. 2003;10:136–141. doi: 10.1097/00062752-200303000-00007. [DOI] [PubMed] [Google Scholar]
- 4.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: Trials and tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 5.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2001;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annual Review of Cell & Developmental Biology. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nature Reviews Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 8.Lambert E, Dasse E, Haye B, Petitfrere E. TIMPs as multifacial proteins. Critical Reviews in Oncology Hematology. 2004;49:187–198. doi: 10.1016/j.critrevonc.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 9.Crocker SJ, Pagenstecher A, Campbell IL. The TIMPs tango with MMPs and more in the central nervous system. Journal of Neuroscience Research. 2004;75:1–11. doi: 10.1002/jnr.10836. [DOI] [PubMed] [Google Scholar]
- 10.Brew K, Dinakarpandian D, Nagase H. Tissue inhibitors of metalloproteinases: Evolution, structure and function. Biochimica et Biophysica Acta. 2000;1477:267–283. doi: 10.1016/s0167-4838(99)00279-4. [DOI] [PubMed] [Google Scholar]
- 11.Zhang JS, Bai S, Tanase C, Nagase H, Sarras MP. The expression of tissue inhibitor of metalloproteinase 2 (TIMP-2) is required for normal development of zebrafish embryos. Development Genes and Evolution. 2003;213:382–389. doi: 10.1007/s00427-003-0333-9. [DOI] [PubMed] [Google Scholar]
- 12.Gill SE, Pape MC, Khokha R, Watson AJ, Leco KJ. A null mutation for tissue inhibitor of metalloproteinases-3 (Timp-3) impairs murine bronchiole branching morphogenesis. Developmental Biology. 2003;261:313–323. doi: 10.1016/s0012-1606(03)00318-x. [DOI] [PubMed] [Google Scholar]
- 13.Fata JE, Leco KJ, Voura EB, Yu HY, Waterhouse P, Murphy G, et al. Accelerated apoptosis in the Timp-3-deficient mammary gland. Journal of Clinical Investigation. 2001;108:831–841. doi: 10.1172/JCI13171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Godenschwege TA, Pohar N, Buchner S, Buchner E. Inflated wings, tissue autolysis and early death in tissue inhibitor of metalloproteinases mutants of Drosophila. European Journal of Cell Biology. 2000;79:495–501. doi: 10.1078/0171-9335-00072. [DOI] [PubMed] [Google Scholar]
- 15.Stetler-Stevenson WG. Matrix metalloproteinases in angiogenesis: A moving target for therapeutic intervention. Journal of Clinical Investigation. 1999;103:1237–1241. doi: 10.1172/JCI6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Derry JM, Barnard PJ. Physical linkage of the A-raf-1, properdin, synapsin I, and TIMP genes on the human and mouse X chromosomes. Genomics. 1992;12:632–638. doi: 10.1016/0888-7543(92)90286-2. [DOI] [PubMed] [Google Scholar]
- 17.Dunham I, Shimizu N, Roe BA, Chissoe S, Hunt AR, Collins JE, et al. The DNA sequence of human chromosome 22.[see comment][erratum appears in Nature 2000 Apr 20;404(6780):904] Nature. 1999;402:489–495. doi: 10.1038/990031. [DOI] [PubMed] [Google Scholar]
- 18.Pohar N, Godenschwege TA, Buchner E. Invertebrate tissue inhibitor of metalloproteinase: Structure and nested gene organization within the synapsin locus is conserved from Drosophila to human. Genomics. 1999;57:293–296. doi: 10.1006/geno.1999.5776. [DOI] [PubMed] [Google Scholar]
- 19.Caterina JJ, Yamada S, Caterina NCM, Longenecker G, Holmback K, Shi J, et al. Inactivating mutation of the mouse tissue inhibitor of metalloproteinases-2(Timp-2) gene alters proMMP-2 activation. Journal of Biological Chemistry. 2000;275:26416–26422. doi: 10.1074/jbc.M001271200. [DOI] [PubMed] [Google Scholar]
- 20.Jaworski DM, Beem-Miller M, Lluri G, Barrantes-Reynolds R. Potential regulatory relationship between the nested gene DDC8 and its host gene tissue inhibitor of metalloproteinase-2. Physiological Genomics. 2007;28:168–178. doi: 10.1152/physiolgenomics.00160.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Z, Juttermann R, Soloway PD. TIMP-2 is required for efficient activation of proMMP-2 in vivo. Journal of Biological Chemistry. 2000;275:26411–26415. doi: 10.1074/jbc.M001270200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Docherty AJ, Lyons A, Smith BJ, Wright EM, Stephens PE, Harris TJ, et al. Sequence of human tissue inhibitor of metalloproteinases and its identity to erythroid-potentiating activity. Nature. 1985;318:66–69. doi: 10.1038/318066a0. [DOI] [PubMed] [Google Scholar]
- 23.Gasson JC, Bersch N, Golde DW. Characterization of purified human erythroid-potentiating activity. Progress in Clinical & Biological Research. 1985;184:95–104. [PubMed] [Google Scholar]
- 24.Stetler-Stevenson WG, Bersch N, Golde DW. Tissue inhibitor of metalloproteinase-2 (TIMP-2) has erythroid-potentiating activity. FEBS Letters. 1992;296:231–234. doi: 10.1016/0014-5793(92)80386-u. [DOI] [PubMed] [Google Scholar]
- 25.Stricklin GP, Welgus HG. Physiological relevance of erythroid-potentiating activity of TIMP. Nature. 1986;321:628. doi: 10.1038/321628a0. [DOI] [PubMed] [Google Scholar]
- 26.Hayakawa T, Yamashita K, Tanzawa K, Uchijima E, Iwata K. Growth-promoting activity of tissue inhibitor of metalloproteinases-1 (TIMP-1) for a wide range of cells. A possible new growth factor in serum. FEBS Letters. 1992;298:29–32. doi: 10.1016/0014-5793(92)80015-9. [DOI] [PubMed] [Google Scholar]
- 27.Guedez L, Mansoor A, Birkedal-Hansen B, Lim MS, Fukushima P, Venzon D, et al. Tissue inhibitor of metalloproteinases 1 regulation of interleukin-10 in B-cell differentiation and lymphomagenesis. Blood. 2001;97:1796–1802. doi: 10.1182/blood.v97.6.1796. [DOI] [PubMed] [Google Scholar]
- 28.Guedez L, Martinez A, Zhao S, Vivero A, Pittaluga S, Stetler-Stevenson M, et al. Tissue inhibitor of metalloproteinase 1 (TIMP-1) promotes plasmablastic differentiation of a Burkitt lymphoma cell line: Implications in the pathogenesis of plasmacytic/plasmablastic tumors. Blood. 2005;105:1660–1668. doi: 10.1182/blood-2004-04-1385. [DOI] [PubMed] [Google Scholar]
- 29.Guedez L, McMarlin AJ, Kingma DW, Bennett TA, Stetler-Stevenson M, Stetler-Stevenson WG. Tissue inhibitor of metalloproteinase-1 alters the tumorigenicity of Burkitt’s lymphoma via divergent effects on tumor growth and angiogenesis. American Journal of Pathology. 2001;158:1207–1215. doi: 10.1016/S0002-9440(10)64070-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guedez L, Stetler-Stevenson WG, Wolff L, Wang J, Fukushima P, Mansoor A, et al. In vitro suppression of programmed cell death of B cells by tissue inhibitor of metalloproteinases-1. Journal of Clinical Investigation. 1998;102:2002–2010. doi: 10.1172/JCI2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jung KK, Liu XW, Chirco R, Fridman R, Kim HR. Identification of CD63 as a tissue inhibitor of metalloproteinase-1 interacting cell surface protein. EMBO Journal. 2006;25:3934–3942. doi: 10.1038/sj.emboj.7601281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu XW, Bernardo MM, Fridman R, Kim HR. Tissue inhibitor of metalloproteinase-1 protects human breast epithelial cells against intrinsic apoptotic cell death via the focal adhesion kinase/phosphatidylinositol 3-kinase and MAPK signaling pathway. Journal of Biological Chemistry. 2003;278:40364–40372. doi: 10.1074/jbc.M302999200. [DOI] [PubMed] [Google Scholar]
- 33.Liu XW, Taube ME, Jung KK, Dong Z, Lee YJ, Roshy S, et al. Tissue inhibitor of metalloproteinase-1 protects human breast epithelial cells from extrinsic cell death: A potential oncogenic activity of tissue inhibitor of metalloproteinase-1. Cancer Research. 2005;65:898–906. [PubMed] [Google Scholar]
- 34.Taube ME, Liu XW, Fridman R, Kim HR. TIMP-1 regulation of cell cycle in human breast epithelial cells via stabilization of p27(KIP1) protein. Oncogene. 2006;25:3041–3048. doi: 10.1038/sj.onc.1209336. [DOI] [PubMed] [Google Scholar]
- 35.Mohammed FF, Smookler DS, Taylor SEM, Fingleton B, Kassiri Z, Sanchez OH, et al. Abnormal TNF activity in Timp3(−/−) mice leads to chronic hepatic inflammation and failure of liver regeneration. Nature Genetics. 2004;36:969–977. doi: 10.1038/ng1413. [DOI] [PubMed] [Google Scholar]
- 36.Qi JH, Ebrahem Q, Moore N, Murphy G, Claesson-Welsh L, Bond M, et al. A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): Inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nature Medicine. 2003;9:407–415. doi: 10.1038/nm846. [DOI] [PubMed] [Google Scholar]
- 37.Jiang Y, Wang M, Celiker MY, Liu YE, Sang QX, Goldberg ID, et al. Stimulation of mammary tumorigenesis by systemic tissue inhibitor of matrix metalloproteinase 4 gene delivery. Cancer Research. 2001;61:2365–2370. [PubMed] [Google Scholar]
- 38.Celiker MY, Wang M, Atsidaftos E, Liu X, Liu YE, Jiang Y, et al. Inhibition of Wilms’ tumor growth by intramuscular administration of tissue inhibitor of metalloproteinases-4 plasmid DNA. Oncogene. 2001;20:4337–4343. doi: 10.1038/sj.onc.1204508. [DOI] [PubMed] [Google Scholar]
- 39.Fernandez CA, Moses MA. Modulation of angiogenesis by tissue inhibitor of metalloproteinase-4. Biochemical and Biophysical Research Communications. 2006;345:523–529. doi: 10.1016/j.bbrc.2006.04.083. [DOI] [PubMed] [Google Scholar]
- 40.Yu WH, Yu S, Meng Q, Brew K, Woessner JF., Jr TIMP-3 binds to sulfated glycosaminoglycans of the extracellular matrix. Journal of Biological Chemistry. 2000;275:31226–31232. doi: 10.1074/jbc.M000907200. [DOI] [PubMed] [Google Scholar]
- 41.Johnson MD, Kim HR, Chesler L, Tsao-Wu G, Bouck N, Polverini PJ. Inhibition of angiogenesis by tissue inhibitor of metalloproteinase. Journal of Cellular Physiology. 1994;160:194–202. doi: 10.1002/jcp.1041600122. [DOI] [PubMed] [Google Scholar]
- 42.Anand-Apte B, Pepper MS, Voest E, Montesano R, Olsen B, Murphy G, et al. Inhibition of angiogenesis by tissue inhibitor of metalloproteinase-3. Investigative Ophthalmology & Visual Science. 1997;38:817–823. [PubMed] [Google Scholar]
- 43.Baker AH, Zaltsman AB, George SJ, Newby AC. Divergent effects of tissue inhibitor of metalloproteinase-1, -2, or -3 overexpression on rat vascular smooth muscle cell invasion, proliferation, and death in vitro. TIMP-3 promotes apoptosis. Journal of Clinical Investigation. 1998;101:1478–1487. doi: 10.1172/JCI1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown PD. Matrix metalloproteinase inhibitors. Angiogenesis. 1998;1:142–154. doi: 10.1023/A:1018373520193. [DOI] [PubMed] [Google Scholar]
- 45.Moses MA, Sudhalter J, Langer R. Identification of an inhibitor of neovascularization from cartilage. Science. 1990;248:1408–1410. doi: 10.1126/science.1694043. [DOI] [PubMed] [Google Scholar]
- 46.Takigawa M, Nishida Y, Suzuki F, Kishi J, Yamashita K, Hayakawa T. Induction of angiogenesis in chick yolk-sac membrane by polyamines and its inhibition by tissue inhibitors of metalloproteinases (TIMP and TIMP-2) Biochemical & Biophysical Research Communications. 1990;171:1264–1271. doi: 10.1016/0006-291x(90)90822-5. [DOI] [PubMed] [Google Scholar]
- 47.Murphy AN, Unsworth EJ, Stetler-Stevenson WG. Tissue inhibitor of metalloproteinases-2 inhibits bFGF-induced human microvascular endothelial cell proliferation. Journal of Cellular Physiology. 1993;157:351–358. doi: 10.1002/jcp.1041570219. [DOI] [PubMed] [Google Scholar]
- 48.Wingfield PT, Sax JK, Stahl SJ, Kaufman J, Palmer I, Chung V, et al. Biophysical and functional characterization of full-length, recombinant human tissue inhibitor of metalloproteinases-2 (TIMP-2) produced in Escherichia coli. Comparison of wild type and amino-terminal alanine appended variant with implications for the mechanism of TIMP functions. Journal of Biological Chemistry. 1999;274:21362–21368. doi: 10.1074/jbc.274.30.21362. [DOI] [PubMed] [Google Scholar]
- 49.Hoegy SE, Oh HR, Corcoran ML, Stetler-Stevenson WG. Tissue inhibitor of metalloproteinases-2 (TIMP-2) suppresses TKR-growth factor signaling independent of metalloproteinase inhibition. Journal of Biological Chemistry. 2001;276:3203–3214. doi: 10.1074/jbc.M008157200. [DOI] [PubMed] [Google Scholar]
- 50.Itoh Y, Ito A, Iwata K, Tanzawa K, Mori Y, Nagase H. Plasma membrane-bound tissue inhibitor of metalloproteinases (TIMP)-2 specifically inhibits matrix metalloproteinase 2 (gelatinase A) activated on the cell surface. Journal of Biological Chemistry. 1998;273:24360–24367. doi: 10.1074/jbc.273.38.24360. [DOI] [PubMed] [Google Scholar]
- 51.Bernardo MM, Fridman R. TIMP-2 (tissue inhibitor of metalloproteinase-2) regulates MMP-2 (matrix metalloproteinase-2) activity in the extracellular environment after pro-MMP-2 activation by MT1 (membrane type 1)-MMP. Biochemical Journal. 2003;374:739–745. doi: 10.1042/BJ20030557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fernandez CA, Butterfield C, Jackson G, Moses MA. Structural and functional uncoupling of the enzymatic and angiogenic inhibitory activities of tissue inhibitor of metalloproteinase-2 (TIMP-2): Loop 6 is a novel angiogenesis inhibitor. Journal of Biological Chemistry. 2003;278:40989–40995. doi: 10.1074/jbc.M306176200. [DOI] [PubMed] [Google Scholar]
- 53.Seo DW, Li H, Guedez L, Wingfield PT, Diaz T, Salloum R, et al. TIMP-2 mediated inhibition of angiogenesis: an MMP-independent mechanism. Cell. 2003;114:171–180. doi: 10.1016/s0092-8674(03)00551-8. [DOI] [PubMed] [Google Scholar]
- 54.Seo DW, Li H, Qu CK, Kim YS, Diaz T, Wei B, et al. Shp-1 mediates the antiproliferative activity of tissue inhibitor of metalloproteinase-2 in human microvascular endothelial cells. Journal of Biological Chemistry. 2006;281:3711–3721. doi: 10.1074/jbc.M509932200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oh J, Seo DW, Diaz T, Wei B, Ward Y, Ray JM, et al. Tissue inhibitors of metalloproteinase 2 inhibits endothelial cell migration through increased expression of RECK. Cancer Research. 2004;64:9062–9069. doi: 10.1158/0008-5472.CAN-04-1981. [DOI] [PubMed] [Google Scholar]
- 56.Oh J, Diaz T, Wei B, Chang H, Noda M, Stetler-Stevenson WG. TIMP-2 upregulates RECK expression via dephosphorylation of paxillin tyrosine residues 31 and 118. Oncogene. 2006;25:4230–4234. doi: 10.1038/sj.onc.1209444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nuttall RK, Sampieri CL, Pennington CJ, Gill SE, Schultz GA, Edwards DR. Expression analysis of the entire MMP and TIMP gene families during mouse tissue development. FEBS Letters. 2004;563:129–134. doi: 10.1016/S0014-5793(04)00281-9. [DOI] [PubMed] [Google Scholar]
- 58.Blavier L, DeClerck YA. Tissue inhibitor of metalloproteinases-2 is expressed in the interstitial matrix in adult mouse organs and during embryonic development. Molecular Biology of the Cell. 1997;8:1513–1527. doi: 10.1091/mbc.8.8.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Perez-Martinez L, Jaworski DM. Tissue inhibitor of metalloproteinase-2 promotes neuronal differentiation by acting as an anti-mitogenic signal. Journal of Neuroscience. 2005;25:4917–4929. doi: 10.1523/JNEUROSCI.5066-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jaworski DM, Perez-Martinez L. Tissue inhibitor of metalloproteinase-2 (TIMP-2) expression is regulated by multiple neural differentiation signals. Journal of Neurochemistry. 2006;98:234–247. doi: 10.1111/j.1471-4159.2006.03855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jaworski DM, Soloway P, Caterina J, Falls WA. Tissue inhibitor of metalloproteinase-2(TIMP-2)-deficient mice display motor deficits. Journal of Neurobiology. 2006;66:82–94. doi: 10.1002/neu.20205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lamoreaux WJ, Fitzgerald ME, Reiner A, Hasty KA, Charles ST. Vascular endothelial growth factor increases release of gelatinase A and decreases release of tissue inhibitor of metalloproteinases by microvascular endothelial cells in vitro. Microvascular Research. 1998;55:29–42. doi: 10.1006/mvre.1997.2056. [DOI] [PubMed] [Google Scholar]
- 63.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nature Medicine. 2000;6:389–395. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 64.Caterina JJ, Yamada S, Caterina NC, Longenecker G, Holmback K, Shi J, et al. Inactivating mutation of the mouse tissue inhibitor of metalloproteinases-2(Timp-2) gene alters proMMP-2 activation. Journal of Biological Chemistry. 2000;275:26416–26422. doi: 10.1074/jbc.M001271200. [DOI] [PubMed] [Google Scholar]
- 65.Blavier L, Lazaryev A, Dorey F, Shackleford GM, DeClerck Y. Matrix metalloproteinases play an active role in Wnt1-induced mammary tumorigenesis. Cancer Research. 2006;66:2691–2699. doi: 10.1158/0008-5472.CAN-05-2919. [DOI] [PubMed] [Google Scholar]
- 66.Rhee JS, Diaz R, Korets L, Hodgson JG, Coussens LM. TIMP-1 alters susceptibility to carcinogenesis. Cancer Research. 2004;64:952–991. doi: 10.1158/0008-5472.can-03-2445. [DOI] [PubMed] [Google Scholar]
- 67.Akahane T, Akahane M, Shah A, Connor CM, Thorgeirsson UP. TIMP-1 inhibits microvascular endothelial cell migration by MMP-dependent and MMP-independent mechanisms. Experimental Cell Research. 2004;301:158–167. doi: 10.1016/j.yexcr.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 68.Akahane T, Akahane M, Shah A, Thorgeirsson UP. TIMP-1 stimulates proliferation of human aortic smooth muscle cells and Ras effector pathways. Biochemical & Biophysical Research Communications. 2004;324:440–445. doi: 10.1016/j.bbrc.2004.09.063. [DOI] [PubMed] [Google Scholar]
- 69.Thorgeirsson UP, Yoshiji H, Sinha CC, Gomez DE. Breast cancer; tumor neovasculature and the effect of tissue inhibitor of metalloproteinases-1 (TIMP-1) on angiogenesis. In Vivo. 1996;10:137–144. [PubMed] [Google Scholar]
- 70.Yamazaki M, Akahane T, Buck T, Yoshiji H, Gomez DE, Schoeffner DJ, et al. Long-term exposure to elevated levels of circulating TIMP-1 but not mammary TIMP-1 suppresses growth of mammary carcinomas in transgenic mice. Carcinogenesis. 2004;25:1735–1746. doi: 10.1093/carcin/bgh181. [DOI] [PubMed] [Google Scholar]
- 71.Yoshiji H, Kuriyama S, Miyamoto Y, Thorgeirsson UP, Gomez DE, Kawata M, et al. Tissue inhibitor of metalloproteinases-1 promotes liver fibrosis development in a transgenic mouse model. Hepatology. 2000;32:1248–1254. doi: 10.1053/jhep.2000.20521. [DOI] [PubMed] [Google Scholar]