

Abstract
M98Q amicyanin is isolated with zinc bound to its type 1 copper-binding site. The influence of the axial ligand of the type 1 copper site on metal specificity is strongest prior to the completion of protein folding and adoption of the final type 1 site geometry. The preference for zinc over copper correlated with the selectivity of apoamicyanin in vitro in the partially folded, rather than the completely folded state. These results suggest that metal incorporation in vivo occurs during protein folding in the periplasm and not to a preformed type 1 site.
1. Introduction
Type 1 copper sites are found in a wide range of redox proteins including azurin and amicyanin in bacteria, plastocyanin in plants, and ceruloplasmin in animals [1]. The active site of type 1 cupredoxins typically consists of three strong equatorial ligands, two nitrogens of two His and a Cys sulfur which form a trigonal plane. An additional axial ligand, usually provided by a Met sulfur yields a distorted tetrahedral geometry. In amicyanin from Paracoccus denitrificans, the copper is coordinated by two Nδ of His53 and His95, S of Cys92, and S of Met98 [2]. Amicyanin exhibits a very high specificity and affinity for Cu2+. We recently characterized an M98Q mutant amicyanin which is isolated with zinc bound rather than copper, but which can be easily reconstituted with copper after denaturation and the removal of zinc [3]. The decreased selectivity for copper over zinc exhibited by M98Q amicyanin provides an opportunity to use this mutant to address a key question in metalloprotein biogenesis. To what extent can the folding process itself dictate metal specificity?
The roles of cofactors, in particular metals, in the stabilization of metalloproteins and in protein folding during metalloprotein biogenesis have been extensively studied [4]. The exact role of the cofactors in the protein folding process remains not well understood. Azurin has been the topic of many such studies [5]. Amicyanin from Paracoccus versutus has also been studied and the effect of mutation of its axial Met ligand to Gln was examined [6]. In those studies the metal specificity and role of the metal during protein folding was studied by first denaturing the protein usually with guanidine HCl. In the present work we also used that approach but in addition exploited the fact that a completely folded apoprotein could be generated by reduction of amicyanin and removal of the bound Cu+ by dialysis against KCN [7]. Significantly, the results obtained using this apoprotein differ from those obtained using apoprotein that was generated by denaturation. This approach and the use of the M98Q amicyanin, which has altered metal binding specificity, has allowed us to differentiate the metal selectivity of the completely folded protein and that prepared by denaturation which we show to be in the partially folded state when it interacts with the metal in vitro.
Incorporation of metal cofactors into metalloproteins during biosynthesis will be affected by ambient metal concentrations and metal delivery via specific chaperones. For type 1 copper proteins there is no evidence for the involvement of chaperones during assembly. Furthermore, in this study native and M98Q amicyanins are expressed in the periplasmic space where the metal content is expected to be similar to the external medium. Given the typical Sec signal sequence that directs amicyanin to the periplasm [8], the protein is believed to cross the membrane in the unfolded state and then fold and assemble in the periplasm. This paper describes in vitro experiments which show that the specificity for metal incorporation in vivo is determined by the residue which provides the axial ligand during protein folding before formation of the type 1 site.
2. Materials and Methods
Metal content of the protein was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) using a Spectro Genesis spectrometer as previously described [3]. Protein samples were incubated with 3 mM EDTA prior to buffer exchange and analysis. Metal content is reported as per amicyanin molecule on the basis of sulfur content which is predicted from the amino acid sequence and which was also determined by ICP-OES of the same samples. Fluorescence measurements were performed using a Perkin-Elmer LS-50B spectrofluorometer. The excitation wavelength was 280 nm and emission spectra were collected with a 5 nm slit width.
Native amicyanin [9] and M98Q amicyanin [3] were expressed in E. coli and purified from the periplasmic fraction of the cells as described previously. Two forms of apoamicyanin which are designated “fully folded” apoamicyanin and “partially folded” apoamicyanin were prepared for metal reconstitution studies. The basis for these designations is discussed later. Fully folded apoamicyanin was prepared from native or copper-reconstituted M98Q amicyanin by reduction with sodium dithionite followed by dialysis against 0.1 M KCN as previously described [7]. After removal of KCN and unbound copper by further dialysis, reconstitution experiments were preformed at room temperature in the presence of the indicated mixtures of CuSO4 and ZnSO4. This species is known to be completely folded because it was shown by x-ray crystallography that the crystal structure of this apoprotein is essentially identical to that of holoamicyanin with copper absent but the type 1 geometry intact [10]. The second form which was designated partially folded apoamicyanin was prepared as described originally by Diederix, et al [6], and was used previously to reconstitute this M98Q amicyanin with copper [3]. Amicyanins were incubated in 10 mM HEPES buffer, pH 8.0, containing 6 M guanidine HCl, 50 mM EDTA and 2 mM dithiothreitol. The unfolded apoproteins were then diluted 50-fold into 10 mM HEPES buffer, pH 8.0, containing 5 mM dithiothreitol. The proteins were then dialyzed against 100 mM ammonium acetate, pH 8.0, at 4° C for 4 hr, followed by dialysis against 250 mM ammonium acetate, pH 8.0, overnight. In reconstitution experiments the dialyzed protein solution was then brought to room temperature in the presence of the indicated mixtures of CuSO4 and ZnSO4.
3. Results and Discussion
As discussed later, the M98Q apoamicyanins generated by the two different methods exhibited different metal uptake selectivity. Therefore, it is important to have some indication of the relative extent of folding of the apoamicyanins. Tryptophan fluorescence is an ideal method for assessing the folding state of amicyanin. Amicyanin contains a single tryptophan residue which is completely buried in the structure [2]. In holoamicyanin the tryptophan fluorescence is partially quenched by the presence of copper [7]. We have previously shown that the apoamicyanin generated by dialysis of reduced amicyanin against KCN exhibits an intense fluorescence emission at 310–330 nm on excitation at 280 nm while in the completely unfolded state (i.e., in 6 M guanidine HCl) the tryptophan fluorescence is almost completely quenched [7]. This apoamicyanin form was shown by x-ray crystallography to have a structure essentially identical to that of holoamicyanin except for the absence of copper [10]. As such, this apoamicyanin form is designated “fully folded” apoamicyanin. These results were reproduced in the current study and compared with the level of fluorescence exhibited by the apoamicyanin form generated by denaturation in guanidine HCl followed by renaturation by removal of guanidine HCl by dialysis (Fig. 1). It is evident that this apoamicyanin form exhibits a level of fluorescence intermediate between that of fully folded apoamicyanin and completely unfolded apoamicyanin. As such, this form is designated “partially folded” apoamicyanin.
Fig. 1.
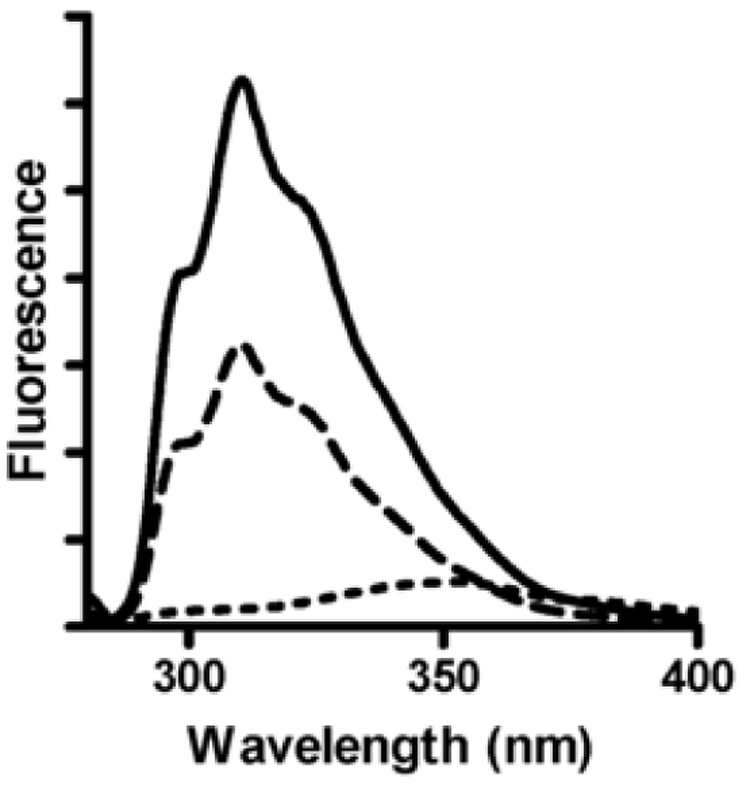
Fluorescence spectra of apoamicyanins. Each spectrum was recorded with the same concentration of protein and buffer backgrounds were corrected. Fully folded apoamicyanin (solid line) was recorded in 100 mM potassium phosphate, pH 7.5, partially folded apoamicyanin (dashed line) was recorded in 250 mM ammonium acetate, pH 8.0, and fully unfolded apoamicyanin (dotted line) was recorded in 10 mM HEPES, pH 8.0, in the presence of 6 M guanidine hydrochloride. Spectra were recorded at 15 °C with an excitation wavelength of 280 nm.
Similar studies were performed with the different forms of M98Q amicyanin and yielded qualitatively similar results (not shown). Quantitative interpretation was complicated by the fact that the apo forms of M98Q amicyanin were more prone to irreversible denaturation than native apoamicyanin. This is reflected in the fact that the reconstitution of M98Q apoamicyanin routinely yielded less than 100% holoprotein; typically around 70% (Table 1).
Table 1.
Metal content and spectroscopic properties of reconstituted amicyanins
| Molar ratio Zn:Cu | Metal Contenta |
Relative A595/A280b |
||
|---|---|---|---|---|
| Native | M98Q | Native | M98Q | |
| Reconstituted from folded apoamicyanin | ||||
| 0:1 | 0.97 Cu | 0.76 Cu | 1.0 | 0.68 |
| 0.00 Zn | 0.00 Zn | |||
| 1:1 | 0.97 Cu | 0.59 Cu | 0.98 | 0.63 |
| 0.20 Zn | 0.22 Zn | |||
| 7:1 | 0.87 Cu | 0.36 Cu | 0.81 | 0.31 |
| 0.40 Zn | 1.09 Zn | |||
| Reconstituted from partially folded apoamicyanin | ||||
| 0:1 | 0.92 Cu | 0.77 Cu | 0.95 | 0.84 |
| 0.00 Zn | 0.00 Zn | |||
| 1:1 | 0.92 Cu | 0.32 Cu | 0.97 | 0.38 |
| 0.69 Zn | 0.72 Zn | |||
| 7:1 | 1.02 Cu | 0.10 Cu | 0.87 | 0.13 |
| 1.12 Zn | 2.15 Zn | |||
| As isolated from cells | ||||
| 1:7c | 0.89 Cu | 0.07 Cu | 1.0 | 0.1 |
| 0.00 Zn | 0.90 Zn | |||
Metal contents are reported as per amicyanin molecule on the basis of sulfur content which is predicted from the amino acid sequence and which was also determined by ICP-OES of the same samples. The standard errors for repetitive measurement of the same samples were < 1%, and the variation in content from different preparations was approximately ± 10%.
A595/A280 values are normalized relative to that of native amicyanin with full occupancy of type 1 copper. All proteins were fully oxidized for UV-vis analysis as verified by treatment with potassium ferricyanide.
This is the ratio present in the growth medium of the E. coli cells from which the proteins are expressed in the periplasm of the cell. It was not possible to grow cells at higher concentrations of copper as this was toxic to the cells.
The relative selectivity of fully folded apoamicyanin and partially folded apoamicyanin for incorporation of Cu2+ or Zn2+ in vitro was examined (Table 1). The amount of tightly bound Cu2+ in the protein was determined by ICP-OES and by UV-visible spectroscopy. In the latter, the extent of formation of the type 1 copper was determined from the extinction coefficient at 595 nm in the visible absorption spectrum [11] of the protein after metal reconstitution. These values for reconstituted amicyanins are compared with the values for the proteins as-isolated from the cells which are grown in a medium which is supplemented with CuSO4. The molar ratio of Cu:Zn present in the growth medium of the E. coli cells from which the proteins are expressed is 7:1, as determined by ICP-OES. Since amicyanin assembles in the periplasm, which is separated from the external medium by a relatively permeable cell wall, the relative concentrations of the metals present during assembly are likely similar to that in the growth medium. Even in the presence of this excess copper, the as-isolated M98Q amicyanin contained primarily zinc with only 0.07 Cu (Table 1).
It should be noted that for the samples of native apoamicyanin that were reconstituted with Cu2+ in vitro in the presence of added Zn2+, a background of bound zinc was observed which was not present at the type 1 copper site, yet was not removed by EDTA treatment. This background zinc was more prominent in the samples which were reconstituted from partially folded apoamicyanin. The basis for the presence of this excess bound zinc is unknown. It is possible that during the unfolding process sites with an affinity for zinc are exposed and bind zinc which then becomes trapped after protein refolding. Zinc may also bind nonspecifically and become trapped within the subpopulation of protein which becomes irreversibly denatured during reconstitution of M98Q amicyanin (discussed earlier). The accuracy and validity of the amount of specifically-bound copper determined by ICP-OES is strongly supported by the close correlation with the A595/A280 ratio relative to that of native amicyanin with full occupancy of type 1 copper (Table 1). This discussion of metal selectivity will focus on the levels of incorporation of copper into the type 1 site in the absence and presence of zinc.
The effect of the extent of protein folding on the selectivity for Cu2+ may be seen in Fig. 2. In this chart the values in Table 1 are normalized to 1.0 for the copper content and A595/A280 relative to the proteins that were reconstituted with stoichiometric CuSO4 in the absence of zinc. We have previously shown that the crystal structure of the fully folded apoamicyanin is essentially identical to amicyanin [10] and that it may be readily reconstituted by the addition of stoichiometric amounts of CuSO4 [7]. Consistent with those results, in the present study native amicyanin exhibits a relatively strong preference for Cu2+ over Zn2+ incorporation in both the fully folded and partially folded states. For M98Q amicyanin a weak preference for Cu2+ over Zn2+ is observed when the metals are added to the fully folded apoprotein. These results show that for M98Q amicyanin, which is isolated as a zinc protein, the folded apoprotein with type 1 geometry established actually does prefer to incorporate copper. In contrast, the preference for Cu2+ is much weaker when the metals are added during protein folding. Thus, the metal selectivity in vivo which is strongly for Zn2+ is more closely reflected by the selectivity exhibited in vitro by partially folded apoamicyanin during refolding rather than by its final folded state (Fig. 3). The addition of metal to partially folded M98Q apoamicyanin does not precisely mimic assembly in vivo, where the selectivity for Zn2+ over Cu2+ is much stronger, and where the metal will have access to the completely unfolded apoprotein. However, the selectivity of the partially folded M98Q apoamicyanin is clearly more similar to in vivo assembly than is the addition of metal to the fully folded apoprotein.
Fig. 2.
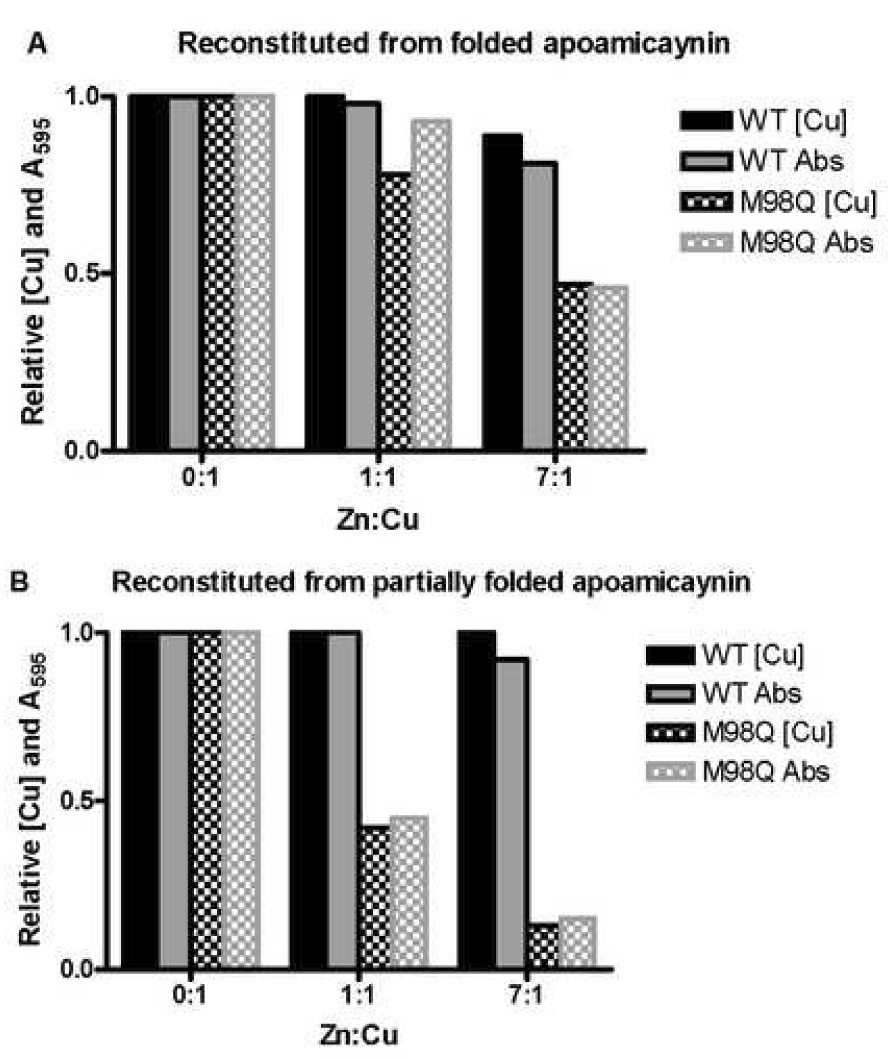
Relative extent of formation of the Cu-occupied type 1 copper site in reconstituted native and M98Q amicyanins. (A) using fully folded apoamicyanin, (B) using partially folded apoamicyanin.
Fig. 3.
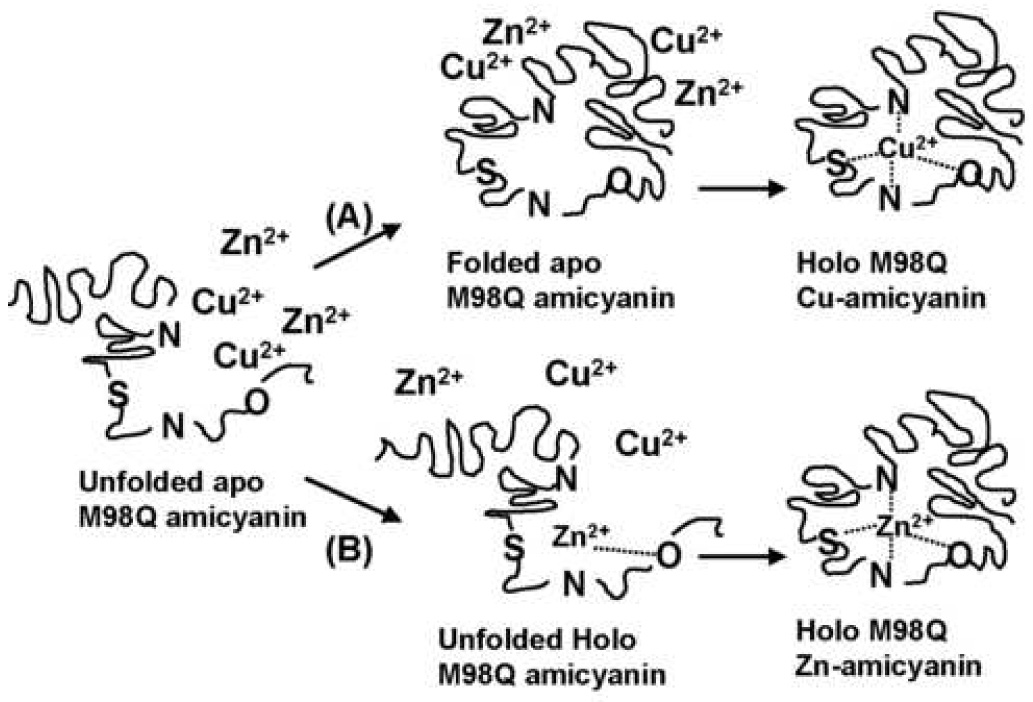
Possible mechanisms of metal incorporation into M98Q amicyanin. (A) Apoprotein enters the periplasm in its fully unfolded state, then completely folds into its final state with the geometry of the type 1 copper site established prior to binding the metal. This would result in formation of primarily copper-containing M98Q amicyanin. (B) Apoprotein enters the periplasm in its fully unfolded state, and then binds metal before completely folding into its final state. This would result in formation of primarily zinc-containing M98Q amicyanin. The data presented here indicate that mechanism B is the one which operates in vivo.
As stated earlier the effects of the analogous M98Q mutation on P. versutus amicyanin have been studied [6]. Despite the fact that these are very similar proteins from closely related organisms they seem to exhibit significant differences with respect to role of the axial Met ligand. We have previously published the crystal structure and paramagnetic NMR analysis of cobalt-substituted P. denitrificans amicyanin [12] and compared it with the paramagnetic NMR data for P. versutus cobalt-amicyanin [13]. The crystal structure of P. denitrificans cobalt-amicyanin indicates that Met98 is no longer ligated to the metal and that a water molecule is recruited from solvent to form the fourth metal ligand. The NMR data in solution also indicated that the side chain of the Met residue interacts less strongly with the metal in P. denitrificans amicyanin than in P. versutus amicyanin. The redox properties of the two amicyanins are also differentially affected by the M98Q mutation. For P. versutus amicyanin a significant reduction in Em value was observed [6] while there was no effect on P. denitrificans amicyanin [14]. Results of Met to Gln mutations of azurin have yielded results more similar to that of P. versutus amicyanin than P. denitrificans amicyanin. It is important to note that the results presented here do not necessarily suggest a disagreement with results of other studies with similar type 1 copper proteins. The differences are because P. denitrificans M98Q amicyanin has distinct properties and because in this study two different forms of apoprotein which are folded to a different extent could be studied and compared. In fact the results presented here are consistent with the report that formation of active holoazurin occurs much faster when copper is introduced at the initiation of refolding of the denatured apoazurin then when it is added to the folded apoprotein [15]. Most studies of the role of the metal in folding of blue copper proteins have used the apo form generated by denaturation in guanidine HCl followed by renaturation. On the basis of the results presented here, caution should be used in assuming the extent of folding of such species.
Two significant conclusions may be drawn from these data. The influence of the axial ligand of the type 1 copper site on metal specificity is strongest prior to the completion of protein folding and adoption of the final type 1 site geometry. This suggests that metal incorporation in vivo most likely occurs during protein folding in the periplasm (pathway B in Fig. 3) and not to a preformed type 1 site (pathway A in Fig. 3).
Acknowledgments
This work was supported by NIH grants GM-41574 (V.L.D.) and GM-56824 (J.P.H.). G.R.B. would like to acknowledge grant support from the W.M. Keck Foundation, Research Corporation CCSA #CC5939, and the Mississippi College Catalysts.
4. Abbreviations
- ICP-OES
inductively coupled plasma optical emission spectroscopy
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Adman ET. Adv. Protein Chem. 1991;42:145–197. doi: 10.1016/s0065-3233(08)60536-7. [DOI] [PubMed] [Google Scholar]
- 2.Cunane LM, Chen Z, Durley RCE, Mathews FS. Acta Cryst. 1996;D52:676–686. doi: 10.1107/S0907444996001072. [DOI] [PubMed] [Google Scholar]
- 3.Carrell CJ, Ma JK, Antholine WE, Hosler JP, Mathews FS, Davidson VL. Biochemistry. 2007;46:1900–1912. doi: 10.1021/bi0619674. [DOI] [PubMed] [Google Scholar]
- 4.Wittung-Stafshede P. Acc. Chem. Res. 2002;35:201–208. doi: 10.1021/ar010106e. [DOI] [PubMed] [Google Scholar]
- 5.Wittung-Stafshede P. Inorg. Chem. 2004;43:7926–7933. doi: 10.1021/ic049398g. [DOI] [PubMed] [Google Scholar]
- 6.Diederix RE, Canters GW, Dennison C. Biochemistry. 2000;39:9551–9560. doi: 10.1021/bi000648o. [DOI] [PubMed] [Google Scholar]
- 7.Husain M, Davidson VL, Smith AJ. Biochemistry. 1986;25:2431–2436. doi: 10.1021/bi00357a020. [DOI] [PubMed] [Google Scholar]
- 8.van Spanning RJ, Wansell CW, Reijnders WN, Oltmann LF, Stouthamer AH. FEBS Lett. 1990;275:217–220. doi: 10.1016/0014-5793(90)81475-4. [DOI] [PubMed] [Google Scholar]
- 9.Davidson VL, Jones LH, Graichen ME, Mathews FS, Hosler JP. Biochemistry. 1997;36:12733–12738. doi: 10.1021/bi971353m. [DOI] [PubMed] [Google Scholar]
- 10.Durley R, Chen L, Lim LW, Mathews FS, Davidson VL. Protein Sci. 1993;2:739–752. doi: 10.1002/pro.5560020506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Husain M, Davidson VL. J. Biol. Chem. 1985;260:14626–14629. [PubMed] [Google Scholar]
- 12.Carrell CJ, Wang X, Jones L, Jarrett WL, Davidson VL, Mathews FS. Biochemistry. 2004;43:9381–9389. doi: 10.1021/bi049635r. [DOI] [PubMed] [Google Scholar]
- 13.Salgado J, Kalverda AP, Diederix RE, Canters GW, Moratal JM, Lawler AT, Dennison C. J. Biol. Inorg. Chem. 1999;4:457–467. doi: 10.1007/s007750050332. [DOI] [PubMed] [Google Scholar]
- 14.Ma JK, Mathews FS, Davidson VL. Bochemistry. 2007;46:8561–8568. doi: 10.1021/bi700303e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pozdnyakova I, Wittung-Stafshede P. Biochemistry. 2001;40:13728–13733. doi: 10.1021/bi011591o. [DOI] [PubMed] [Google Scholar]